
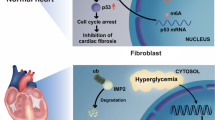
Abstract
Absent in melanoma 2(AIM2) exacerbates atherosclerosis by inflammasome assembly. However, AIM2-mediated inflammation in diabetic cardiomyopathy remains incompletely understood. Here we investigate the role of AIM2 in high glucose (HG)- and diabetes-induced inflammatory cardiomyopathy. By RNA-seq, we found that AIM2 were significantly upregulated in HG-induced macrophages, upregulation of AIM2 in cardiac infiltrating macrophages was confirmed in a high-fat diet (HFD)/streptozotocin (STZ)-induceddiabetic mouse model . Therefore, AIM2 knockout mice were constructed. Compared to WT mice, HFD/STZ-induced cardiac hypertrophy and dysfunction were significantly improved in AIM2-/- mice, despite no changes in blood glucose and body weight. Further, AIM2 deficiency inhibited cardiac recruitment of M1-macrophages and cytokine production. Mechanistically, AIM2-deficient macrophgaes reduced IL-1β and TNF-α secretion, which impaired the NLRC4/IRF1 signaling in cardiomyocytes, and reduced further recruitment of macrophages, attenuated cardiac inflammation and hypertrophy, these effects were confirmed by silencing IRF1 in WT mice, and significantly reversed by overexpression of IRF1 in AIM2-/- mice. Taken together, our findings suggest that AIM2 serves as a novel target for the treatment of diabetic cardiomyopathy.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Among the numerous risks of complications for diabetes mellitus, diabetic cardiomyopathy (DCM) has received considerable attention due to the aging worldwide in recent years [1]. Approximately 12% of diabetic patients develop myocardial dysfunction, also known as DCM [2]. The main pathology of DCM is myocardial remodeling, which includes cardiac hypertrophy and fibrosis but often occurs in the absence of coronary artery disease [3, 4]. It increases the difficulty of predicting DCM, because the development of heart failure takes a long time when cardiac ischemic symptoms occur. Therefore, the understanding of its underlying mechanism is urgent for the target therapy of DCM.
Macrophage activation has been considered so far as a main risk factor for high-fat diet (HFD)-induced DCM [5, 6], and inflammation cytokines such as interleukin 6 (IL-6) and IL1-1β play an important role in macrophage-mediated cardiac inflammation during the development of DCM. However, there are several clinical trials targeting proinflammatory cytokines have been disappointing so far [7, 8], therefore a question has been raised that understanding the upstream regulators, and the mechanism regulating cytokine production during the development of DCM may find a breakthrough for new therapeutic approaches.
The recognition of pathogen-associated molecular patterns (PAMPs) by their pathogen recognition receptors (PRRs) triggers the signaling pathways and initiates immune response, which is a crucial step in developing inflammation and cytokine release [9]. Absent in melanoma (AIM2), a member of the interferon-inducible HIN-200 protein family, a cytosolic double-stranded DNA sensor, plays a critical role in PAMPs/PRRs-mediated activation of inflammasomes and innate immune response [10]. Previous studies have revealed that AIM2 accelerates the atherosclerotic plaque progressions through double-strand DNA sensing-dependent PAMPs recognition pattern [11, 12]. AIM2 plays a similar role in the process of inflammasome assembly as NLRP3 [13]. AIM2 is activated by PAMPs/PRR recognition pattern, which forms a platform for recruiting ASC, activates serine protease caspase-1, and further leads to the maturation and secretion of IL-1β [10, 14].
Macrophages are enriched with PRRs, therefore the recruitment of macrophages is one of the main participants in inducing PAMPs/PRRs recognition [15], which promote macrophage polarization and pyroptosis, thereby enhancing antigen presenting, immune response, and inflammation expansion [11, 16]. AIM2 is highly expressed in macrophages, as an initiator triggers the formation of NRLP3/ASC inflammasome, which in turn promotes macrophage maturation, differentiation, polarization and pyroptosis [12, 17]. However, the role of AIM2 in PAMPs/PRRs-induced DCM and the potential mechanism and relationship between AIM2 activation and macrophages recruitment and cytokine release remains to be explored.
In the present study, we investigated the role of AIM2 in DCM with HFD/streptozotocin (STZ)-induced diabetic mouse model. We observed that AIM2 was significantly upregulated in STZ-induced diabetic hearts, and STZ-induced cardiac hypertrophy and dysfunction were alleviated in AIM2 gene knockout (Aim2-/-) mice. Deletion of AIM2 attenuated M1-polarization and cardiac infiltration of macrophages, which in turn reduced cardiac inflammation and DCM progression by blocking the NLRC4 inflammasome assembly and IRF1 signaling pathway in diabetic cardiomyocytes.
Methods
Animal Models
All animal care and intervention procedures in this study were approved by the University Committee on Laboratory Animals, in accordance with the guidelines of the China Council on Animal Care. C57BL/6J-Aim2em1C mice were purchased from Cyagen Biosciences (Guangzhou, China). Exons 2-3 of mouse AIM2 gene was deleted using the CRISPR/Cas9-sgRNA system, and the exons deletion in mice was confirmed by PCR analysis. The Aim2em1C mice were obtained from Aim2+/− heterozygotes, therefore, the Aim2+/− heterozygotes were crossed back to the C57BL/6J to breeding the Aim2−/− mice and their Aim2+/+ littermates as negative controls. All mice were housed in standard cages in a room with conditions of maintained temperature (~24 °C) and a 12 h/12 h light/dark cycle. Some groups of mice were fed with a high-fat diet (HFD, D12451, Shanghai Puluteng Biotechnology Co., Ltd., Shanghai, China) containing 40% fat with an energy level of 4.2 kcal/g. After one week of HFD feeding, all mice were randomly divided into 4 groups: the wild-type (WT, Aim2+/+) control, the AIM2−/− control, the WT with STZ and the AIM2−/− with STZ group. STZ (S0130, Sigma, St. Louis, MO, USA) was dissolved in citrate buffer, 0.1mol/L, pH4.5, sterile. STZ (citrate buffer served as a control) was intraperitoneally injected once a day at a dose of 40 mg/kg body weight (BW) for five consecutive days. All mice were fed with HFD continuously for 12 weeks, and body weights and blood glucose were recorded weekly during the experiment.
