
Abstract
The NLRP3 inflammasome is assembled and activated in certain types of myeloid cells upon sensing microbe-derived toxins or host-derived danger signals. Activation of the NLRP3 inflammasome by endogenous ligands has been discovered in various disorders, including metabolic syndrome, type 2 diabetes, atherosclerosis, gout, reperfusion injury of the heart, neurodegeneration, such as Alzheimer’s disease, chronic kidney diseases, and macular degeneration of the eyes. Despite the potential significance of the NLRP3 inflammasome in the pathogenesis of several diseases, details on the activation mechanism of the NLRP3 inflammasome by a variety of stimulators have yet to be reported. Emerging evidence suggests that mitochondrial events are associated with NLRP3 activation in disease conditions. Mitochondrial dysfunction acts upstream of NLRP3 activation by providing reactive oxygen species (ROS) to trigger NLRP3 oligomerization or by inducing α-tubulin acetylation to relocate mitochondria to the proximity of NLRP3. In addition, mitochondria work as a platform for inflammasome assembly. Mitochondrial events may also lie downstream of NLRP3 activation. While the molecular mechanisms of mitochondrial dysfunction associated with NLRP3 activation are still unclear, they may involve the perturbation of mitochondria by K+ efflux and subsequent intracellular disequilibrium. Thus, mitochondria and NLRP3 machinery appear to be closely interwoven at multiple levels.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An inflammasome is a cytoplasmic multi-protein complex composed of a sensor protein, an adaptor protein called apoptosis-associated speck-like protein, containing a caspase-recruitment domain (ASC) and pro-caspase-1, a cysteine protease (Schroder and Tschopp 2010). Inflammasomes can be classified into four major sub-families depending on the sensor molecule, such as NACHT, LRR, and PYD domains-containing protein 3 (NLRP3), NLRP1, NLR Family, CARD domain containing 4 (NLRC4), and absent in melanoma 2 (AIM2) (Schroder and Tschopp 2010). These sensor molecules are activated by diverse stimulators ranging from microbial products to host-derived danger signals, leading to the assembly of the inflammasome complex through homotypic interaction of pyrin domain (PYD) and caspase activation and recruitment domain (CARD) (Rathinam et al. 2012). Once assembled, inactive pro-caspase-1 in the inflammasome complex is auto-processed by proximity to active caspase-1, which subsequently induces the maturation of pro-interleukin-1β (pro-IL-1β) or pro-IL-18 to their active forms (Rathinam et al. 2012; Schroder and Tschopp 2010) (Fig. 1). The assembly and activation of the inflammasome complex thereby leads to the pro-inflammatory process through the production of IL-1β or IL-18. Among the inflammasomes, much attention has been given to the NLRP3 inflammasome due to its potential contribution to the pathogenesis of infectious, metabolic, and neurodegenerative diseases (Franchi et al. 2012; Henao-Mejia et al. 2014; Heneka et al. 2014).

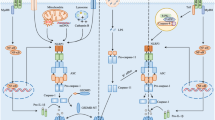
Contribution of mitochondria to NLRP3 inflammasome activation. Signal 1 confers a priming signal leading to the induction of NLRP3 and pro-IL-1β. Signal 2 allows NLRP3 activation through multiple mechanisms. Mitochondria-damaging conditions or NLRP3 stimulators, such as ATP or nigericin, induce mitochondrial dysfunction, which contributes to NLRP3 inflammasome activation through release of mtDAMP, such as mtDNA and mtROS, or by inducing abnormal mitochondrial movement. Mitochondria could also serve as a platform for the assembly of NLRP3 inflammasome through mitochondrial proteins, such as MAVS or Mfn2, and the mitochondrial lipid cardiolipin in response to viral infections or other NLRP3 activators. LRR leucine-rich repeat, NACHT NAIP, C2TA, HET-E and TP-1, PYD pyrin domain, CARD caspase activation and recruitment domain, TIR Toll/interleukin-1 receptor
NLRP3 activation is accompanied by complex cellular changes that might be causally related to inflammasome activation. Mitochondrial changes could be particularly important as inducers of NLRP3 activation amongst such intracellular events. In this review, we will focus on recent findings on the regulation of NLRP3 activation associated with mitochondrial dysfunction in both physiological and pathological contexts.
NLRP3 inflammasome
Expression
The expression of NLRP3 is mainly restricted to myeloid lineage cells (Yazdi et al. 2010). In particular, conventional dendritic cells and monocytes exhibit the highest basal expression of NLRP3 (Guarda et al. 2011). Robust expression of NLRP3 has also been observed in macrophages and neutrophils. Meanwhile, no expression of NLRP3 has been found in some myeloid cells, such as plasmacytoid dendritic cells or eosinophils (Guarda et al. 2011). NLRP3 was hardly expressed in lymphoid cells, including splenic B cells, T cells, and NK cells. Therefore, the activation of NLRP3 inflammasome may occur in myeloid cells already present in tissue or in recruited myeloid cells. Nonetheless, there is some evidence suggesting that the activation of NLRP3 inflammasome occurs in non-myeloid cell types, such as keratinocytes, T cells, and skeletal muscle cells (Martin et al. 2016; Rawat et al. 2010; Watanabe et al. 2007). It is noteworthy to mention that the expression of NLRP3 can be also induced by the engagement of toll-like receptors (TLRs) or other receptors (Bauernfeind et al. 2009) (Fig. 1). Further investigations will give more insights into the importance of cell type-specific activation of NLRP3 inflammasome.
Among the components of inflammasomes, ASC protein was first identified as a target of methylation-induced silencing-1 (TMS-1), denoting that the expression of ASC is controlled by epigenetic regulation (Conway et al. 2000). The expression of ASC was indeed silenced in many cancer cells due to an aberrant methylation of CpG island of ASC1 promoter regions (Salminen et al. 2014). However, the basal expression of ASC is relatively high in myeloid cells. Thus, it will be intriguing to investigate whether epigenetic regulation is able to affect the inflammatory responses through modulation of the expression of ASC in myeloid cells. Furthermore, the cell- or tissue-specific function of ASC was also suggested in non-myeloid cells, such as keratinocytes or helper T cells (Martin et al. 2016). Further studies will shed light on the significance of inflammasome-dependent and -independent role of ASC in tumorigenesis and autoimmune diseases. Overall, the expressions of NLRP3 and ASC are primarily restricted to myeloid cell types, such as monocytes and macrophages. In this regard, resident or recruited myeloid cells are mainly responsible for elevations in NLRP3 inflammasome activation in inflamed tissues.
NLRP3 stimulators
Although NLR was first discovered as an intracellular receptor for microbial products (Inohara et al. 2000), the direct ligand for NLRP3 has yet to be discovered. Instead, a wide range of stimulators have been shown to induce NLRP3-dependent caspase-1 activation (Rathinam et al. 2012). For instance, microbial products, such as nigericin, maitotoxin, hemolysin, or bacterial RNA, induce the robust secretion of IL-1β from lipopolysaccharide (LPS)-primed bone marrow-derived macrophages (BMDMs) in an NLRP3-dependent manner (Kanneganti et al. 2006; Mariathasan et al. 2006). In addition, endogenous substances (ATP, palmitic acid, amyloid β, monosodium urate crystals, cholesterol crystals) and exogenous crystalline particles (asbestos, silica, alum) could also act as specific triggers for the activation of NLRP3 inflammasome (Henao-Mejia et al. 2014). Besides these conventional NLRP3 stimulators, intracellular LPS from vacuolar or cytosolic bacteria has been recently shown to trigger caspase-11-dependent non-canonical activation of caspase-1. Of interest, non-canonical inflammasome activation mediated by caspase-11 also requires NLRP3 (Kayagaki et al. 2011), suggesting that caspase-11-mediated downstream targets could affect NLRP3 activation. For example, a recent study showed that the cleavage of gasdermin D by caspase-11 is required for non-canonical NLRP3 inflammasome activation by an unknown mechanism (Kayagaki et al. 2015). Further studies are necessary to clarify how caspase-11 signaling could function as an upstream event of NLRP3 activation in response to intracellular LPS stimulation.
