
Abstract
Several fungi have recently been described as capable to recombine and drive large genetic diversity in clinical samples and in the environment. Among the genotyping methods, microsatellite analysis is frequently reported as preferred for studying local epidemiology, but single nucleotide polymorphisms represent the best markers for evaluation of recombination, linkage and aneuploidy. The future of typing analyses may reside in strategies capable of cataloging the whole genome and complete microbial diversity. The present review focuses the current strategies employed for fungal genotyping and evaluation of genetic diversity, and the challenges of next generation sequencing with regard to this topic. Typing methods establish the genetic identity of fungal isolates and allow clarification of outbreaks and transmission of strains between individuals, comparison of chronic colonization versus patients carrying unrelated strains, detection of co-evolution of pathogenic and/or drug-resistant strains. The next advances in molecular mycology may revolutionize clinics and redesign concepts of microbial evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fungi play a crucial role in nature, representing typical ubiquitous microorganisms that survive and flourish with minimal nutrients. They are grouped in a fascinating and heterogenic kingdom, with expected numbers of more than 1.5 million species. Fungi are extremely useful for our industries and society; nevertheless, they can be harmful for humans and animals. In these last cases, fungal species can be associated with diseases, such as mucositis, allergies, hypersensitivity, or highly problematic invasive and systemic infections. The incidence of invasive fungal diseases is particularly high among allogeneic and autologous stem cell transplants, hematological patients and solid organ transplants, the degree and duration of immunosuppression being a critical factor for disease development [1, 2]. The patients tend to stay longer periods at clinical units due to treatments; therefore, the risk for hospital-acquired fungal infections can be much higher [3–5]. Fungi responsible for frequent infections in patients are mainly Candida and Aspergillus species, but emerging fungal species of Fusarium, Scedosporium, Trichosporon, Malassezia, and Mucorales may also represent serious threats to high-risk patients.
Candida species are commonly recovered from environmental samples, but also from human mucosal surfaces of the gastrointestinal and genitourinary tracts. The prevalence of Candida spp. is higher in pregnant, diabetic, elderly, or immunocompromised individuals, and patients receiving antibiotic or corticosteroid treatment [6]. Of clinical importance in humans are Candida glabrata, Candida tropicalis and Candida parapsilosis, but Candida albicans is principally the most prevalent, successful and pathogenic species [6, 7].
Aspergillus species are airborne and saprophytic molds that grow without any particular nutritional requirement. The species belonging to section Fumigati—principally Aspergillus fumigatus—are frequently associated with human disorders and may be responsible for severe infections in hematological and transplant patients [2, 8]. Section Fumigati contains an amorphous Aspergillus species and teleomorphic Neosartorya species [9]; sequencing analyses of the genes actin, calmodulin, ITS, rodlet A and/or β-tubulin are recommended for correct molecular identification of these species [9–11]. Conidia are usually widespread in the environment, and their inhalation facilitates the acquisition of fungal organisms that may progress to disease.
It is unquestionable that correct identification of fungal species is critical for clinical and environmental microbiology and still represents a huge challenge today. This topic, however, will not be detailed in this manuscript. The present review focuses on the strategies for genotyping and evaluating genetic diversity in distinct fungi, and the challenges that come with the recent molecular advances of next generation sequencing.
Fungal Reproduction Systems and Generation of Genetic Diversity
In nature, fungi may be sexual or asexual, or a mix of both. One fifth of the fungi have been thought to be asexual or clonal, but there is recent evidence supporting that even those species may be capable to recombine [12]. Authors claim that species that are genuinely asexual may be rare in nature, there being only a few rotifers that are accepted as truly asexual [13]. Some fungal species may have clearly defined sexual and asexual cycles, such as Aspergillus nidulans, while others face sex more rarely or it may be absent, e.g., Penicillium marneffei [14]. Asexuality does not represent a success for long-term reproduction, but it is frequent among fungi because it is requires less energy. The low number of individuals produced by sexual reproduction, the constant need to look for a mate (opportunities for sexual recombination may be limited) and the conflicts that may arise between individuals with different genetic backgrounds are reasons enough to drive microbial eukaryotes to asexual reproduction [15]. But sex may also bring important advantages, by accelerating the adaptation to adverse environments and purging deleterious mutations [15–17]. Adverse mutations can bring problems to the survival of a species, and diploid states easily cover the effect of deleterious mutations for continued existence of organisms. Fungi evolved heterothallism (mating requires two compatible partners to occur) or homothallism (self-fertile involving a single individual). Both yeast and molds have evolved specific pheromones and receptors to distinguish and identify other mating types. Recombination and chromosomal rearrangements may be common under stressful conditions, being responsible for the evolution of opportunistic pathogens, such as Candida albicans [12, 16]. The sexual reproductive mode has recently been confirmed in several Aspergillus and Penicillium species, opening new perceptions on fungi described as asexual for decades [17, 18, 19•]. Questions remain on the role of sexual mode for the emergence of strains with decreased susceptibility to the antifungals and/or increased pathogenicity, but the diversity driven from this strategy is huge and should be measured.