Echocardiographic Examination
The left ventricular systolic-diastolic function of mice was examined by echocardiographic assessment using a preclinical ultrasound system (Vevo 2100, FUJIFILM Visual Sonics, Canada). Briefly, mice were anesthetized with inhaled 2% isoflurane and were placed on a heating board, with maintained body temperature at 37 °C, heart rate at ~550 beats/min. The intercept angle between the Doppler beam line and flow direction was controlled within 50°-60°, and M-mode echocardiographic images were captured. Conventional parameters of echocardiography were collected including the left ventricular end-diastolic diameter (LVEDd), left ventricular end-systolic diameter (LVEDs), and left ventricular ejection fraction (EF). EF (%) = (LVEDV-LVESV)/ LVESV ×100%. All the measurements were double-blind.
Hematoxylin-Eosin (H&E) and Wheat Germ Agglutinin (WGA) Staining
Histopathological changes of the cross-sectional area of heart tissues (n=5 per group) were observed by H&E and WGA staining. After induction with HFD for 12 weeks, all mice were euthanized using 5% isoflurane inhalation followed by cervical dislocation, and then thoracotomy to obtain the hearts. The isolated heart tissues were washed with PBS twice to remove residual blood, and fixed with 4% paraformaldehyde. After paraffin embedding, cardiac tissue was cut into 5 μm-thick sections. Then, all sections were xylene dewaxed and alcohol gradient dehydration followed by stained with H&E or WGA dyes. After staining, we measured the mean cardiomyocyte surface area (CSA, μm2) in a high-magnification(40×) image using an inverted fluorescence microscope, with five randomly selected areas in the left ventricle tissue. The color photographs were converted into black/white mode and circled around the cell boundaries using Image J.1.48V software. For each, the CSA was quantified and analyzed to determine cardiac hypertrophy.
Cell Culture and Transwell Examination
The mouse-derived RAW264.7 cell line, H9c2 cardiomyocytes cell line, and AC16 cells, derived from the adult human ventricular tissues, were purchased from the Cell bank of Shanghai Institute of Biochemistry and Cell Biology (Chinese Academy of Sciences, China). Both cells were cultured with Dulbecco's modified Eagle medium (DMEM, Gibco, Grand Island, USA) supplemented with 10% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Gibco), and cells were cultured in a humidified incubator at 37°C, supplemented with 5% CO2 . After digesting with 0.25% trypsin, 2 × 105 RAW 264.7 cells were seeded in a 6-well plate. After culturing for 2 days, the culture medium was changed with 0.5%FBS DMEM, and the cells were transfected with recombined lentiviruse-AIM2 siRNA (20 μL, 5×108 U/ml) or negative control siRNA (20 μL, 5×108 U/ml) for 24 h. Then, the RAW264.7 cells were cultured in high glucose (25 mM D-glucose) medium or glucose-free medium, and the secretion of inflammatory cytokines were detected by ELISA in the culture supernatants. For coculture transwell system, 5 × 104 RAW 264.7 cells were seeded in the bottom layer of a 0.4 μm pore insert of 24-well transwell plates, and H9c2 or AC16 cardiomyocytes were cultured in upper layer of transwell plates with coated-matrigel. After the RAW 264.7 cells stimulating by 2.5 ng/ml IFN-γ and 200 ng/ml LPS for 48h, the cell morphology and immunofluorescence staining of anti-NLRC4 were examined in cultured H9c2 or AC16 cardiomyocytes.
In Vitro Lentiviral Vectors Construction
For AIM2 inducible knockdown, shRNA oligo sequences were designed as follows: 5’-ACUUUACAUUGAGAAAAAGGA-3’; the antisense: 5’-CUUUUUCUCAAUGUAAAGUGA-3'. The siRNA oligos were then synthesized and subcloned into the pLKO-Tet-On-shRNA-control vector (#98398, Addgene, USA) between AgeI and EcoRI restriction endonuclease sites. The recombined pLKO-Tet-On vector together with pSPAX2 and pMD2G vectors were transfected into HEK293T cells. After transfection for 72 hours, viruse packaging in the supernatant were collected by a polyethersulfone syringe filter. The filtered supernatant was ultracentrifugated at 20,000 g, 4°C for 2 h, and the viral pellets were titrated by QuickTiter™ HIV Lentivirus Quantitation Kit (VPK-108-H, Cell Biolabs Inc).