These observations collectively suggest that NLRP3 could sense various intra- or extracellular pathogens, danger signals, or death-associated molecular patterns (DAMP) inducing pro-inflammatory responses. Nevertheless, it is still poorly understood what kind of common ligand or molecular event is responsible for the conversion of inactive NLRP3 into active form in response to such diverse stimulators.
Pathophysiological role of the NLRP3 inflammasome in response to bacterial infection or endogenous substances
Upon microbial infections, inflammasome-mediated IL-1β or IL-18 production mainly provides an innate immune protection. For example, the NLRP3 inflammasome confers an important host innate defense against several microbial pathogens, such as Streptococcus pneumonia, Group B Streptococcus, Citrobacter rodentium, Salmonella typhimurium, or influenza A virus (Broz et al. 2010; Costa et al. 2012; Liu et al. 2012). On the other hand, deficiency of NLRP3 mitigated the severity of infection caused by some pathogens, such as Staphylococcus aureus (Kebaier et al. 2012). In contrast to the potential role of NLRP3 in host defense, NLRP3 inflammasome has been frequently implicated in the progression of several diseases, including metabolic disorders or neurodegeneration. Mice lacking NLRP3, ASC or caspase-1 clearly showed an attenuated clinical manifestations of several disorders, including gout, atherosclerosis, type 2 diabetes (T2D), or Alzheimer’s disease (Duewell et al. 2010; Martinon et al. 2006; Vandanmagsar et al. 2011). Despite the importance of NLRP3 inflammasome in diverse physiological and pathological context, the underlying molecular mechanisms by which NLRP3 inflammasome is activated in such conditions remain unclear. With this, we summarize current progress in the understanding of the NLRP3 activation mechanism.
Mechanism of NLRP3 inflammasome activation
Two-step process
Unlike other inflammasomes, such as NLRC4 or AIM2, the NLRP3 inflammasome can be activated by a wide range of stimulators, including bacterial or viral infection, endogenous metabolites, or particulate substances. So far, a two-step process has been widely accepted as the activation mechanism of the NLRP3 inflammasome (Schroder and Tschopp 2010). The first step, a priming step, is triggered by engagement of TLRs with their ligands (signal 1), leading to the NF-κB-dependent transcription of NLRP3 and pro-IL-1β. Then, many of the stimulators (signal 2) mentioned above, promote the assembly and activation of the NLRP3 inflammasome in a manner dependent on K+ efflux, lysosomal rupture, or mitochondrial reactive oxygen species (mtROS) (Rathinam et al. 2012) (Fig. 1). The activation of the NLRP3 inflammasome is now considered much more complicated than previously expected, and a two-step model is not always applicable in some cases. Despite the diversity of NLRP3-activating signals, all NLRP3 stimulators ultimately lead to the molecular association of NLRP3 with ASC and the formation of ASC speck-like aggregates. This ASC speck formation is now widely used as a common indicator of inflammasome activation (Fig. 2).

Formation of ASC speck-like aggregates by NLRP3 activation. Mouse bone marrow-derived macrophages were untreated (a) or primed with 0.25 μg/ml LPS for 3 h, followed with treatment of 2.5 mM ATP for 30 min (b). The immunofluorescent staining was then performed using anti-ASC antibody. The arrow indicates the speck formation of ASC. DAPI represents nuclear fluorescence. Scale bar, 10 μm
Priming or licensing step by signal 1
Initial studies suggested that a priming step is required for the enhanced transcription of NLRP3 and pro-IL-1β. However, emerging evidence indicates that signal 1 for the priming step actually does more than transcription activation (Ghonime et al. 2014; Juliana et al. 2012). The non-transcriptional priming step, required for activation of the NLRP3 inflammasome, involves interleukin-1 receptor-associated kinase 1 (IRAK1), extracellular signal-regulated kinase (ERK), and caspase-8, which are downstream targets of TLR pathways (Allam et al. 2014; Fernandes-Alnemri et al. 2013; Ghonime et al. 2014; Gurung et al. 2014). The molecular mechanism of IRAK1 or ERK action in NLRP3 activation is still ambiguous, although it possibly involves the protein modification of NLRP3 or upstream targets of NLRP3. For instance, the deubiquitination of NLRP3 has been proposed as an essential priming step for NLRP3 activation (Juliana et al. 2012). Of note, NLRP3 deubiquitination was induced by LPS priming and also by signal 2 stimulators, such as ATP (Juliana et al. 2012). This observation indicates that it is not always appropriate to distinguish signal 1 or a priming step from signal 2. Indeed, priming signal is sufficient to promote the secretion of IL-1β in certain type of cells, such as monocytes and dendritic cells (He et al. 2013; Viganò et al. 2015). Furthermore, a recent study proposed that LPS-stimulated NF-κB signaling could suppress NLRP3 inflammasome activation through induction of p62, an autophagy receptor, which helps eliminate damaged mitochondria by mitophagy (Zhong et al. 2016). Altogether, these studies suggest a more complicated role of a priming step by signal 1. Thus, it will be necessary to investigate the impact of a priming step in NLRP3 activation at the molecular level in a more discerning way.
Activating step by signal 2
As mentioned above, the NLRP3 inflammasome is assembled and activated by extremely diverse stimulators. So far, K+ efflux has been considered as the most common and essential phenomenon for promotion of NLRP3 activation triggered by diverse NLRP3 stimulators (Muñoz-Planillo et al. 2013). Indeed, the inhibition of K+ efflux blocks NLRP3 inflammasome activation regardless of treatment with NLRP3 stimulators. However, it remains elusive how K+ efflux is induced by NLRP3-activating signal 2 and what K+ efflux does in the activation of NLRP3. One possibility that accounts for the effect of K+ efflux could be the conformational change of NLRP3 due to a decrease in intracellular K+, leading to increased electrostatic interactions between NLRP3 (Compan et al. 2012). Recent studies showed that NIMA-related kinase 7 (NEK7) is a critical upstream regulator of NLRP3 (He et al. 2016; Shi et al. 2016). Intriguingly, K+ efflux still occurred in NEK7-deficient BMDMs, despite the abrogation of inflammasome activation. Moreover, the inhibition of K+ efflux reduced the association of NLRP3 with NEK7, which suggests that NEK7 is a downstream of K+ efflux (He et al. 2016). Further studies on NEK7 and NLRP3 interaction will be required to clarify the molecular mechanism and the impact of K+ efflux on alterations of NLRP3.
In addition to K+ efflux, stimulus-specific events could also be required for the activation of NLRP3 inflammasome. For example, treatment with crystals or particulate matters induces the destabilization of lysosome, leading to the cytoplasmic release of lysosomal proteases, such as cathepsin B. Lysosomal protease in the cytoplasm is thought to confer activating signal for NLRP3 (Hornung et al. 2008), while some papers report no effect of cathepsin B deficiency on NLRP3 activation (Zhong et al. 2013). It is plausible that lysosomal destabilization or rupture by particulate substances induces the release of additional lysosomal contents, such as Ca2+ or other proteases. The molecular details for how lysosomal destabilization or lysosomotrophic agents affect the assembly and activation of NLRP3 inflammasome await further elucidation.
Another important consideration for the NLRP3 activation process is the role of extracellular osmolality. Compan et al. showed that hypotonic solution, which causes cell swelling and subsequent regulatory volume decrease (RVD), promotes the activation of NLRP3 inflammasome in a manner dependent on K+ efflux or transient receptor potential (TRP) channels (Compan et al. 2012). It was suggested that, in addition to K+ efflux, cell volume change, which also could be induced by other NLRP3 stimulators, might be an important event for NLRP3 activation, possibly through an increase in intracellular Ca2+. In contrast, a recent study suggested that hyperosmotic stress activates NLRP3 and NLRC4 inflammasome, leading to the induction of inflammatory Th17 response (Ip and Medzhitov 2015). These observations raise the possibility that NLRP3 inflammasome is a potential sensor for alterations in environmental osmolality, thereby promoting inflammatory responses. Whether cell volume change accounts for the common mechanism of NLRP3 activation requires further experimentation.