Methods for Evaluation of Genetic Diversity
Over the past decades, molecular biology techniques had revolutionized clinical and environmental microbiology. Molecular tools may greatly improve our understanding of fungal evolution, diversity and epidemiology. Presently, there is no single method advised for genotyping, or evaluation of genetic diversity or fungal population studies. Nevertheless, it is widely accepted that some methods are more capable to discriminate strains, such as microsatellite analysis being useful for local epidemiology and diversity testing on these collections. Each genotyping method or strategy presents obvious advantages and limitations that must be fully acknowledge when applied to studies (Table 1).
Electrophoretic Methods
Electrophoretic methods have long been used for microbial strain typing; these methods were previously considered the “gold standard” for genotyping bacteria and fungi [20, 21]. They were frequently used in the past decades, due to represent inexpensive approaches that did not require complete genome sequence of fungi. Random amplified polymorphic DNA (RAPD), restriction fragment length polymorphism (RFLP), and pulsed field gel electrophoresis (PFGE) were widely used for genotyping of distinct species [21–24]. Although these methods are useful and cheap to be used for local epidemiology, they lack reproducibility and the final results are hardly comparable and transferable between laboratories. Whole-genome mapping was recently applied to a subtyping analysis of bacteria, and is a promising tool in microbiology [25]. This new restriction fragment method represents an improved alternative to RFLP and PFGE, offering a restriction map of the complete genome.
Single and Multiple Genes Sequencing Analyses
Gene sequencing analysis can be useful for distinction of strains. Internal transcriber spacer regions 1 and 2 are frequently used for identification of fungi, but these regions accumulate several polymorphisms that can be used to separate fungal strains [26–28]. Other genetic regions can occasionally be used; nevertheless, the value of a single gene is limited and the discriminatory power can be low. Therefore, multilocus sequence analysis (MLSA) and multilocus sequence typing (MLST) have been suggested as excellent alternatives for genotyping fungal species. MLSA and MLST are powerful tools for analysis of population structure and molecular epidemiology based on the genomic analysis of multiple genes. The last method, MLST, represents a standard technique that shows highly reproducible, unambiguous data and the possibility to transfer information between laboratories worldwide through a shared database (http://www.mlst.net/) [29]. Variable genetic diversity values have been reported by testing MLST in C. albicans and other Candida species, as well as in A. fumigatus [30–33]. Genotyping profiles obtained in studies conducted worldwide can be consulted at MLST databases available online; e.g., to date, 2,217 profiles have been described for C. albicans, 152 profiles for C. krusei, 389 for C. tropicalis, and 43 for A. fumigatus. MLSA and MLST strategies can become expensive and time-consuming methodologies when employed in the screening of large collections of clinical and/or environmental isolates. Other schemes based on single nucleotide polymorphisms (SNPs) have been proposed as alternatives to simplify these methodologies that target multiple genes with relevant information.
Single Nucleotide Polymorphisms (SNPs)
The SNaPshot mini-sequencing–based assay represents a fast, sensitive and robust technique executed per extension primer in the presence of the fluorescently labeled dideoxynucleotides [34]. Advantages of this assay include the possibility of targeting small PCR products (partially degraded DNA may be targeted), the absence of stutter products and the flexibility for multiplexing, as well as capability to accept the addition or replacement of markers. The limited number of markers and alleles resulting from these assays may restrict some genomic analyses. Few SNP assays have been proposed for genotyping of fungi. The diversity of Pandora neoaphidis could be evaluated by 13 SNPs located in six genomic regions; the method discriminated 15 profiles among 23 isolates representing a good genotyping strategy [35]. An assay with 100 SNPs was described for Cryptococcus gattii, for description of molecular subtypes VGIIa, VGIIb, and VGIIc and genetic diversity analysis [36•]. The SNaPAfu assay represents a panel of 20 SNPs previously identified on MLST genes that was developed for Aspergillus fumigatus detection, identification and genotyping in clinical samples [37••]. A total of 27 SNP markers were also proposed for identification and genotyping of Podosphaera plantaginis, revealing mixed infections as a main force for pathogen evolution [38]. Recently, PCR melting analyses have been tested in yeast, showing good discrimination results [39, 40], but these methods represent indirect evaluation of the sequencing data as being more susceptible to errors. SNPs represent excellent markers for evaluation of recombination, linkage and aneuploidy and their value was recently shown in Saccharomyces cerevisiae [41].