In Vivo Lentiviral-Mediated Cardiac-Specific siRNA Delivery and IRF1 Overexpression
The siRNA oligo sequences against IRF1 were designed as follows: 5’-AAUUAAUCUGCAUCUCUAGCC-3’; the antisense: 5’-CUAGAGAUGCAGAUUAAUUCC-3’. The siRNA oligos were then constructed into the pLKD-CMV-eGFP-U6-vector (Obio Technologies, Inc. Shanghai, China) A plasmid carrying a non-sense sequence was set as control. For siRNA lentivirus packaging, either Lenti-IRF1-siRNA or Lenti-control was cotransfected with Mission lentiviral packing mix, and the vector carrying IRF1 siRNA oligo sequences was recombined into the pLenti-CMV-EGFP-P2A-MCS vector (Obio Technologies, Inc. Shanghai, China). For IRF1 overexpression (IRF1-OE), the full-length of IRF1 plasmid was purchased from OriGene Technologies (MR225563, the ORF sequence and the cloning site of IRF1 were shown in Supplementary file-1), and the IRF1-OE plasmid was reconstructed into the pSLenti-CMV-EGFP-3xFLAG-WPRE (Obio Technologies, Inc. Shanghai, China) using the BamHI/Mlu I endonuclease cleave sites. Both viral stocks were titrated by QuickTiter™ HIV Lentivirus Quantitation Kit (VPK-108-H, Cell Biolabs Inc). For in vivo cardiac siRNA delivery, mice were anesthetized with 2% isoflurane, and the hearts were exposed via a left thoracotomy at the fifth intercostal space. Lenti-IRF1-siRNA or Lenti-control was administered by direct injection in the left ventricular free wall (three injection sites, 10 μL/site, 5×109 IU/mL). IRF1 gene silencing or IRF1 overexpression was confirmed by Western blot after virus injection for 4 days.
Fluorescence-Activated Cell Sorter (FACS) Analysis
Mice were euthanized and hearts were extensively flushed with sterile PBS to remove residual blood and peripheral cells. Cardiac tissue was excised, minced with fine scissors, and digested with collagenaseII (1.5 mg/ml Worthington Biochemical Corporation) and DNase (0.25 mg/ml Worthington Biochemical Corporation) at 37°C for 30 minutes as described previously (21). After sufficient digestion, tissue suspension was then homogenized by pipetting and subsequently filtered through a 40-μm cell strainer. Erythrocytes were lysed, and cells were washed and fixed in 10% buffered formalin according to the supplier’s instructions, and then incubated with anti-Fcγ receptors block antibody (BioLegend) for 15 min before being labeled with fluorescently conjugated antibodies. After the single-cells suspension was washed and stained with different fluorescently conjugated antibodies including: CD11b-FITC (#130-098-085, Miltenyi Biotec,), Ly6G-PerCP/Cyanine5.5 (#127615, BioLegend), CD68-Alexa Fluor® (ab312903, Abcam) and CD206-APC (#141707, BioLegend) for 30 min, and then flow cytometry and sorting were performed on a FACSAria™ flow cytometer (BD Biosciences), and the data were analyzed by FlowJo 7.6.1 software (Tree Star Inc., Ashland, OR, USA).
Immunofluorescence Staining
Frozen heart tissue was embedded in OCT compound and made into 5 μm thickness cryosection slices, which were dried at room temperature for 30 min, fixed by 4% PFA for 10 mins, and then rinsed with 1x PBS for three times. The cryosections were permeabilized with 0.1% TritonX-100 (Beyotime, Shanghai, China) for 5 min and blocked with 1.5% bovine serum albumin (BSA) for 1 h at room temperature, and then incubated with immunofluorescence antibodies, including anti-AIM2 mAb (1:500, MA5-38442, Clone3C4G11, ThermoFisher Scientific, Waltham, MA, USA); Anti-CD68 pAb (1:200; #25747 Proteintech, Wuhan, China); Anti-NLRC4 mAb (1:500, PA5-88997, ThermoFisher Scientific, Waltham, MA, USA); Anti-βactin pAb (1:200; ab8227, Abcam, Cambridge, UK). Stained section images were visualized using a fluorescence microscope with an oil immersion objective (60×, Leica, Wetzlar, Germany). The individual images were saved as tiff files, and opened with ImageJ 1.48V software. Then, converted color images to single channel images to make the background black, thereby better distinguishing fluorescent stained cells or areas. To analyze the average fluorescence intensity, we clicked on Analyze - Tools - ROI Manager in the menu bar, and added image that needs to be analyzed, set the measurement parameters, and exported the data to Excel files.
Western Blot
Total protein was extracted from heart tissue by RIPA lysis buffer (P0013C, Beyotime, Shanghai, China), protein concentration was examined by the BCA assay kit (P0010, Beyotime, Shanghai, China). Next, protein lysis (~20 μg) was run to 10% SDS-PAGE and then transferred on PVDF membrane (Sangon, Shanghai, China). The PVDF membrane was washed and blocked with 1×TBST solution contained 3% skimmed milk powder for one hour, and then incubated with the primary antibodies (including anti-AIM2 mAb, 1:1000; #53491 Cell Signal Technology; Anti-NLRC4 mAb (1:500, PA5-88997, ThermoFisher Scientific, Waltham, MA, USA); anti-IRF1 mAb, 1:1000; #8478 Cell Signal Technology; anti-Nf-kB mAb, 1:1000; #8242 Cell Signal Technology) at 4 °C overnight. The blots in PVDF membrane were washed by 1×TBST for 3 times and incubated with the HRP-conjugated antibodies for 30 min at room temperature. The blots were exposed using the ECL chemiluminescence reagent (ThermoFisher Scientific, Waltham, MA, USA) and visualized using ChemiDoc Imaging Systems (Bio-Rad, Laboratories, Inc. USA).