Mitochondrial control of NLRP3 inflammasome activation
Role of mitochondrial damage
Mitochondrial events appear to be closely related to NLRP3 inflammasome activation. Zhou et al. has reported that mitochondrial damage can activate the NLRP3 inflammasome through mtROS (Zhou et al. 2011) (Fig. 1). Following studies also supported their notion that the scavenging of mtROS efficiently suppresses activation of the NLRP3 inflammasome (Ip and Medzhitov 2015; Kim et al. 2014b). Consistent with these observations, impaired mitophagy has been shown to enhance activation of the NLRP3 inflammasome due to a failure to clear damaged mitochondria. In addition to the mtROS, mitochondrial DNA (mtDNA), released into cytoplasm from the damaged mitochondria, has also been proposed to act as a mitochondrial danger signal, promoting the activation of NLRP3 through its direct association with NLRP3 (Nakahira et al. 2011) (Fig. 1). These findings support that the dysfunction of mitochondria could facilitate the assembly of the NLRP3 inflammasome.
Although recent evidence suggest that mitochondrial dysfunction is closely associated with activation of the NLRP3 inflammasome (Misawa et al. 2013; Nakahira et al. 2011), there is a lack of understanding of the role of NLRP3 activators in triggering mitochondrial damage. One possible mechanism could be an increase in intracellular Ca2+ levels. It has been shown that NLRP3 stimulators, such as ATP, can induce Ca2+ influx and promote mitochondrial damage, leading to the production of mtROS and the dissipation of mitochondrial membrane potential (Lee et al. 2012; Murakami et al. 2012). Furthermore, a recent study showed that K+ efflux mediates the influx of Ca2+, which causes mitochondrial Ca2+ overload, resulting in mitochondrial dysfunction (Yaron et al. 2015). In contrast, another study proposed that K+ efflux induces NLRP3 activation in a Ca2+ influx-independent manner (Katsnelson et al. 2015). Further studies will be required to validate the mechanistic basis and significance of Ca2+ flux in NLRP3 inflammasome activation and mitochondrial dysfunction. Recently, another type of mitochondrial change related to NLRP3 inflammasome activation was reported. Wolf et al. demonstrated that N-acetylglucosamine derived from peptidoglycan of G+ bacteria was able to activate NLRP3 by dissociating hexokinase from mitochondria outer membrane (Wolf et al. 2016), suggesting an intimate relationship between metabolic and inflammatory processes.
There are also conflicting results regarding the causality between mitochondrial damage and NLRP3 activation. A study proposed that inflammasome activation causes mitochondrial damage (Yu et al. 2014), suggesting that mitochondrial damage might be the consequence of inflammasome activation, rather than the cause. Additionally, the direct target of mtROS in NLRP3 activation is still unknown. There is a report suggesting that ROS scavengers like N-acetyl-lysine (NAC) block the transcription of NLRP3 and pro-IL-1β, but have no effect on the direct activation of NLRP3 by classical NLRP3 agonists (Bauernfeind et al. 2011). In this regard, how mitochondrial events and danger signals derived from the damaged mitochondria affect the activation of NLRP3 needs to be further elucidated.
Role of mitochondrial protein associated with NLRP3
Besides mitochondrial damage mentioned above, mitochondria alone have been shown to contribute to the activation of NLRP3 inflammasome by acting as a platform for the assembly of inflammasomes. In particular, mitochondrial anti-viral signaling protein (MAVS) or mitofusin 2 (Mfn2) was suggested to recruit NLRP3 to mitochondria in response to viral infection or NLRP3 stimulators (Ichinohe et al. 2013; Subramanian et al. 2013) (Fig. 1). Of more interest, MAVS could form prion-like aggregates via engaging RNA-trapped retinoic acid-inducible gene I (RIG-I) in RIG-I-like receptor (RLR) signaling (Hou et al. 2011). These MAVS aggregates may also facilitate the oligomerization of NLRP3 to assemble the inflammasome complex. Subramanian et al. reported that knockdown of MAVS expression reduced NLRP3-dependent IL-1β secretion upon classical NLRP3 agonists, including ATP or nigericin (Subramanian et al. 2013). In contrast, other studies showed that MAVS is not required for activation of NLRP3 inflammasome by classical stimulators (Park et al. 2013). Thus, the role of MAVS in NLRP3 activation in response to non-viral stimulations is still controversial. In addition to mitochondrial proteins, mitochondrial lipid cardiolipin was also reported to interact with NLRP3, promoting its activation (Iyer et al. 2013) (Fig. 1). Based on these results, it is likely that mitochondria could serve as a signaling platform for the assembly of NLRP3 inflammasome. However, more evidence will be necessary to firmly establish the mechanism and the role of recruitment of inflammasome components to mitochondria upon stimulation with classical NLRP3 activators.
Mitochondrial dynamics
Mitochondria are dynamic organelles and undergo continuous fusion and fission. Mitochondria maintain their integrity or homeostasis by eliminating damaged mitochondria through mitochondrial fission and mitophagy (Liesa et al. 2009). Growing evidence suggests that mitochondrial dynamics have an impact on innate immune responses, particularly the anti-viral response (Yasukawa et al. 2009). Recent studies also suggest that impaired mitochondrial dynamics affect NLRP3 inflammasome activation. For instance, the knockdown of Mfn2, a critical protein for mitochondrial fusion, reduced IL-1β secretion by RNA virus infection, suggesting that mitochondrial fusion is required for robust activation of the NLRP3 inflammasome (Ichinohe et al. 2013). In accordance with this finding, aberrant mitochondrial elongation caused by a knockdown of Drp1 led to an increased sensitivity to the classical NLRP3 stimulators (Park et al. 2015). These observations suggest that elongated mitochondria could act as a favorable platform for the assembly of NLRP3 inflammasome. Conversely, another study showed reduced IL-1β secretion in Drp1-knockdown macrophages in responses to poly I:C stimulations (Wang et al. 2014). Given that mitochondrial fusion and fission are closely associated with the removal of damaged mitochondria, further systemic approaches will be required to address the role of mitochondrial dynamics and mitochondrial dysfunction in NLRP3 inflammasome activation.
Mitochondrial transport
In neurons, mitochondrial transport is critical for cellular homeostasis, and defects in mitochondrial transport have been closely associated with the pathogenesis of several neurological disorders (Sheng and Cai 2012). However, the physiological roles of mitochondrial transport in innate immune responses have not been studied in detail. Misawa et al. showed that NLRP3-stimulating conditions induced the mitochondrial transport to endoplasmic reticulum (ER) through the microtubule network (Misawa et al. 2013). Movement of mitochondria imposed the apposition of ASC, which is expressed on mitochondria, with NLRP3 localized on ER. Of note, the authors showed that NLRP3 agonists induce mitochondrial damage leading to decreases in intracellular levels of NAD+. Thereby, a reduction in NAD+ might drive the movement of mitochondria through SIRT2 inactivation and α-tubulin acetylation. It is thus possible that mitochondrial dysfunction could contribute to activation of NLRP3 by triggering aberrant mitochondrial movement (Fig. 1). It will be interesting to further delineate whether the transport of organelles associated with inflammasome activation is a common critical factor for the assembly of the NLRP3 inflammasome.
Disorders involving inflammasome activation associated with mitochondrial dysfunction
Metabolic syndrome and diabetes
Metabolic syndrome or T2D is a representative metabolic disorder characterized by inflammation in metabolic tissues, such as adipose tissue, the liver, and pancreatic islets (metainflammation). Thus, infiltration of macrophages, dendritic cells, T cells, B cells, and NK cells is frequently observed in those tissues, which is accompanied by induction of various cytokines and chemokines. For low-grade tissue inflammation, innate immune receptors and their ligands are necessary. Regarding the innate immune receptors for metainflammation, Vandanmagsar et al. reported a crucial role of NLRP3 in obesity-induced inflammation and insulin resistance. Therein, the NLRP3 inflammasome was activated in visceral adipose tissue and the liver of obese mice. Furthermore, glucose intolerance and metainflammation associated with obesity were improved by targeted disruption of NLRP3 (Vandanmagsar et al. 2011). Such changes in innate immunity were accompanied by reductions in CD4+ or CD8+ effector memory cells, although without significant changes in numbers of naïve T cells. As to the activator of NLRP3 inflammasome associated with metabolic syndrome or T2D, Wen et al. showed that palmitic acid, the most abundant saturated fatty acid in vivo, can activate NLRP3 inflammasomes (Wen et al. 2011) (Table 1). They reported that palmitic acid, together with LPS, a TLR4 agonist, leads to AMPK inhibition, decreased autophagy, and subsequently increased mtROS due to delayed mitochondrial turnover, which eventually activates NLRP3. These results suggest that NLRP3 inflammasome activation in association with mitochondrial dysfunction occurs in metabolic syndrome or T2D. Ceramide generated from fatty acids may also play a part in inflammasome activation (Vandanmagsar et al. 2011) (Table 1). Another recent paper supported the participation of mtROS due to defective mitophagy in the NLRP3 inflammasome activation by palmitic acid in conjunction with LPS. In this case, mitochondrial dysfunction was ascribed to the dysregulation of Rheb, a small GTPase that may induce mitophagy through an mTOR-independent pathway, and that of KIF5B, a microtubule motor protein (Yang et al. 2014) (Table 1). A role of mtROS in inflammasome activation in peripheral blood leukocytes from T2D patients has also been shown (Lee et al. 2013). However, since mitochondria- or ROS-independent NLRP3 activation has also been reported (Lawlor and Vince 2013), further works will be required to confirm the role of mitochondrial dysfunction or defective mitophagy in NLRP3 activation associated with metabolic syndrome or T2D.