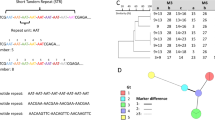
Microsatellite or Short Tandem Repeat (STR) Analyses
Microsatellite typing is frequently reported as the preferred methodology for studying local epidemiology, due to its high discriminatory power (reaching 0.995 in several panels), ease of data analyses, accessible cost and inter-laboratorial reproducibility [42]. Tri-nucleotide, tetra- nucleotide and/or penta-nucleotide motifs are ideal in order to minimize typing ambiguities caused by dinucleotide repeat motifs, which are more prone to lead to DNA polymerase slippage during amplification [43, 44•]. Microsatellite analyses can be practical and cheaper due to the ease of combining markers in multiplex systems [32, 43]. The reported lack of consistency between microsatellite results coming from different laboratories due to configuration of sequencing devices [45] can be easily compensated and corrected by the availability of allelic ladders [43, 46]. Nevertheless, no shared database exists for relevant fungi. Another limitation of microsatellites comes from their reduced power to detect recombination in populations. Multiple microsatellite panels are presently described for the most relevant clinical fungi, such as C. albicans, C. parapsilosis, C. glabrata, A. fumigatus, A. flavus, A. niger, A. terreus, and other environmental molds [32, 43, 47–53]. Occasionally, microsatellites can be useful for identification of closely related species [53–55].
Next Generation Sequencing and Other Strategies
Whole genome sequencing (WGS) is frequently described as the definitive strategy for cataloging microbial diversity, due to its capability to characterize the complete genome and numerous genetic polymorphisms (SNPs). WGS may link epidemiology to pathogen biology, genome evolution and structure, gene characterization in terms of resistance and virulence, and information from noncoding and coding regions across the entire genome [56]. In addition, it may provide relevant information regarding linkage, and recombination and chromosomal aberrations useful for pathogen population genetics [41]. Low frequency events such as horizontal gene transfer may be detected in fungi, as these phenomenon are responsible for functional innovations, niche specification, niche shift, virulence or drug resistance [57]. WGS may also allow the identification and genotyping of fungi difficult to culture under laboratorial conditions [56], revealing an enormous potential and vast applicability. Nevertheless, next generation sequencing requires heavy computer resources and specialized bioinformaticians, and the costs are still too high to be used in clinical routine [58•]. In the near future, with lower costs and practical analyses, WGS can probably replace current genotyping methods currently in use, being helpful for real-time surveillance.
There are other strategies that should be mentioned, as their potential for fungal genotyping is high and they might exemplify good strategies for strain characterization. A SNP microarray of 79 markers located on noncoding and coding regions was described as an alternative to MLST genotyping of C. albicans [59]. Nevertheless, the potential of high-resolution microarrays is much higher, as it detects numerous genomic alterations and can be used for full strain characterization. A panel was reported for the detection of 39,000 SNP alleles and 20,000 copy number variation loci across the C. albicans genome [60••]. The SNP/CGH array allows rapid and accurate detection of large-effect genome changes, but this strategy is not affordable or easily accessible to all researchers. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) employs high resolution power to evaluate the molecular weight of the various molecular products, but its applicability in laboratories is still limited by the device price and the constant need for samples with high purity [61]. MALDI-TOF MS is an automated approach that gives the results quite rapidly, and will benefit in the future from the growth of databases [62].
Advantages of Typing Systems for Clinical Studies
Typing methods may establish the genetic identity of clinical isolates, allowing the genetic characterization and distinction of strains, these data being particularly useful in clinical outbreaks with transmission of strains from one individual to others [63, 64]. Genotyping strategies allow the detection of the same fungal strain at different body locations, or clarification if patients carry unrelated strains [32, 65]. Patients with recurrent infections caused by C. albicans may present the same strain over several episodes, or the disease may be caused by distinct strains [32, 66]. Yeast genetic data may allow the detection of the source of infection, as well as be used to confirm the capability and effectiveness of antifungal treatments in such patients [23]. Occasionally, it can be found reports of patients co-infected by multiple strains of the same fungal species [32, 67••, 68]. The impacts of such presence and co-evolution are still not well known, but may facilitate the emergence of more pathogenic or drug-resistant strains. In addition, the co-presence of numerous strains may “hide” problematic strains among less-disturbing others [68]. Typing systems have been used for studying fungal evolution in cystic fibrosis (CF) and non-CF patients chronically colonized by molds [68, 69].