Quantitative Real-Time-PCR
Total RNA from heart tissue was purified using Trizol reagent (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. The concentration, purity, and amount of total RNA were quantified using the Nano-Drop ND-1000 Ultraviolet Spectrophotometer. The enriched mRNA was fragmented and reverse transcription into cDNA was performed using a PrimeScript™ 1st Strand cDNA Synthesis Kit (Takara Biomedical Technology, Beijing Co., Ltd). cDNA fragments were amplified with specific primers and SYBR green indicator (4913914001, Roche, USA) on a Real-Time PCR System (Bio-Rad, Laboratories, Inc. USA). Quantitative PCR for each pair of primers was repeated three times. The cDNA levels of target transcripts were analyzed by using the ΔΔCT method and normalized to the house-keeping β-actin. The primers used for gene expression were listed in Supplementary Table 1.
RNA Sequencing Data Analysis
Total RNA from control or HG-induced macrophages were isolated and purified using TRIzol reagent (Invitrogen, ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. RNA integrity was assessed by an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) and checked by agarose gel electrophoresis. Then the enriched mRNA was digested into short fragments and reversly transcribed into cDNA using NEBNext® Ultra™ RNA Library Prep Kit (NEB #7530, New England Biolabs, MA, USA). The purified double-stranded cDNA fragments were end repaired, base A added, and ligated to Illumina sequencing adapters. The ligation reaction was purified with the AMPure XP Beads (1.0X), and the cDNA fragments library was sequenced using Illumina HiSeqTM 2500 by Gene Denovo Biotech (Guangzhou, China).
DEGs and Pathway Enrichment Analysis
Differential expressed RNAs were analyzed by DESeq2 software between two groups. The transcripts with the parameter of false discovery rate (FDR) lower than 0.05 and fold change ≥ 1.5 were considered significantly different. Then, the differentially expressed genes (DEGs) were chosen for Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The significantly enriched KEGG pathway in DEGs comparing to the genome background were defined by hypergeometric test. In pathway analysis plots, we reported the name of genes just for those listed at top-10 enriched terms and with an FDR < 0.05, which were displayed in the heatmap.
Cardiac Tissue ATP assay
ATP concentrations were measured using a luciferase–luciferin ATP assay kit (Beyotime, Shanghai, China) following the manufacturer’s instructions. Proteins were exacted from diabetic hearts or control hearts, and protein concentration was examined by the BCA assay kit (23225, Thermo Scientific, MA, USA). Dilute the ATP stock solution in a 1:10 ratio, and approximately per 20 μg of protein lysis was added with 100 μL ATP working solution, after incubating at room temperature for 5 min, the RLU value was measured by a luminometer. The relative ATP content was expressed as relative luminescence units (RLU)/ mg protein.
Enzyme-Linked Immunosorbent Assay (ELISA)
Cytokine levels of IL-1β, TNF-α, IL-10 and IL-13 in heart tissue homogenate were measured using the ELISA kits according to the manufacturer's protocols (Beyotime, Shanghai, China). The concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in serum samples were measured by chemistry colorimetric method using commercial kits (C009-1, C010-1, Nanjing Jiancheng, Bioengineering Institute) according to the manufacturer’s instructions.
Statistical Analyses
Unless otherwise noted, all quantitative data in this study were expressed as the mean ± SEM from at least four independent experiments. Comparisons between two groups, significance was determined using the 2-tailed Student's t-test; for comparisons more than two groups, significance was determined using one-way or two-way ANOVA with Tukey's post hoc test. A P value < 0.05 was considered significant difference. All statistical tests were performed using the Prism software (GraphPad 8.0 Software, Inc., La Jolla, CA).
Results
AIM2 is Significantly Upregulated in STZ-Induced Diabetic Heart
Considering that macrophage plays a crucial role in diabetic inflammation and cardiomyopathy, we first examined the changes in gene transcriptional profiles between high glucose (HG, 25 mM D-glucose) and PBS control induced RAW264.7 cell lines. By RNA sequencing (RNA-seq) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, the significantly enriched KEGG pathway in DEGs comparing to the genome background were determined, and we found that TNF, Nod-like receptor, Toll-like receptor and RIG-I-like receptor signaling pathways were all significantly upregulated in HG-induced macrophages compared with those in PBS-treated control macrophages (Supplementary Fig. 1). According to the bubble plot pathway analysis, we further reported the DEGs for those listed at top-10 enriched terms and with an FDR < 0.05 (Fig. 1a). We revealed that genes involved in inflammasome complex assembly, such as Nlrp3, Nlrc4, Nlrp1b and Naip2, were all markedly increased in HG-induced macrophages (Fig. 1a). In addition, cell death pathway related genes were significantly upregulated, including apoptosis associated caspase8 and caspase12, pyroptosis associated Gsdmd and Gsdme (Fig. 1a). Furthermore, AIM2, the cytosolic double-stranded DNA sensor, was also significantly increased HG-induced macrophages (Fig. 1a). Interestingly, the transcriptional changes of AIM2, as well as inflammasome complex, Nlrp3, Nlrc4, and pyroptosis molecule Gsdme, were confirmed in heart tissues of HFD-induced STZ mice (Fig. 1b-e). To confirm the role of AIM2 in diabetic heart, we further determined the protein level change of AIM2 in STZ mice after HFD feeding for 12 weeks. The result showed that AIM2 protein level was also significantly increased in STZ-induced diabetic heart compared with control heart (Fig. 1f-g). To confirm the upregulation of AIM2 in diabetic heart was due to cardiac infiltrating macrophages, we performed immunofluorescence staining on the cross-section of myocardium. AIM2 positive cells were markedly increased in myocardial tissue of STZ mice, and a good colocalization of AIM2 with CD68 indicated that the infiltrating macrophages were the main source of increased AIM2 in the diabetic heart (Fig. 1h). In addition, cardiac hypertrophy as indicated by the increase in cardiac cross sectional area (CSA) was enhanced (Fig. 1i), but cardiac tissue ATP production was decreased in diabetic hearts (Fig. 1j), these data suggested that increased cardiac infiltrating macrophages and AIM2 production might be the reason for cardiac remodeling and cardiac energy metabolic disorder in HFD-induced STZ mice.