Besides insulin target tissues, such as adipose tissue or liver, the role of NLRP3 inflammasome activation in pancreatic islets producing insulin has been demonstrated as well. Thus, mice with Nlrp3 deletion had improved metabolic profiles after a high-fat diet (HFD), which was ascribed to the reduced IL-1β production in NLRP3-knockout islets (Zhou et al. 2010). Hyperglycemia has also been reported to induce IL-1β release causing islet inflammation, which could be due to increased ROS, TXNIP induction, dissociation from thioredoxin, and increased interaction of TXNIP with NLRP3 and its activation (Table 1). However, the role for TXNIP in NLRP3 activation was not confirmed in subsequent experiments (Masters et al. 2010). Apart from the above-mentioned metabolites inducing NLRP3 activation in rodent models of T2D, there is one more candidate causing inflammasome activation in human T2D. Human islet amyloid polypeptide (hIAPP) can form oligomer and amyloid, which accounts for deposition of islet amyloid in up to 90 % of T2D patients (Kahn et al. 1999). In contrast, rodent IAPP does not form oligomer or amyloid. hIAPP oligomer, but not rodent IAPP, could activate NLRP3 inflammasome (Masters et al. 2010), suggesting greater contribution of islet inflammasome activation in human T2D, compared to rodent T2D (Table 1). The role of mitochondrial events in hIAPP oligomer-induced beta cell apoptosis has been reported (Gurlo et al. 2010); however, it is not clear whether mitochondrial events contribute to hIAPP-induced NLRP3 activation.
Since mitochondrial function and turnover of dysfunctional mitochondria are critically dependent on autophagy or mitophagy (Misawa et al. 2013), inflammasome activation associated with mitochondrial dysfunction would be accentuated in autophagy-deficient macrophages, which may lead to aggravation of metabolic profile associated with obesity and lipid injury. Indeed, recent papers have shown that insulin resistance and metainflammation are exacerbated in mice with systemic autophagy insufficiency or myeloid-specific autophagy deficiency (Lee et al. 2016; Lim et al. 2014).
Atherosclerosis
Atherosclerosis is a well-known metabolic disease with clear evidence for activation of innate immunity. Besides previously a reported role of TLR activation in atherosclerosis (Michelsen et al. 2004), involvement of NLRP3 activation by cholesterol crystals has been shown in this disorder (Duewell et al. 2010) (Table 1). Lysosomal injury after phagocytosis of cholesterol crystals appears to be responsible for NRLP3 activation. In addition to cholesterol crystals, minimally modified low-density lipoprotein (mmLDL) crystal may also participate in the priming and activation of NLRP3 (Duewell et al. 2010) (Table 1). The role for mitochondrial dysfunction in NLRP3 activation associated with atherosclerosis has been suggested in an in vitro experiment showing involvement of LOX-1, a major receptor for oxidized LDL, in the ROS production, mtDNA damage, and NLRP3 activation (Ding et al. 2014) (Table 1). However, in vivo evidence for the role of LOX-1 in NLRP3 activation associated with mitochondrial dysfunction has not been presented. A recent paper reported that fatty acid-mediated mitochondrial uncoupling facilitated NLRP3-independent IL-1α release in vitro (but not IL-1β) and atherosclerosis in vivo (Freigang et al. 2013), which is different from the previously reported role of IL-1β due to inflammasome activation in atherosclerosis (Razani et al. 2012). Further studies are warranted to confirm the role of mitochondrial events associated with inflammasome activation in atherosclerosis.
Gout
Gout is one of the classical examples of inflammasome activation as a pathogenic mechanism. Monosodium urate (MSU) crystals can activate NLRP3 in gout (Martinon et al. 2006) through lysosomal injury (Maejima et al. 2013) (Table 1). A paper reported that quenching of ROS from dysfunctional mitochondria or knockdown of VDAC1 inhibited NLRP3 activation by MSU, suggesting a crucial role for mitochondria in NLRP3 activation by MSU (Zhou et al. 2011). Another paper showed that resveratrol, a natural polyphenol compound and sirtuin modulator with an anti-inflammatory activity, ameliorated NLRP3 activation by MSU by suppressing spatial rearrangement of mitochondria caused by accumulation of acetylated α-tubulin in vitro. Resveratrol also inhibited the development of peritonitis after MSU administration (peritoneal gout) in vivo (Misawa et al. 2015), supporting the role of NLRP3 activation associated with mitochondrial dysfunction in gout.
Sepsis
Since Nod-like receptor (NLR) family members, including NLRP3, recognize diverse bacteria and IL-1β is an important mediator of septic process (Wiersinga 2011), the NLRP3 inflammasome is likely to participate in the clinical manifestation of sepsis. Consistently, IL-1β release, probably due to inflammasome activation, was noted in a cecal ligation and puncture (CLP) model, which was aggravated in autophagy-deficient mice (Nakahira et al. 2011). In vitro data have also suggested that exacerbated inflammasome activation after treatment with LPS and ATP in autophagy deficiency is due to cytoplasmic release of mtDNA in response to inflammasome activator, which may also occur in similar in vivo conditions (Nakahira et al. 2011) (Table 1). A role for UCP2-induced fatty acid synthase in NLRP3 activation during sepsis induced by CLP or LPS administration was also reported (Moon et al. 2015) (Table 1). These results suggest a potential role of NLRP3 activation in association with mitochondrial dysfunction in the clinical manifestation of sepsis, which is further supported by a paper reporting that nitric oxide (NO) downregulates NLRP3 activation by stabilization of mitochondria in vitro and protects mice from sepsis after LPS administration (Mao et al. 2013). A recent paper also reported that elimination of dysfunctional mitochondria through p62-mediated mitophagy is an important mechanism regulating NLRP3 activation in inflammatory processes (Zhong et al. 2016). This notion could explain paradoxical aggravation of septic shock in mice with macrophage-specific deletion of IKKβ, a critical mediator of inflammatory process, since p62 is a target of IKKβ/NF-κB (Greten et al. 2007). Another recent paper demonstrated that sestrin2, a stress-inducible protein and a leucine sensor, suppresses sepsis through mitophagy and subsequent downregulation of NLRP3 activation (Kim et al. 2016).
A novel mechanism of non-canonical inflammasome activation by intracellular LPS has recently been discovered, which may have relevance to host responses to LPS that manages to infiltrate into host cells. This event involves direct recognition of intracellular LPS by caspase-11, but not by TLR4 (Hagar et al. 2013; Kayagaki et al. 2013) (Table 1). The role of mitochondrial events in the non-canonical inflammasome activation by intracellular LPS was shown in Rip2-knockout cells infected with Citrobacter rodentium. In those cells, accumulation of dysfunctional mitochondria and enhanced ROS, leading to JNK activation and subsequent caspase-11 induction, were observed (Lupfer et al. 2014).