Genetic Diversity of A. fumigatus in Cystic Fibrosis Patients
Over several years, the presence of A. fumigatus strains has frequently been reported in CF patients with and without allergic bronchopulmorary aspergillosis (ABPA) [68, 69]. Since the early 1990s, itraconazole has been given to CF patients as oral adjunctive therapy against ABPA, and since then, it has been reported as a safe and effective alternative to glucocorticoid therapy without additional toxicity [70, 71]. In these patients are commonly described microevolution events resulting from small genetic changes and alteration of a few genetic markers; microvariation events are frequent among the isolates exposed to azole antifungals (personal unpublished data). Although sometimes useful, the administration of azole antifungals to CF patients with ABPA has been controversial. The emergence of multiple azole resistance and the fact that some patients remain colonized by the same strains even after the administration of itraconazole may bring into question the real price and efficacy of such treatments [68, 69]. Studies focusing on the evolution, mutation rate and diversity of A. fumigatus may help to clearly define the best timing for azole antifungal treatments.
Routes of Transmission and Prevention of Outbreaks
It is a fact that some strains may be endemic in some hospitals and can evolve in the hospital setting [33, 72]. Nevertheless, it is critical to distinguish epidemic from occasional strains and to determine the origins of infection. The knowledge of hospital molecular epidemiology and the transmission routes of these microbial agents in clinical environments are extremely important for infection prevention and protection of patients admitted in such units. An efficient hospital infection control program is largely dependent upon the availability of preventive procedures and routine application of tools/protocols for genotyping and molecular characterization of microorganisms [73]. Many outbreaks of infection occur through lack of prevention measures and lack of information on the source and transmission routes of pathogens. Continuous monitoring of the conditions of health care institutions is critical, particularly regarding the detection and genotyping of microorganisms in useful time and implementation of contingency or preventive plans. Indoor air and water quality should be monitored for accurate evaluation of the clinical environment and for fast intervention if necessary. The employment of highly discriminating typing methods enables tracking a fungal agent to its origin. A. fumigatus is well known by its persistence and presence in air and water samples collected from clinical wards, even highly restricted environments such as operating theatres and neutropenic patient units. Outbreaks due to particular Aspergillus strains have been largely reported [5, 50, 64, 74], and clinical directors are advised to act in order to restrict such incidents. Nonetheless solutions can be difficult to find. The genetic diversity of A. fumigatus was studied in hospital units with and without infection control policies and protections. Lower values of indoor fungi are found in protected wards, but, curiously, similar high genetic diversity indices were found in all wards [72]. Aspergillus conidia present an extraordinary ability to spread into and outside clinical units; they survive under environmentally adverse conditions and resist physical and chemical treatments [75]. International regulations that normalize indoor air quality are still lacking, as do standardized protocols for bio-contaminant sampling. A safer environment is expected in community hospitals with the consequent improvements in healthcare quality and costs.
Advantage of Genetic Diversity for Environmental Studies
The topic of evolutionary mycology and phylogenetics is facing fascinating alterations and improvements as a result of the modern developments of molecular biology. These technologies can be used to detect the presence and predominance of specific strains in particular geographical areas. Such reports are useful for tracing fungal microbes and their sources/origins. Details of microbial epidemiology and strain distribution in multiple environments can represent valuable information with great scientific and technological potential. These data directly influence other sciences, such as forensic sciences, that are focused on the description of regional, endemic and/or foreign strains [76]. The intentional and accidental carriage of fungal strains between countries and world regions is an increasing subject, disturbing some authorities and travelers. This fact has additional importance when we realize that some fungal strains can have important and destructive impacts on agriculture crops and on the storage of agriculture products. In addition, the presence of fungal strains can be associated with the damage or preservation of buildings and construction materials.
It is challenging to understand the origin of microbial diversity, the occurrence of recombination events, and in the last instance, to uncover the complete biogeography of fungal strains. Climate changes may reveal an even more dynamic map for fungi, particularly the endemic fungi. Certain diseases, such as histoplasmosis, coccidioidomycosis, blastomycosis, and penicilliosis (caused by Penicillium marneffei), have been restricted to some countries or world regions, but may be disseminated to other regions by human travelers or modifications of the local climate [77, 78]. Different evolution and adaptation patterns may be expected for these fungi, and molecular ecologists can certainly help with the development of accurate and suitable evolution models.