A critical role of AIM2 in cardiac inflammation of HFD/STZ-mediated diabetic mice. The WT mice were injected with STZ (40 mg/kg) or citrate buffer (control) once a day for three consecutive days, and fed with high-fat diet for 12 weeks. a Heatmap showed those significantly upregulated genes in HG(25 mM)-induced RAW264.7 cells. b-e The upregulation of AIM2, GSDME, NLRC4 and NLRP3 were confirmed in HFD/STZ-induced diabetic hearts. f-q The protein level of AIM2 was determined and quantified in cardiac tissues of HFD/STZ-induced mice and control mice. h Representative immunofluorescence staining of AIM2 (green color) and CD68 (red color) in the heart tissues from HFD/STZ-induced mice and control mice, scale bar = 20 μm. i The cross sectional area (μm2) of cardiomyocytes was quantified. j ATP level in cardiac tissues of HFD/STZ-induced mice and control mice (n=10). Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01; *** P<0.001
AIM2 Deficiency Alleviates STZ-Induced DCM and Cardiac Dysfunction
To investigate the hypothesis whether upregulation of AIM2 in HFD/STZ hearts contribute to the development of DCM, we constructed the C57BL/6J-Aim2em1C mice in which AIM2 is conventional knockout (AIM2-/-) mice. HE staining of liver tissue sections showed no morphological or pathological changes in AIM2-/- mice after HFD/STZ for 12 weeks (Supplementary Fig. 2), consistently, both ALT and AST were not significantly changed in AIM2-/- mice compared to WT mice (Supplementary Fig. 3). The fasting blood glucose levels were rapidly increased in both AIM2-/- and WT mice within 2 weeks after STZ injection, and no significant difference was observed between AIM2-/- mice and WT mice (Fig. 2a). Despite HFD feeding, STZ-induced type-I diabetes mellitus, both AIM2-/- and WT mice showed a significant loss of weight compared with their control counterparts (Fig. 2b). Echocardiography showed that deletion of AIM2 significantly improved cardiac left ventricular (LV) function in HFD/STZ mice, however, AIM2 deficiency did not affect LV function under physiological conditions compared with their WT littermates (Fig. 2d), as indicated by decreased LVIDs (2.724 mm ± 0.105 mm vs. 2.954 mm ± 0.155 mm, P<0.001), and LVIDd (3.322 mm ± 0.197 mm vs. 3.636 mm ± 0.240 mm, P=0.0463), but increased EF value (59.682% ± 4.877% vs. 52.468% ± 5.675%, P=0.0432) (Fig. 2i-k). We next performed historical analysis, In line with the results of HFD/STZ-induced LV dysfunction and remodeling in WT mice, HE staining showed that HFD/STZ caused a robust cardiac enlargement in WT mice, cardiac hypertrophy was observed through local myocardial tissue magnification, and increase in cross sectional area (CSA) was also confirmed by WGA staining (Fig. 2c, middle panel). In addition, cardiac fibrosis was also markedly increased in HFD/STZ-induced mice compared with that in WT control mice (Fig. 2c, bottom panel). However, AIM2 deficiency significantly attenuated HFD/STZ-induced cardiac hypertrophy, reduced fibrosis area (Fig. 2e-f), meanwhile, loss of AIM2 reduced the mRNA levels of ANP and β-MHC (Fig. 2g-h), these data suggested that deletion of AIM2 could improve DCM symptom, including pathological hypertrophy, cardiac fibrosis and cardiac dysfunction in HFD/STZ-induced diabetic mice.

Diabetic cardiac hypertrophy and dysfunction were attenuated in HFD/STZ-induced AIM2-/- mice. AIM2-/- mice and WT control were randomly divided into four groups: WT+Ctrl, AIM2-/-+Ctrl, WT+STZ, AIM2-/-+STZ, STZ or citrate buffer (control) were injected within the first 2 weeks. a Blood glucose level and (b) body weight of mice from each group were recorded every two weeks. c Mice were sacrificed after HFD feeding for 12 weeks, the cross section of the hearts were stained by HE (the top panel, scale bar = 1 mm.), WGA dye (the middle panel), or Masson’s trichrome staining (the bottom panel), representative images and enlarged images of each group of mice were displayed, scale bar = 20 μm. d Representative images of M-mode echocardiography for each group of mice were shown. e The cross sectional area (μm2) of cardiomyocytes was quantified. f The percentage of fibrosis area was quantified using Image J2X. g-h The mRNA levels of ANP and β-MHC were detected by real-time PCR. i LVIDs, j LVIDd, and (k) LVEF were assessed by software (n=5). Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01; *** P<0.001
Macrophage M1 Polarization and Cardiac Inflammation were Attenuated in STZ-Induced AIM2-/- Mice
Considering that AIM2 positive macrophages are significantly increased in HFD/STZ-induced diabetic hearts, and the activation and recruitment of macrophages play an important role in promoting inflammation and remodeling in diabetic hearts, therefore we thought to measure macrophage polarization and infiltration, and cardiac inflammation in AIM2 deficient diabetic hearts. Flow cytometric analysis revealed that the proportion of cardiac infiltrating CD11b+Ly6Ghigh neutrophils was significantly decreased in STZ-induced AIM2-/- mice after HFD feeding for 12 weeks compared with those in STZ-induced WT mice (Fig. 3a-b). The sorting of macrophages were further gated by anti-CD68 antibody. Compared with WT mice, the proportion of cardiac infiltrating macrophages significantly reduced in STZ-induced AIM2-/- mice (Fig. 3c-d). Notably, macrophages transited a M1-like phonotype (the subpopulation of CD68+CD86+ cells) in WT mice (Fig. 3e-f), while it transited a M2-like phonotype (the subpopulation of CD68+CD206+ cells) in AIM2-/- mice (Fig. 3g-h). To confirm the impact of AIM2 deficiency on the phenotype of cardiac infiltrating macrophages, we further determined the transcriptional levels of M1/M2 marker genes of macrophages. Consistently, cardiac expressions of iNOS and TNF-α were significantly decreased (Fig. 3i-j), but Arg-1 and IL-10 were significantly increased in HFD/STZ-induced AIM2-/- mice compared with those in WT mice (Fig. 3k-l), indicating that loss of AIM2 prevented M1 polarization while promoted M2 polarization of macrophages in diabetic hearts. Next, we examined the levels of proinflammatory cytokines in cardiac tissues. As expected that, loss of AIM2 reduced the secretion of IL-1β and TNF-α (Fig. 3m-n), but increased the secretion of IL-10 and IL-13 in diabetic hearts (Fig. 3o-p). Collectively, these data suggested that AIM2 deficiency suppressed M1 polarization and cardiac recruitment of macrophages, meanwhile attenuated cardiac inflammation in diabetic hearts.