Neurodegeneration
Accumulating reports suggest that there is a potential link between the NLRP3 inflammasome and neurodegenerative disorders, such as Alzheimer’s disease (Heneka et al. 2014). Fibrillar amyloid-β, considered as the most important molecule in the pathogenesis of Alzheimer’s disease, was shown to promote the activation of NLRP3 inflammasome in brain microglia (Halle et al. 2008) (Table 1). Supporting this finding, deficiency of NLRP3 markedly reduced the processing of brain caspase-1 and the cognitive impairments of APP/PS1 mice (Heneka et al. 2013). Parkinson’s disease could be another neurodegenerative disorder associated with inflammasome activation. Aggregated α-synuclein, a critical factor in the pathogenesis of Parkinson’s disease, was reported to promote caspase-1-dependent IL-1β secretion in human monocytes (Codolo et al. 2013) (Table 1). Furthermore, administration of interleukin-1 receptor antagonist (IL-1ra, Anakinra) has significantly attenuated the loss of dopaminergic neurons in a rat model of Parkinson’s disease (Koprich et al. 2008). Consistent with these results, clinical data show a significant increase in IL-1β and IL-18 in the cerebrospinal fluid of Parkinson’s disease patients, but not in their serum, compared with control individuals (Zhang et al. 2016). While amyloid-β oligomer or α-synuclein has been reported to induce mtROS (Freeman et al. 2013; Parajuli et al. 2013), the role of mitochondrial events has not been clearly demonstrated in inflammasome activation in Alzheimer’s disease or Parkinson’s disease. Further studies will shed light on the linkage of NLRP3 inflammasome associated with mitochondrial dysfunction to neurodegenerative or neurodevelopmental disorders.
Colitis
The exact pathogenesis of inflammatory bowel disease is not clearly understood, though it is a major health threat in developed countries. Susceptibility genes include ATG16L1, IRGM, ULK1 and NOD2, suggesting dysregulation of innate immunity, autophagy, or related processes as etiologic factors (Hoefkens et al. 2013). A role of NLRP3 inflammasome therein was also suggested in genetic studies or animal experiments (Schoultz et al. 2009; Seo et al. 2015). A couple of papers described NRLP3 activation in colonic tissue after sodium dextran sulfate (DSS) administration, a commonly used animal model of inflammatory bowel disease. In this model, NLRP3 activation was associated with enhanced mtROS production (Table 1). Furthermore, mtROS-specific quencher or a natural compound downregulating mtROS generation ameliorated experimental colitis (Dashdorj et al. 2013; Guo et al. 2014a), supporting the role of mitochondrial event and associated inflammasome activation in the pathogenesis of colitis. On the other hand, a protective role of NLRP3 inflammasome against experimental colitis has also been reported, which could be due to the suppression of colon epithelial injury by IL-18 (Zaki et al. 2010).
Heart
Inflammasome activation has been shown to play a role in heart disease models such as ischemia/reperfusion (I/R) injury which could be mediated by ROS or ATP (Kawaguchi et al. 2011; Sandanger et al. 2013) (Table 1). In an animal model of diabetic cardiomyopathy, NLRP3 silencing with lentiviral expression of NLRP3-miRNA improved cardiac function. Therein, mitochondrial morphological abnormalities associated with diabetic cardiomyopathy such as destruction of myofibrils, mitochondrial swelling or disrupted cristae were ameliorated by NLRP3 silencing (Luo et al. 2014), suggesting that mitochondrial events are downstream of NLRP3 activation. However, since a protective role of NLRP3 activation in cardiac I/R model has also been reported (Sandanger et al. 2016), further studies will be required to fully understand the clinical implication of NLRP3 inflammasome in cardiac diseases.
Kidney
Kidney diseases, such as hyperuricemic nephropathy, are well recognized examples of disorders caused by inflammasome activation. Aldosterone, a contributing factor in the development of chronic kidney disease (CKD), was shown to activate inflammasome by enhancing the expression of NLRP3, ASC, pro-caspase-1, and pro-IL-1β (Ding et al. 2016) (Table 1). Genetic deletion of NLRP3 ameliorated renal injury by aldosterone, indicating the role of NLRP3 inflammasome in this condition. Aldosterone also induced mtROS generation in this condition. Furthermore, quenching of mtROS reversed NLRP3 activation by aldosterone (Ding et al. 2016), which indicates the role of mitochondrial events in NLRP3 activation. Renal tubular injury by albumin overload, a model of kidney damage caused by proteinuria, was also shown to be mediated by NLRP3 activation (Table 1). In these conditions, NLRP3 knockout attenuated not only kidney injury by albumin but also mitochondrial damage (Zhuang et al. 2014), suggesting that NLRP3 activation contributes to kidney damage by proteinuria and that mitochondrial events occur downstream of NLRP3 activation.
Allergy
NLRP3 inflammasome activated by extracellular ATP has been reported to contribute to the development of asthma (Idzko et al. 2007) (Table 1). Participation of NLRP3 activation has also been observed in airway hyperreactivity associated with obesity (Kim et al. 2014a). Furthermore, administration of IL-1β neutralizing antibody ameliorated clinical and histological manifestations of bronchial asthma, supporting the pathogenic role of inflammasome in this disorder (Kim et al. 2014b). mtROS was increased in tissues of asthmatic animal models, and an mtROS inhibitor improved diverse features and inflammasome activation in asthma, substantiating the role of mitochondrial dysfunction associated with NLRP3 activation in this disease (Kim et al. 2014b).
Eye
Inflammation and ROS damage are important components of macular degeneration (MD). A crucial role of NLRP3 inflammasome in MD induced by deficit of DICER RNase or Alu RNA exposure has been reported (Tarallo et al. 2012) (Table 1). In this model, NRLP3 activation was preceded by mtROS production and blocked by quenching of mtROS, suggesting a causative role of mitochondrial events in NLRP3 activation. Another paper reported mitochondrial damage associated with NLRP3 activation in retinal pigment epithelium of age-related MD (Wang et al. 2016). In contrast, a protective role of NLRP3 activation by drusen, a yellowish deposit in a layer of the retina, in the development of MD has also been reported (Table 1), which was attributed to the inhibition of retinal damage by IL-18 (Doyle et al. 2012).
Aging
Inflammation theory of aging posits low-grade tissue inflammation as one of the hallmarks of aging, which was termed “inflammaging” (Franceschi et al. 2007). Inflammasome activation could be a cause of ‘inflammaging’, as several waste or oxidized materials and amyloidogenic protein aggregates that accumulate in aged or senescent tissues can act as activating signals for canonical or non-canonical NLRP3 inflammasome (Salminen et al. 2012; Zanoni et al. 2016) (Table 1). A paper clearly showed the role for NLRP3 inflammasome in thymic aging or immune senescence (Youm et al. 2012). Deterioration of mitochondrial function is another hallmark of aging, the mitochondrial theory of aging (Linnane et al. 1989). Thus, it is likely that mitochondrial dysfunction contributes to NLRP3 activation in aged tissues by releasing mtROS or mitochondrial DAMP (mtDAMP) (Kapetanovic et al. 2012) (Table 1). Since both maintenance of mitochondrial function and clearance of amyloidogenic proteins protein aggregates rely on autophagy, decline of autophagic activity in aging (Sun et al. 2015) could profoundly affect inflammaging, presumably as an underlying cause of activation (Green et al. 2011). Further studies will be needed to clarify the role of inflammasome activation and autophagy deficiency in aging, since there are not much data from long-term aging studies, particularly those of mammalian system.
Inflammasome inhibitors as potential therapeutic agents
Given the role of inflammasome in the disease conditions mentioned above, much effort has been made to develop inflammasome inhibitors against those diseases.
Glyburide
Glyburide, a well-known anti-diabetic drug belonging to sulfonylurea, has been found to inhibit ATP-sensitive K+ channels by binding to sulfonylurea receptor (SUR). Lamkanfi et al. reported that glyburide inhibits NLRP3 inflammasome activation, which may have relevance to its anti-diabetic action (Lamkanfi et al. 2009). Inflammasome-inhibitory action of glyburide was downstream of P2X7, an extracellular ATP-gated ion channel involved in inflammasome activation, but was not related to ATP-sensitive K+ channels or ATPase activity of NLRP3. Likely because of inhibitory activity on NLRP3 inflammasome, glyburide was found to delay mortality in LPS-treated mice (Lamkanfi et al. 2009). Glyburide has also been shown to inhibit hIAPP-induced inflammasome activation (Masters et al. 2010). Consistently, an intermediate in the glyburide synthesis without cyclohexylurea moiety, 16673-34-0, was reported to limit ischemia–reperfusion injury of the heart by inhibiting NLRP3 inflammasome (Marchetti et al. 2014).