Current Challenges for Fungal Genetic Diversity
A new era is starting, with the application of second and third generation massive sequencing analyses to molecular mycology (Table 2). The amount of data coming from these new devices is huge, and there is no doubt that proper and innovative bioinformatics tools are needed. Therefore, extensive and central databases that are practical to use, store and download data and that allow interaction between researches remain urgent. It will be difficult for single researchers or groups dealing with massive genomic data; its association and population genetics studies thus needing shared databases in order to deposit and amplify within the scientific community the potential coming from such untreated data. MLST success in microbiology is partly justified by the ease to deposit and share data between laboratories. But previous available data and tools should not be neglected. Communication and transference of genomic information between distinct databases should be reinforced. MLST@SNaP is a recent software that allows the conversion of MLST sequences to SNP data and vice-versa [79•]. This and similar tools are critical to increase the application of technologies, close gaps worldwide, and facilitate the interaction of researchers and distinct genomic platforms.
It is also urgent to understand the interaction between microorganisms (belonging or not to the same species), communities and transmission of microorganisms with increased pathogenicity or antifungal resistance [80]. Studies in protected clinical environments are also needed, in order to develop strategies that contribute to improving the health and well-being of patients, especially those subjected to immunosuppressive treatments and longer stays at hospitals. Clinical controls, recommendations and guidelines should also be constantly developed and made available for public consulting, particularly regarding the topics of prevention and protection within the community and detailed information for health units.
Conclusions
The most successful genotyping methods share certain features, such as being unambiguous, portable, highly discriminatory and facilitating the analyses and exchange of results internationally (through online freely accessible databases). The advances of next generation sequencing may progressively turn complete fungal genomes into being even more feasible and inexpensive. Democratization of broad spectrum sequencing platforms may revolutionize molecular diagnosis and redesign concepts of microbial evolution. These technologies will certain enable the simultaneous detection, identification and molecular characterization of complete communities existing in biological samples. This fact allows better knowledge of our bodies and of the environment where we live, and may reveal strong evolutionary forces of microbial interaction. Exciting times lay ahead in molecular mycology.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Pagano L, Caira M, Candoni A, et al. The epidemiology of fungal infections in patients with hematologic malignancies: the SEIFEM-2004 study. Haematologica. 2006;91:1068–75.
Singh N, Paterson DL. Aspergillus infections in transplant recipients. Clin Microbiol Rev. 2005;18:44–69.
Araujo R, Cabral JP, Rodrigues AG. Air filtration systems and restrictive access conditions improve indoor air quality in clinical units: Penicillium as a general indicator of hospital indoor fungal levels. Am J Infect Control. 2008;36:129–34.
Alberti C, Bouakline A, Ribaud P, et al. Relationship between environmental fungal contamination and the incidence of invasive aspergillosis in haematology patients. J Hosp Infect. 2001;48:198–206.
Vonberg R, Gastmeier P. Nosocomial aspergillosis in outbreak settings. J Hosp Infect. 2006;63:246–54.
Montagna M, Lovero G, Borghi E, et al. Candidemia in intensive care unit: a nationwide prospective observational survey (GISIA-3 study) and review of the European literature from 2000 through 2013. Eur Rev Med Pharmacol Sci. 2014;18:661–74.
Pfaller M, Neofytos D, Diekema D, et al. Epidemiology and outcomes of candidemia in 3648 patients: data from the Prospective Antifungal Therapy (PATH Alliance®) registry, 2004-2008. Diagn Microbiol Infect Dis. 2012;74:323–31.
Araujo R, Carneiro A, Costa-Oliveira S, et al. Fungal infections after haematology unit renovation: evidence of clinical, environmental and economical impact. Eur J Haematol. 2008;80:436–43.
Samson R, Hong S, Peterson SW, et al. Polyphasic taxonomy of Aspergillus section Fumigati and its teleomorph Neosartorya. Stud Mycol. 2007;59:147–203.
Yaguchi T, Horie Y, Tanaka R, et al. Molecular phylogenetics of multiple genes on Aspergillus section Fumigati isolated from clinical specimens in Japan. Jpn J Med Mycol. 2007;48:37–46.
Serrano R, Gusmão L, Amorim A, Araujo R. Rapid identification of Aspergillus fumigatus within the section Fumigati. BMC Microbiol. 2011;11:82.
Butler G. Fungal sex and pathogenesis. Clin Microbiol Rev. 2010;23:140–59.
Schurko A, Neiman M, Logsdon Jr JM. Signs of sex: what we know and how we know it. Trends Ecol Evol. 2009;24:208–17.
Fisher M, Hanage WP, de Hoog S, et al. Low effective dispersal of asexual genotypes in heterogeneous landscapes by the endemic pathogen Penicillium marneffei. PLoS Pathog. 2005;1:e20.
Hadany L, Comeron JM. Why are sex and recombination so common? Ann N Y Acad Sci. 2008;1133:26–43.