Deletion of AIM2 attenuated cardiac infiltrating M1-like macrophages and inflammatory response in HFD/STZ-induced mice. a-b Flow cytometric analysis of the proportions of CD11b+Ly6Glow and CD11b+Ly6Ghigh neutrophils that infiltrating in the hearts of STZ-induced AIM2-/- mice and WT mice. c-d The CD11b+Ly6Glow subset was further sorted by anti-CD68, and the proportions of Ly6GlowCD68high macrophages were quantified. e-h The proportions CD86+CD68+ (M1-like) and CD206+CD68+ (M2-like) subsets were sorted and quantified. i-l The expressions of M1-marker genes iNOS and TNF-α, M2-marker genes Arg-1 and IL-10 were determined by real-time PCR. m-p The cytokines expressed in diabetic hearts and control hearts of AIM2-/- mice were detected by ELISA assays. Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01; *** P<0.001
AIM2 Deficiency Suppresses NLRC4 Inflammasome Activation in Diabetic Heart
To confirm the role of AIM2 in the regulation of macrophage polarization, we knocked down AIM2 gene in cultured murine RAW264.7 cells using specific siRNA. The RAW264.7 macrophages were incubated with high glucose (HG, 25 mM D-glucose) or PBS for 24 hours, and then the M1/M2 markers were examined by RT-PCR. Results showed that the expressions of iNOS, TNF-α, Arg-1 and IL-10 were not changed significantly between AIM2-siRNA cells and siRNA-NC cells with PBS treatment (Fig. 4a-d). However, iNOS, TNF-α, Arg-1 and IL-10 were all markedly increased in RAW264.7 macrophages after stimulating with HG for 24 hours (Fig. 4a-d). But knocking down AIM2 significantly reduced the levels of iNOS and TNF-α (Fig. 4a-b), and increased the levels of Arg-1 and IL-10 in HG-induced RAW264.7 macrophages (Fig. 4c-d). Consistently, knocking down of AIM2 did not change the baseline levels of inflammatory cytokines in PBS-treated RAW264.7 cells, but AIM2 inhibition markedly decreased the supernatant levels of IL-1β and TNF-α, increased the supernatant levels of IL-10 and IL-13 induced by HG in cultured RAW264.7 macrophages (Fig. 4e-h). Next, we co-cultured H9c2 cardiomyocytes with HG-induced RAW264.7 macrophages for 48 hours. We found that hypertrophic response as measured by the cell surface area was significantly enhanced in H9c2 cardiomyocytes co-cultured with HG-induced RAW264.7 cells, but it was markedly reduced after knocking down AIM2 in HG-induced RAW264.7 cells (Fig. 4i-j). Similarly, cardiac hypertrophy was also observed in human-derived AC16 cardiomyocytes that co-cultured with HG-induced RAW264.7 macrophages, and this effect disappeared after co-culturing with AIM2 deficient macrophages (Supplementary Fig. 4). Of note, the expression of NLRC4 was significantly downregulated both in H9c2 and AC16 cardiomyocytes that co-cultured with HG-induced AIM2-deficient RAW264.7 macrophages as compared to those co-cultured with HG-induced WT macrophages (Fig. 4i, k), suggesting that loss of AIM2 in macrophages hindered the NLRC4 inflammasome assembly in cardiomyocytes with which being contacted.