MCC950
MCC950 was developed as a diarylsulfonylurea-containing compound. MCC950 inhibits canonical NLRP3 inflammasome activation, NLRP3-dependent pyroptosis, and also non-canonical inflammasome activation in vitro by blocking ASC oligomerization. MCC950 exhibits the ability to attenuate the severity of experimental autoimmune encephalomyelitis, and was found to be effective in a mouse model of cryopyrin-associated periodic syndromes (CAPS) with NLRP3 mutations in vivo (Coll et al. 2015).
Parthenolide
Parthenolide is a sesquiterpene lactone with potent anti-cancer and anti-inflammatory activities. Juliana et al. reported that Parthenolide inhibits inflammasome activation by inhibiting ATPase activity of NLRP3, which was independent of its well-known inhibitory action on IKK/NF-κB (Juliana et al. 2010). Other NF-κB inhibitors that can also suppress NLRP3 inflammasome activation include Bay 11-7082 and isoliquiritigenin (ILG), while other NF-kB inhibitors do not inhibit NLRP3 inflammasome activation (Honda et al. 2014; Juliana et al. 2010). ILG also ameliorates metabolic inflammation, steatosis, and insulin resistance in mice fed HFD (Honda et al. 2014).
NZ
Olean-28,13β-olide 2 (NZ) was designed as a modification of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (CDDO), a triterpenoid showing anti-inflammatory properties. NZ was shown to suppress activation of NLRP3 inflammasome and TLR4-NF-κB in LPS-treated RAW264.7 macrophages (Xiang et al. 2015), while the mechanism of NLRP3 inflammasome activation in RAW264.7 cells lacking ASC is not clear (Pelegrin et al. 2008).
Polyenylpyrrole
Polyenylpyrroles from Gymnoascus reessli, a soil fungus, exert anti-bacterial and anti-tumor activity. Recently, polyenylpyrroles, such as auxarconjugatin A or its derivatives, were reported to block NLRP3 inflammasome activation by inhibiting ROS production and MAPK activation (Hua et al. 2013).
Rapamycin
Since autophagy is closely related to the modulation of inflammasome activation, autophagy enhancers may be able to suppress NLRP3 activation. Indeed, rapamycin, a classical activator of autophagy inhibiting mTOR, was reported to suppress IL-1β release in response to classical inflammasome activators, such as ATP or alum (Harris et al. 2011).
Andrographolide
Andrographolide is a natural diterpenoid with anti-bacterial and anti-viral activities. A recent paper reported that andrographolid can prevent DSS-induced colitis and colitis-associated cancer after azoxymethane (AOM)-DSS administration, which was attributed to the inhibition of NLRP3 inflammasome activation due to enhanced mitophagy (Guo et al. 2014b).
Resveratrol
Resveratrol (3,5,4′-trihydroxy-trans-stilbene), is a polyphenol that is abundant in grapes or berries and can activate sirtuin. Resveratrol has been shown to ameliorate NLRP3 activation by MSU and to inhibit the development of ‘peritoneal gout’ after MSU administration through downregulation of α-tubulin acetylation (Misawa et al. 2015), as described in the previous section.
Aloe
Aloe vera, a traditional herbal medicine, is known to have anti-inflammatory action. A paper has reported that Aloe vera suppresses IL-1β release in response to LPS due to downregulation of NLRP3, procaspase-1, and P2X7 in human monocyte-derived macrophages (Budai et al. 2013).
NO
NO donors, such as SNAP or GSNO, have been reported to suppress NLRP3 inflammasome activation through inhibition of ASC pyroptosome formation, which may be related to the downregulation of inflammation by NO (Mao et al. 2013).
Benzoimidazole
Fc11a-2, a novel benzoimidazole derivative discovered in a screening of synthetic chemical library, was reported to inhibit NLRP3 inflammasome activation by suppressing caspase-1 autocleavage and its release from NLRP3/ASC complex. Fc11a-2 attenuates the severity of colitis after DSS administration in vivo (Liu et al. 2013).
ω-3 fatty acid
ω-3 polyunsaturated fatty acids, such as docosahexaenoic acid (DHA) or eicosapentaenoic acid (EPA), are known to possess anti-inflammatory activity. Yan et al. showed that DHA and EPA suppress NLRP3 inflammasome activation through GPR120 and GPR40 in vitro and thereby attenuate metabolic inflammation or insulin resistance in mice fed HFD in vivo (Yan et al. 2013).
Dopamine
Dopamine is a neurotransmitter modulating diverse neuroendocrine processes, movement, and behavior. A recent paper reported that dopamine can attenuate NLRP3 inflammasome activation by producing cAMP and inducing subsequent autophagy-dependent degradation of NLRP3 through K48 ubiquitination (Yan et al. 2015). Dopamine also attenuated LPS-induced systemic inflammation and MSU-induced peritonitis in vivo.
Concluding remarks
Mitochondria are considered the power plants of cells, producing ATP and intermediary metabolites. Now we know that mitochondria actively engage in vital cellular and organismal processes, such as cell death, degeneration, aging, and ‘mitokine’ responses (Durieux et al. 2011; Kim et al. 2013; Tyynismaa et al. 2010). Recent investigations have revealed that mitochondria are hubs for innate immune processes, such as NLRP and RLR signaling. Mitochondria also provide ligands for innate immunity, such as mtDNA or formyl peptides. Furthermore, mitochondria control adaptive immunity by modulating metabolism of T or B cells. The interaction between mitochondria and NLRP3 inflammasome has a profound impact in various physiological states and pathological conditions, such as metabolic syndrome, atherosclerosis, gout, neurodegeneration, allergy, and diseases of the heart, kidneys, and eyes. Ongoing and future studies of the complex relationship between mitochondria and inflammasome or inflammation as a whole in these conditions will provide a new horizon for understanding of the advantages and sequelae of the symbiosis between mitochondria and host and will spur the development of novel therapeutic strategies against diverse diseases and aging.