Ni M, Feretzaki M, Sun S, et al. Sex in Fungi. Annu Rev Genet. 2011;45:405–30.
Dyer P, O'Gorman CM. A fungal sexual revolution: Aspergillus and Penicillium show the way. Curr Opin Microbiol. 2011;14:649–54.
O'Gorman C, Fuller HT, Dyer PS. Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature. 2009;457:471–4.
Böhm J, Hoff B, O'Gorman CM, et al. Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc Natl Acad Sci U S A. 2013;110:1476–81. Description of the first time of sexual reproduction in the environmental mold Penicillium chrysogenum.
Li W, Raoult D, Fournier P-E. Bacterial strain typing in the genomic era. FEMS Microbiol Rev. 2009;33:892–916.
Rho J, Shin JH, Song JW, et al. Molecular investigation of two consecutive nosocomial clusters of Candida tropicalis candiduria using pulsed-field gel electrophoresis. J Microbiol. 2004;42:80–6.
Khadraoui N, Kallel K, Bouchami O, et al. Pulsed Field Gel Electrophoresis types of Candida albicans isolates from an intensive care unit in a Tunisian hospital. Ann Biol Clin. 2011;69:289–94.
Saghrouni F, Ben Abdeljelil J, Boukadida J, Ben Said M. Molecular methods for strain typing of Candida albicans: a review. J Appl Microbiol. 2013;114:1559–74.
Gaitanis G, Bassukas ID, Velegraki A. The range of molecular methods for typing Malassezia. Curr Opin Infect Dis. 2009;22:119–25.
Miller J. Whole-genome mapping: a new paradigm in strain-typing technology. J Clin Microbiol. 2013;51:1066–70.
Woo P, Leung SY, To KK, et al. Internal transcribed spacer region sequence heterogeneity in Rhizopus microsporus: implications for molecular diagnosis in clinical microbiology laboratories. J Clin Microbiol. 2010;48:208–14.
Chae H, Jang GE, Kim NH, et al. Classification of Cryptococcus neoformans and yeast-like fungus isolates from pigeon droppings by colony phenotyping and ITS genotyping and their seasonal variations in Korea. Avian Dis. 2012;56:58–64.
Wang J, Ndoye M, Zhang JB, et al. Population structure and genetic diversity of the Fusarium graminearum species complex. Toxins. 2011;3:1020–37.
Jolley K, Chan M-S, Maiden M. mlstdbNet—distributed multi-locus sequence typing (MLST) databases. BMC Bioinforma. 2004;5:86.
Bain J, Tavanti A, Davidson AD, et al. Multilocus sequence typing of the pathogenic fungus Aspergillus fumigatus. J Clin Microbiol. 2007;45:1469–77.
Odds F, Jacobsen MD. Multilocus sequence typing of pathogenic Candida species. Eukaryot Cell. 2008;7:1075–84.
Sampaio P, Gusmão L, Correia A, et al. New microsatellite multiplex PCR for Candida albicans strain typing reveals microevolutionary changes. J Clin Microbiol. 2005;43:3869–76.
Viviani M, Cogliati M, Esposto MC, et al. Four-year persistence of a single Candida albicans genotype causing bloodstream infections in a surgical ward proven by multilocus sequence typing. J Clin Microbiol. 2006;44:218–21.
Makridakis N, Reichardt J. Multiplex automated primer extension analysis: simultaneous genotyping of several polymorphisms. Biotechniques. 2001;31:1374–80.
Fournier A, Widmer F, Enkerli J. Development of a single-nucleotide polymorphism (SNP) assay for genotyping of Pandora neoaphidis. Fungal Biol. 2010;114:498–506.
Gillece J, Schupp JM, Balajee SA, et al. Whole genome sequence analysis of Cryptococcus gattii from the pacific northwest reveals unexpected diversity. PLoS One. 2011;6:e28550. Genetic diversity of Cryptococcus gattii was studied by whole genome sequence analysis; a group of single nucleotide polymorphims was chosen for characterization of this species.
Caramalho R, Gusmão L, Lackner M, et al. SNaPAfu: a novel single nucleotide polymorphism multiplex assay for Aspergillus fumigatus direct detection, identification and genotyping in clinical specimens. PLoS One. 2013;8:e75968. A set of 20 single nucleotide polymorphisms is proposed for the first time for detection, identification and genotyping of Aspergillus fumigatus; the markers located in coding regions are suggested for population structure studies of A. fumigatus.
Tollenaere C, Susi H, Nokso-Koivisto J, et al. SNP design from 454 sequencing of Podosphaera plantaginis transcriptome reveals a genetically diverse pathogen metapopulation with high levels of mixed-genotype infection. PLoS One. 2012;7:e52492.