Deletion of AIM2 reduced NLRC4 inflammasome formation in cardiomyocytes cocultured with HG-induced macrophages. a-d The RAW264.7 macrophages were stimulated with high glucose (HG, 25 mM D-glucose) or PBS for 24 hours, the mRNA levels of iNOS, TNF-α, Arg-1 and IL-10 were determined by real-time PCR. e-h The secretion of IL-1β, TNF-α, IL-10 and IL-13 were determined by ELISA. i Inflammasome formation in cardiomyocytes that cocultured with HG-induced RAW264.7 macrophages was detected by anti-NLRC4 immunofluorescence staining. j The cell surface area of cardiomyocytes from cocultured with AIM2-siRNA macrophages or with NC-siRNA macrophages was assessed. k The average immunofluorescence intensity of anti-NLRC4 was determined in cardiomyocytes from cocultured with AIM2-siRNA macrophages or with NC-siRNA macrophages (n=5). Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01; *** P<0.001
AIM2-Mediated NLRC4 Inflammasome Activation in Cardiomyocytes Accelerates DCM Through Positively Regulating the IRF1 Signaling Pathway
In vivo, we confirmed that NLRC4, IRF1 and Nf-kB were all significantly upregulated in heart tissues of HFD/STZ-induced diabetic model (Fig. 5a-d). However, deletion of AIM2 markedly inhibited the activation of NLRC4-IRF1-Nf-kB cascade in diabetic hearts (Fig. 5a-d). Recent studies showed that a key role of IRF1 in regulating macrophage polarization, inflammasome activation, downstream signal transduction and finally evoking inflammatory response [18, 19], we therefore knocked down IRF1 in vivo by lentivirus-mediated siRNA against IRF1. Considering that siRNA alone transfection is unstable in vivo, we recombined the IRF1 siRNA oligo into the pLenti-CMV-EGFP-P2A-MCS vector. The transfection efficiency was verified by tracing the GFP immunofluorescence signal in the heart (Supplementary Fig. 5a). Although GFP signal was weakened at 12 weeks after pLenti-IRF1 siRNA injection, the inhibitory effect on IRF1 mRNA remained significant compared to the 0 day period without injection (Supplementary Fig. 5b). Also, the effect on IRF1 protein inhibition was confirmed by Western blot (Supplementary Fig. 6). After STZ stimulation and HFD feeding for 12 weeks, cardiac function was assessed by cardiac ultrasound. Compared with control Lenti-siRNA transfected mice, a significant decrease in LVIDs and LVIDd but a marked increase in LVEF was observed in IRF1 deficient mice (Fig. 5i-l). Similar to the results of cardiac function, knocking down of IRF1 in vivo also improved cardiac remodeling by reducing cardiac hypertrophy, which was indicated by HE staining, WGA staining and CSA measurement (Fig. 5e-f). In line with this, both the mRNA levels of ANP and β-MHC were significantly downregulated in IRF1 siRNA transfected STZ mice compared with those in control siRNA transfected mice (Fig. 5g-h). Furthermore, by in situ immunofluorescence staining, we found that IRF1 deficiency not only reduced the cardiac expressions of AIM2 and CD68, but also attenuated the colocalization of AIM2 and CD68 in the diabetic heart (Fig. 5m), suggesting that suppression of IRF1 also attenuated cardiac infiltration of macrophages and AIM2-mediated inflammasome formation in the heart. Indeed, we observed that knocking down of IRF1 and deletion of AIM2 had similar effects in reducing cardiac inflammation. The secretion of IL-1β and TNF-α were increased, but the secretion of IL-10 and IL-13 were reduced in the cardiac tissues of IRF1 deficient mice compared to those with control siRNA injection (Fig. 5n-q). Collectively, these data indicated that IRF1 played an important role in signal transduction after activating the NLRC4 inflammasome in cardiomyocytes, and IRF1 inhibition effectively reversed AIM2-mediated cardiac hypertrophy, dysfunction and remodeling by blocking NLRC4 inflammasome signaling induced by AIM2-dependent macrophage M1 polarization and cardiac infiltration.

Inhibition of IRF1 signaling improved HFD/STZ-induced cardiac inflammation, dysfunction and remodeling. a The protein levels of IRF1, Nf-κB and NLRC4 were detected in heart tissue of STZ (or citrate buffer)-induced WT and AIM2-/- mice. b-d Plot-histogram showed the quantification of the protein levels of IRF1, Nf-κB and NLRC4, n=4 # P<0.05 vs. WT+STZ group. e Representative of HE or WGA staining heart tissue images showed the cross section hearts from HFD/STZ mice transfected with IRF1 siRNA or control siRNA, scale bar = 20 μm. f The cross sectional area (μm2) of cardiomyocytes was quantified. g-h The mRNA levels of ANP and β-MHC were detected by real-time PCR. i M-mode echocardiography of STZ mice transfected with IRF1 siRNA or control siRNA, and (j-l) LVIDs, LVIDd, and LVEF were assessed by software. m Representative IF staining of AIM2 (green color) and CD68 (red color) in the heart tissues from STZ mice transfected with IRF1 siRNA or control siRNA, scale bar = 20 μm. n-q The levels of IL-1β, TNF-α, IL-10 and IL-13 expressed in heart tissue were determined by ELISA (n=5). Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01; *** P<0.001
IRF1 Overexpression Abolished AIM2 Deficiency-Mediated Protective Effects on HFD/STZ-Induced Myocardial Inflammation and Hypertrophy
To confirm whether IRF1 is a downstream molecule of AIM2 signaling that critically involved in NLRC4 inflammasome activation in diabetic heart, we further overexpressed IRF1 in vivo by reconstructing the full-length of IRF1 gene into the pSLenti-CMV-EGFP-3xFLAG-WPRE vector. Overexpression of IRF1 aggravated cardiac dysfunction in AIM2-/- mice induced by HFD/STZ, indicated by increased LVIDs and LVIDd but decreased LVEF value (Fig. 6a-d). Meantime, Overexpression of IRF1 caused marketable cardiac hypertrophy (Fig. 6e, g), both levels of ANP and β-MHC were significantly increased in heart tissues of AIM2-/- mice (Fig. 6h-i). Furthermore, cardiac infiltrating CD68+ macrophages was increased and NLRC4 inflammasome assembly was re-enhanced in AIM2 deficient hearts after IRF1 overexpression (Fig. 6f), these data suggested that IRF1 is a key downstream mediator of AIM2-mediated cardiac inflammation in HFD/STZ-induced diabetic hearts.