References
Allam R, Lawlor KE, Yu EC, Mildenhall AL, Moujalled DM, Lewis RS, Ke F, Mason KD, White MJ, Stacey KJ, Strasser A, O’Reilly LA, Alexander W, Kile BT, Vaux DL, Vince JE (2014) Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep 15:982–990
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E (2009) NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183:787–792
Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez GVH (2011) Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol 187:613–617
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755
Budai MM, Varga A, Milesz S, Tőzsér J, Benkő S (2013) Aloe vera downregulates LPS-induced inflammatory cytokine production and expression of NLRP3 inflammasome in human macrophages. Mol Immunol 56:471–479
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS One 8:e55375
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, Vetter I, Dungan LS, Monks BG, Stutz A, Croker DE, Butler MS, Haneklaus M, Sutton CE, Núñez G, Latz E, Kastner DL, Mills KH, Masters SL, Schroder K, Cooper MA, O’Neill LA (2015) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21:263–269
Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, Montero MT, Herranz AS, Bazán E, Reimers D, Mulero V, Pelegrín P (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37:487–500
Conway KE, McConnell BB, Bowring CE, Donald CD, Warren ST, Vertino PM (2000) TMS1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res 60:6236–6242
Costa A, Gupta R, Signorino G, Malara A, Cardile F, Biondo C, Midiri A, Galbo R, Trieu-Cuot P, Papasergi S, Teti G, Henneke P, Mancuso G, Golenbock DT, Beninati C (2012) Activation of the NLRP3 inflammasome by group B streptococci. J Immunol 188:1953–1960
Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, Kim SS (2013) Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med 11:178
Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Deng X, Fan Y, Xiang D, Mehta JL (2014) LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: implications in atherogenesis. Cardiovasc Res 103:619–628
Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F (2016) Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget 7:17479–17491
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG, Humphries P (2012) NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 18:791–798
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E (2010) NLRP3 inflammasomes are required for atherosclerosis and activated by cholesterol crystals. Nature 464:1357–1362
Durieux J, Wolff S, Dillin A (2011) The cell-non-autonomous nature of elecdtron transport chain-mediated longevity. Cell 144:79–91
Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES (2013) TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol 191:3995–3999
Franceschi C, Capri M, Monti D, Giunta S, Olivieri F, Sevini F, Panourgia MP, Invidia L, Celani L, Scurti M, Cevenini E, Castellani GC, Salvioli S (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mech Age Dev 128:92–105
Franchi L, Muñoz-Planillo R, Núñez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13:325–332
Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143
Freigang S, Ampenberger F, Weiss A, Kanneganti TD, Iwakura Y, Hersberger M, Kopf M (2013) Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol 14:1045–1053
Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, Gavrilin MA, Wewers MD (2014) Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol 192:3881–3888
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112
Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Göktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O’Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M (2007) NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130:918–931
Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, Tschopp J (2011) Differential expression of NLRP3 among hematopoietic cells. J Immunol 186:2529–2534
Guo W, Liu W, Jin B, Geng J, Li J, Ding H, Wu X, Xu Q, Sun Y, Gao J (2014a) Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int Immunopharmacol 24:232–238
Guo W, Sun Y, Liu W, Wu X, Guo L, Cai P, Wu X, Wu X, Shen Y, Shu Y, Gu Y, Xu Q (2014b) Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 10:972–985
Gurlo T, Ryazantsev S, Huang CJ, Yeh MW, Reber HA, Hines OJ, O’Brien TD, Glabe CG, Butler PC (2010) Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol 176:861–869
Gurung P, Anand PK, Malireddi RK, Vande Walle L, Van Opdenbosch N, Dillon CP, Weinlich R, Green DR, Lamkanfi M, Kanneganti TD (2014) FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J Immunol 192:1835–1846
Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–1253
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-b. Nat Immunol 8:857–865
Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, Kornfeld H, Fitzgerald KA, Lavelle EC (2011) Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem 286:9587–9597
He Y, Franchi L, Nunez G (2013) TLR agonists stimulate Nlrp3-dependent IL-1beta production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J Immunol 190:334–339
He Y, Zeng MY, Yang D, Motro B, Núñez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530:354–357
Henao-Mejia J, Elinav E, Thaiss CA, Flavell RA (2014) Inflammasomes and metabolic disease. Annu Rev Physiol 76:57–78
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT (2013) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493:674–678
Heneka MT, Kummer MP, Latz E (2014) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14:463–477
Hoefkens E, Nys K, John JM, Van Steen K, Arijs I, Van der Goten J, Van Assche G, Agostinis P, Rutgeerts P, Vermeire S, Cleynen I (2013) Autophagy. Genetic association and functional role of Crohn disease risk alleles involved in microbial sensing, autophagy, and endoplasmic reticulum (ER) stress 9:2046–2055
Honda H, Nagai Y, Matsunaga T, Okamoto N, Watanabe Y, Tsuneyama K, Hayashi H, Fujii I, Ikutani M, Hirai Y, Muraguchi A, Takatsu K (2014) Isoliquiritigenin is a potent inhibitor of NLRP3 inflammasome activation and diet-induced adipose tissue inflammation. J Leukocyte Biol 96:1087–1100
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856
Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (2011) MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 146:448–461
Hua KF, Chou JC, Lam Y, Tasi YL, Chen A, Ka SM, Fang Z, Liu ML, Yang FL, Yang YL, Chiu YC, Wu SH (2013) Polyenylpyrrole derivatives inhibit NLRP3 inflammasome activation and inflammatory mediator expression by reducing reactive oxygen species production and mitogen-activated protein kinase activation. PLoS One 8:e76754
Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y (2013) Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. 110:17963–17968
Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, Hoogsteden HC, Luttmann W, Ferrari D, Di Virgilio F, Virchow JCJ, Lambrecht BN (2007) Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 8:913–919
Inohara N, Ogura Y, Chen FF, Muto A, Nuñez G (2000) Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J Biol Chem 276:2551–2554
Ip WK, Medzhitov R (2015) Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun 6:6931
Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper-Adrian V, Han R, Qiao L, Eisenbarth SC, Nauseef WM, Cassel SL, Sutterwala FS (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39:311–323
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP, Alnemri ES (2010) Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem 285:9792–9802
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287:36617–36622
Kahn SE, Andrikopoulos S, Verchere CB (1999) Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 48:241–253
Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Núñez G (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440:233–236
Kapetanovic R, Bokil NJ, Sweet MJ (2012) Innate immune perturbations, accumulating DAMPs and inflammasome dysregulation: A ticking time bomb in ageing. Ageing Res Rev 24:40–53
Katsnelson MA, Rucker LG, Russo HM, Dubyak GR (2015) K + efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194:3937–3952
Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, Hongo M, Noda T, Nakayama J, Sagara J, Taniguchi S, Ikeda U (2011) Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123:594–604
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479:117–121
Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszyński A, Forsberg LS, Carlson RW, Dixit VM (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341:1246–1249
Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671
Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL, Duncan JA (2012) Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis 205:807–817
Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim Y-N, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee M-S (2013) Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19:83–92
Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, DeKruyff RH, Umetsu DT (2014a) Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med 20:54–61
Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, Lee YC (2014b) NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis 5:e1498
Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, Moon J, Kim K, Miyawaki A, Lee MG, Shin J, Kim YS, Kim CH, Ryter SW, Choi AM, Rhee SG, Ryu JH, Yoon JH (2016) SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy in press
Koprich JB, Reske-Nielsen C, Mithal P, Isacson O (2008) Neuroinflammation mediated by IL-1beta increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. J Neuroinflammation 5:8
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the cryopirin/nalp3 inflammasome. J Cell Biol 187:61–70
Lawlor KE, Vince JE (2013) Ambiguities in NLRP3 inflammasome regulation: is there a role for mitochondria? Biochim Biophys Acta 1840:1433–1440
Lee GS, Subramanian N, Kim A, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2 + and cAMP. Nature 492:123–127
Lee H-M, Kim J-J, Kim HJ, Shong M, Ku BJ, Jo E-K (2013) Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 62:194–204
Lee H-Y, Kim J, Quan Y, Lee J-C, Kim M-S, Km S, Bae J-W, Hur KY, Lee MS (2016) Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy in press
Liesa M, Palacín M, Zorzano A (2009) Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89:799–845
Lim Y-M, Lim H-J, Hur KY, Quan W, Lee H-Y, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, Komatsu M, Lee M-S (2014) Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 5:4934
Linnane AW, Marzuki S, Ozawa T, Tanaka M (1989) Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet 8639:642–645
Liu Z, Zaki MH, Vogel P, Gurung P, Finlay BB, Deng W, Lamkanfi M, Kanneganti TD (2012) Role of inflammasomes in host defense against Citrobacter rodentium infection. J Biol Chem 287:16955–16964