Krawczyk B, Leibner-Ciszak J, Mielech A, et al. PCR melting profile (PCR MP)-a new tool for differentiation of Candida albicans strains. BMC Infect Dis. 2009;9:177.
Costa J, Garcia-Hermoso D, Olivi M, Cabaret O, et al. Genotyping of Candida albicans using length fragment and high-resolution melting analyses together with minisequencing of a polymorphic microsatellite locus. J Microbiol Methods. 2010;80:306–9.
Wilkening S, Tekkedil MM, Lin G, et al. Genotyping 1000 yeast strains by next-generation sequencing. BMC Genomics. 2013;14:90.
Vanhee L, Symoens F, Jacobsen MD, et al. Comparison of multiple typing methods for Aspergillus fumigatus. Clin Microbiol Infect. 2009;15:643–50.
Araujo R, Pina-Vaz C, Rodrigues AG, et al. Simple and highly discriminatory microsatellite-based multiplex PCR for Aspergillus fumigatus strain typing. Clin Microbiol Infect. 2009;15:260–6.
Ananda G, Walsh E, Jacob KD, et al. Distinct mutational behaviors differentiate short tandem repeats from microsatellites in the human genome. Genome Biol Evol. 2013;5:606–20. An excellent study on the dynamics of microsatellite markers in eukaryotes comparing distinct repeat motifs.
Pasqualotto A, Denning DW, Anderson MJ. A cautionary tale: lack of consistency in allele sizes between two laboratories for a published multilocus microsatellite typing system. J Clin Microbiol. 2007;45:522–8.
de Valk H, Meis JF, Bretagne S, et al. Interlaboratory reproducibility of a microsatellite-based typing assay for Aspergillus fumigatus through the use of allelic ladders: proof of concept. Clin Microbiol Infect. 2009;15:180–7.
Sabino R, Sampaio P, Rosado L, et al. New polymorphic microsatellite markers able to distinguish among Candida parapsilosis sensu stricto isolates. J Clin Microbiol. 2010;48:1677–82.
Neal C, Richardson AO, Hurst SF, et al. Global population structure of Aspergillus terreus inferred by ISSR typing reveals geographical subclustering. BMC Microbiol. 2011;11:203.
Abbes S, Sellami H, Sellami A, et al. Candida glabrata strain relatedness by new microsatellite markers. Eur J Clin Microbiol Infect Dis. 2012;31:83–91.
Hadrich I, Makni F, Ayadi A, Ranque S. Microsatellite typing to trace Aspergillus flavus infections in a hematology unit. J Clin Microbiol. 2010;48:2396–401.
Bahkali A, Abd-Elsalam KA, Guo JR, et al. Characterization of novel di-, tri-, and tetranucleotide microsatellite primers suitable for genotyping various plant pathogenic fungi with special emphasis on fusaria and Mycospherella graminicola. Int J Mol Sci. 2012;13:2951–64.
Benichou S, Dongo A, Henni DE, et al. Isolation and characterization of microsatellite markers from the phytopathogenic fungus Alternaria dauci. Mol Ecol Resour. 2009;9:390–2.
Esteban A, Leong SL, Hocking AD, et al. Utility of microsatellite markers and amplified fragment length polymorphism in the study of potentially ochratoxigenic black aspergilli. Curr Microbiol. 2008;57:348–55.
Almeida L, Araujo R. Highlights on molecular identification of closely related species. Infect Genet Evol. 2013;13:67–75.
Araujo R, Amorim A, Gusmão L. Diversity and specificity of microsatellites within Aspergillus section Fumigati. BMC Microbiol. 2012;12:154.
Robinson E, Walker TM, Pallen MJ. Genomics and outbreak investigation: from sequence to consequence. Genome Med. 2013;5:36.
Fitzpatrick D. Horizontal gene transfer in fungi. FEMS Microbiol Lett. 2012;329:1–8.
Sabat A, Budimir A, Nashev D, et al. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill. 2013;18:20380. Review on the genotyping and molecular epidemiology strategies presently available in clinical microbiology for bacteria and fungi.
Lott T, Scarborough RT. Development of a MLST-biased SNP microarray for Candida albicans. Fungal Genet Biol. 2008;45:803–11.
Abbey D, Hickman M, Gresham D, Berman J. High-resolution SNP/CGH microarrays reveal the accumulation of loss of heterozygosity in commonly used Candida albicans strains. G3. 2011;1:523–30. Description of microarrays including 39,000 SNP alleles and 20,000 copy number variation loci across the genome of C. albicans suitable for complete genomic characterization of this yeast.