Liu W, Guo W, Wu J, Luo Q, Tao F, Gu Y, Shen Y, Li J, Tan R, Xu Q, Sun Y (2013) A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol 85:1504–1512
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, Zhang M, Zhang Y, An F (2014) NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One 9:e104771
Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, Kanneganti TD (2014) Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLoS Pathog 10:e1004410
Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, Yoshimori T (2013) Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 32:2336–2347
Mao K, Chen S, Chen M, Ma Y, Wang Y, Huang B, He Z, Zeng Y, Hu Y, Sun S, Li J, Wu X, Wang X, Strober W, Chen C, Meng G, Sun B (2013) Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res 23:201–212
Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, Federici M, Van Tassell BW, Zhang S, Abbate A (2014) A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovas Pharmacol 63:316–322
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440
Martin BN, Wang C, Zhang CJ, Kang Z, Gulen MF, Zepp JA, Zhao J, Bian G, Do JS, Min B, Pavicic PG, El-Sanadi C, Fox PL, Akitsu A, Iwakura Y, Sarkar A, Wewers MD, Kaiser WJ, Mocarski ES, Rothenberg ME, Hise AG, Dubyak GR, Ransohoff RM, Li X (2016) T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol 17:583–592
Martinon F, Petrilli V, Mayor A, Tardivel A, Tshopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, Mullooly GM, Mielke LA, Harris J, Coll RC, Mills KHG, Mok KH, Newsholme P, Nunez G, Yodoi J, Kahn SE, Lavelle EC, O’Neill LAJ (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1b in type 2 diabetes. Nat Immunol 11:897–904
Michelsen KS, Wong WH, Shah PK, Zhang W, Yano J, Doherty TM, Akira S, Rajavashisth TB, Arditi M (2004) Lack of Toll-like receptor 4 or myeloid differntiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA 101:10679–10684
Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14:454–460
Misawa T, Saitoh T, Kozaki T, Park S, Takahama M, Akira S (2015) Resveratrol inhibits the acetylated α-tubulin-mediated assembly of the NLRP3-inflammasome. Int Immunol 27:425–434
Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW, Nakahira K, Choi AMK (2015) UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Diabetes Invest 125:665–680
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G (2013) K + efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38:1142–1153
Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci USA 109:11282–11287
Nakahira K, Haspel JA, Rathinam VAK, Lee S-J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AMK (2011) Autophagy proteins regulates innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 8:222–231
Parajuli B, Sonobe Y, Horiuchi H, Takeuchi H, Mizuno T, Suzumura A (2013) Oligomeric amyloid β induces IL-1β processing via production of ROS: implication in Alzheimer’s disease. Cell Death Dis 4:e975
Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, Yu JW, Alnemri ES (2013) The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191:4358–4366
Park S, Won JH, Hwang I, Hong S, Lee HK, Yu JW (2015) Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci Rep 5:15489
Pelegrin P, Barroso-Gutierrez C, Surprenant A (2008) P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol 180:7147–7157
Rathinam VA, Vanaja SK, Fitzgerald KA (2012) Regulation of inflammasome signaling. Nat Immunol 13:333–342
Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A, Hoffman EP, Nagaraju K (2010) Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol 176:2891–2900
Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF (2012) Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15:533–544
Salminen A, Ojala J, Kaarniranta K, Kauppinen A (2012) Mitochondrial dysfunction and oxidative stress activates inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci 69:2999–3013
Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K (2014) Epigenetic regulation of ASC/TMS1 expression: potential role in apoptosis and inflammasome function. Cell Mol Life Sci 71:1855–1864
Sandanger Ø, Ranheim T, Vinge LE, Bliksøen M, Alfsnes K, Finsen AV, Dahl CP, Askevold ET, Florholmen G, Christensen G, Fitzgerald KA, Lien E, Valen G, Espevik T, Aukrust P, Yndestad A (2013) The NLRP3 inflammasome is up-regulated in cardiac fibroblasts and mediates myocardial ischaemia-reperfusion injury. Cardiovasc Res 99:164–174
Sandanger Ø, Gao E, Ranheim T, Bliksøen M, Kaasbøll OJ, Alfsnes K, Nymo SH, Rashidi A, Ohm IK, Attramadal H, Aukrust P, Vinge LE, Yndestad A (2016) NLRP3 inflammasome activation during myocardial ischemia reperfusion is cardioprotective. Biochem Biophys Res Com 469:1012–1020
Schoultz I, Verma D, Halfvarsson J, Törkvist L, Fredrikson M, Sjöqvist U, Lördal M, Tysk C, Lerm M, Söderkvist P, Söderholm JD (2009) Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn’s disease in Swedish men. Am J Gastroenterol 104:1180–1188
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832
Seo SU, Kamada N, Muñoz-Planillo R, Kim YG, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, Mobley HL, Inohara N, Núñez G (2015) Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity 42:744–755
Sheng ZH, Cai Q (2012) Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Neurosci 13:77–93
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, Lyon S, Scott L, Quan J, Sun Q, Russell J, Arnett S, Jurek P, Chen D, Kravchenko VV, Mathison JC, Moresco EM, Monson NL, Ulevitch RJ, Beutler B (2016) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17:250–258
Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153:348–361
Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmström KM, Fergusson MM, Yoo YH, Combs CA, Finkel T (2015) Measuring in vivo mitophagy. Mol Cell 60:685–696
Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Núñez G, Baffi JZ, Kleinman ME, Ambati J (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149:847–859
Tyynismaa H, Carroll CJ, Raimundo N, Ahola-Erkkilä S, Wenz T, Ruhanen H, Guse K, Hemminki A, Peltola-Mjøsund KE, Tulkki V, Oresic M, Moraes CT, Pietiläinen K, Hovatta I, Suomalainen A (2010) Mitochondrial myopathy induces a starvation-like response. Hum Mol Genet 19:3948–3958
Vandanmagsar B, Youm Y-H, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit WD (2011) The NLRP3 inflammasome instigate obesity-induced inflammation and insulin resistance. Nat Med 15:179–188
Viganò E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A (2015) Human caspase-4 and caspase-5 regulate the one-step non-canonical inflammasome activation in monocytes. Nat Commun 6:8761
Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, Zhou R (2014) RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol 15:1126–1133
Wang Y, Hanus JW, Abu-Asab MS, Shen D, Ogilvy A, Ou J, Chu XK, Shi G, Li W, Wang S, Chan CC (2016) NLRP3 upregulation in retinal pigment epithelium in age-related macular degeneration. Iint J Mol Sci 17:e73
Watanabe H, Gaide O, Pertrilli V, Martinon F, Contassot E, Roques S, Kummer JA, Tshopp J, French LE (2007) Activation of the IL-1b-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 127:1256–1263
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12:408–415
Wiersinga WJ (2011) Current insights in sepsis: from pathogenesis to new treatment targets. Curr Opin Crit Care 17:480–486
Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, Cho HC, Popescu N, Coggeshall KM, Arditi M, Underhill DM (2016) Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166:624–636
Xiang P, Chen T, Mou Y, Wu H, Xie P, Lu G, Gong X, Hu Q, Zhang Y, Ji H (2015) NZ suppresses TLR4/NF-κB signalings and NLRP3 inflammasome activation in LPS-induced RAW264.7 macrophages. Inflamm Res 64:799–808
Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R (2013) Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38:1154–1163
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, Zhou R (2015) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160:62–73
Yang S, Xia C, Li S, Du L, Zhang L, Zhou R (2014) Defective mitophagy driven by dysregulation of rheb and KIF5B contributes to mitochondrial reactive oxygen species (ROS)-induced nod-like receptor 3 (NLRP3) dependent proinflammatory response and aggravates lipotoxicity. Redox Biol 3:63–71
Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F, Tian Y, Glenn HL, Meldrum DR (2015) K(+) regulates Ca(2 +) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis 6:e1964
Yasukawa K, Oshiumi H, Takeda M, Ishihara N, Yanagi Y, Seya T, Kawabata S, Koshiba T (2009) Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci Signal 2:ra47
Yazdi AS, Drexler SK, Tschopp J (2010) The role of the inflammasome in nonmyeloid cells. J Clin Immunol 30:623–627
Youm YH, Kanneganti TD, Vandanmagsar B, Zhu X, Ravussin A, Adijiang A, Owen JS, Thomas MJ, Francis J, Parks JS, Dixit VD (2012) The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Reports 1:56–68
Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, Horng T (2014) Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc Natl Acad Sci USA 111:15514–15519
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391
Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, Donado CA, Shao F, Wu H, Springstead JR, Kagan JC (2016) An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352:1232–12236
Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, Shi J, Ming J, Tian B (2016) Cdk5-dependent activation of neuronal inflammasomes in parkinson’s disease. Mov Disord 31:366–376
Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L (2013) TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 4:1611
Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M (2016) NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164:896–910
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–141
Zhou R, Yazdi AS, Menu P, Tshopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–226
Zhuang Y, Ding G, Zhao M, Bai M, Yang L, Ni J, Wang R, Jia Z, Huang S, Zhang A (2014) NLRP3 inflammasome mediates albumin-induced renal tubular injury through impaired mitochondrial function. J Biol Chem 289:25101–25111
Acknowledgements
This study was supported by Bio & Medical Technology Development Program Fund of the National Research Foundation (NRF-2015M3A9B6073846, NRF-2015M3A9B6073856), and the NRF grant (NRF-2013R1A2A2A01067985). M-SL is the recipient of the Global Research Laboratory Grant of the National Research Foundation of Korea (K21004000003-12A0500-00310) and the Ulsan National Institute of Science and Technology Research Fund (2014M3A9D8034459).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Yu, JW., Lee, MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch. Pharm. Res. 39, 1503–1518 (2016). https://doi.org/10.1007/s12272-016-0827-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0827-4