Haff L, Smirnov I. Single-nucleotide polymorphism identification assays using a thermostable DNA polymerase and delayed extraction MALDI-TOF mass spectrometry. Genome Res. 1997;7:378–88.
Bader O. MALDI-TOF-MS-based species identification and typing approaches in medical mycology. Proteomics. 2013;13:788–99.
Ben Abdeljelil J, Saghrouni F, Emira N, et al. Molecular typing of Candida albicans isolates from patients and health care workers in a neonatal intensive care unit. J Appl Microbiol. 2011;111:1235–49.
Kidd S, Ling LM, Meyer W, et al. Molecular epidemiology of invasive aspergillosis: lessons learned from an outbreak investigation in an Australian hematology unit. Infect Control Hosp Epidemiol. 2009;30:1223–6.
Kam A, Xu J. Diversity of commensal yeasts within and among healthy hosts. Diagn Microbiol Infect Dis. 2002;43:19–28.
Odds F, Davidson AD, Jacobsen MD, et al. Candida albicans strain maintenance, replacement, and microvariation demonstrated by multilocus sequence typing. J Clin Microbiol. 2006;44:3647–58.
McManus B, Coleman DC. Molecular epidemiology, phylogeny and evolution of Candida albicans. Infect Genet Evol. 2014;21:166–78. Recent review on the epidemiology and population structure of Candida albicans; the evolution and genetic diversity of the fungus is clearly described.
Amorim A, Guedes-Vaz L, Araujo R. Susceptibility to five antifungals of Aspergillus fumigatus strains isolated from chronically colonised cystic fibrosis patients receiving azole therapy. Int J Antimicrob Agents. 2010;35:396–9.
Howard S, Cerar D, Anderson MJ, et al. Frequency and evolution of Azole resistance in Aspergillus fumigatus associated with treatment failure. Emerg Infect Dis. 2009;15:1068–76.
Hennig S, Waterhouse TH, Bell SC, et al. A d-optimal designed population pharmacokinetic study of oral itraconazole in adult cystic fibrosis patients. Br J Clin Pharmacol. 2007;63:438–50.
Stevens D, Kan VL, Judson MA, et al. Practice guidelines for diseases caused by Aspergillus. Infectious Diseases Society of America. Clin Infect Dis. 2000;30:696–709.
Araujo R, Amorim A, Gusmão L. Genetic diversity of Aspergillus fumigatus in indoor hospital environments. Med Mycol. 2010;48:832–8.
Marcel J, Alfa M, Baquero F, et al. Healthcare-associated infections: think globally, act locally. Clin Microbiol Infect. 2008;14:895–907.
Peláez T, Muñoz P, Guinea J, et al. Outbreak of invasive aspergillosis after major heart surgery caused by spores in the air of the intensive care unit. Clin Infect Dis. 2012;54:e24–31.
Araujo R, Gonçalves Rodrigues A, Pina-Vaz C. Susceptibility pattern among pathogenic species of Aspergillus to physical and chemical treatments. Med Mycol. 2006;44:439–43.
Araujo R, Amorim A, Gusmão L. Microbial forensics: do Aspergillus fumigatus strains present local or regional differentiation? Forensic Sci Int: Genet. 2009;2:297–9.
Brown J, Benedict K, Park BJ, Thompson 3rd GR. Coccidioidomycosis: epidemiology. Clin Epidemiol. 2013;5:185–97.
Ramos-e-Silva M, Lima CM, Schechtman RC, et al. Systemic mycoses in immunodepressed patients (AIDS). Clin Dermatol. 2012;30:616–27.
Soares I, Araujo R. MLST@SNaP: user-friendly software for simplification of multilocus sequence typing and dissemination of microbial population analyses. Methods Ecol Evol. 2014;5:491–4. Innovative software is proposed for conversion of multilocus sequence typing results in single nucleotide polymorphisms data and vice-versa; integration of multiple genomic platforms should be prioritized for more extensive epidemiological studies.
Vandeputte P, Ferrari S, Coste AT. Antifungal resistance and new strategies to control fungal infections. Int J Microbiol. 2012;2012:713687.
Acknowledgments
R Araujo was supported by Fundação para a Ciência e a Tecnologia (FCT) Ciência 2007 and by the European Social Fund. IPATIMUP is an associate laboratory of the Portuguese Ministry of Science, Technology and Higher Education, and is partially supported by FCT.
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
R Araujo declares no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by R Araujo involving animal and/or human subjects were performed after approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Araujo, R. Towards the Genotyping of Fungi: Methods, Benefits and Challenges. Curr Fungal Infect Rep 8, 203–210 (2014). https://doi.org/10.1007/s12281-014-0190-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12281-014-0190-1