Overexpression of IRF1 aggravated HFD/STZ-induced cardiac inflammation, dysfunction and hypertrophy in AIM2-/- mice. a M-mode echocardiography of HFD/STZ mice transfected with Lenti-IRF1-OE vector or control vector, b-d LVIDs, LVIDd, and LVEF were assessed and anlyzed. e Representative HE or WGA staining images of heart tissues of AIM2-/- mice transfected with control or IRF1-OE vector, scale bar = 20 μm. f Representative IF staining of NLRC4 (green color) and CD68 (red color) in myocardium of HFD/STZ mice transfected with control or IRF1-OE vector, scale bar = 20 μm. g The cardiac CSA was quantified, and (h-i) the mRNA levels of ANP and β-MHC were determined. Data were expressed as Mean ± SEM. * P<0.05; ** P<0.01
Discussion
Emerging evidence has revealed that PAMPs/PRRs recognition pattern-mediated inflammasome activation in macrophages plays a crucial role in the development of DCM and heart failure [10]. In this study, we reported that AIM2, a scaffold that responsible for the inflammasome assembly, was essential for HFD/STZ-induced DCM and cardiac remodeling. Deletion of AIM2 hinders M1 pro-inflammatory polarization and cardiac infiltration of macrophages in diabetic hearts, thereby reducing assembly of the NLRC4 inflammasome and activation of the IRF1 signaling in cardiomyocytes, which further inhibits pathological cardiac hypertrophy and the development of diabetic heart failure (Fig. 6).
AIM2, a key regulator of the inflammasome assembly, triggering by double DNA sensing (a common PAMPs), plays an important role in the regulation of macrophage maturation, differentiation, polarization and pyroptosis [12, 17, 20]. By RNA-seq, we found that membrane receptors associated with PAMPs/PRRs-mediated innate immunity, including Nod-like receptor, Toll-like receptor and RIG-I-like receptor, were activated in HG-induced macrophages. Importantly, we also found the upregulation of AIM2, as well as inflammasome complex assembly molecules such as Nlrp3 and Nlrc4 in HG-induced macrophages, and the transcriptional changes of these genes were confirmed in heart tissues of HFD/STZ-induced diabetic hearts. Consistent with the data of AIM2 in atherosclerosis that reported by Paulin N. et al.[12], here we confirmed that AIM2 was specially increased in cardiac infiltrating macrophages in a diabetic mouse model. To explore whether AIM2 upregulation affects cardiac glucose metabolism [21], we constructed the AIM2 gene knockout mice. The blood glucose level and loss of weight were comparable between STZ-induced WT and AIM2-/- mice, indicating that AIM2 deficiency did not change cardiac glucose metabolism under the HFD feeding conditions. However, deletion of AIM2 did improve cardiac hypertrophy, LV dysfunction and remodeling HFD/STZ-induced diabetic hearts
Considering that macrophage M1 polarization promoted inflammasome assembly and inflammation expansion, we therefore studied the characteristics of AIM2 on macrophage polarization and its relationship with diabetic hypertrophy. By flow cytometry analysis, we found that cardiac infiltrating immune cells including neutrophils (CD11b+Ly6Ghigh subset) and macrophages (CD68+Ly6Glow subset) were significantly decreased in AIM2 deficient diabetic hearts [22]. Further, the proportion of M1-like macrophages (CD86+CD68+Ly6Glow subset) was reduced, but M2-like macrophages (CD206+CD68+Ly6Glow subset) was relatively increased in AIM2 deficient diabetic hearts. In line with this, analysis with the M1/M2 polarization marker genes confirmed that deletion of AIM2 inhibited M1 polarization of macrophages, meantime reduced the secretion of pro-inflammatory cytokines, IL-1β and TNF-α. In addition, activation of AIM2 promoted M1 polarization of macrophages was also observed in LPS-mediated acute lung injury [23], and facilitated tumor rejection in renal carcinoma through inflammasome signaling activation [20].
IL-1β played a critical role in cardiac inflammation and remodeling. Anti-inflammatory therapy targeting the IL-1β signaling with Canakinumab led to a significantly lower rate of cardiovascular adverse events [24]. IL-1β secretion promoted taget cell pyroptosis or macrophage self pyroptosis [25, 26]. Inhibition of inflammasome formation and pyroptosis reversed remodeling and improved cardiac repair in DCM [25, 27]. In addition, exosomes-mediated M2 polarization of macrophages antagonized inflammation and attenuated doxorubicin-induced cardiomyopathy through reducing IL-1β production [28]. Here, we found that AIM2 activated the proinflammatory state of HG-mediated macrophages and increased production of TNF-α and IL-1β, which further activated the NLRC4 inflammasome and its assembly in cardiomyocytes. Formation of NLRC4 inflammasome further triggered the downstream IRF1/Nf-kB signaling pathway, and finally resulted in cardiac adverse hypertrophy and remodeling. IRF1 is an endogenous mediator of myocardial ischemia reperfusion injury [29], and IRF1 activation is essential for cardiac hypertrophy in response to pressure overload [30]. Treatment with IRF1 siRNA attenuated cardiac fibrosis and diastolic dysfunction by reducing pyroptotic markers, IL-1β and IL-18 production [3]. In our study, we also observed that treatment with IRF1 siRNA in vivo effectively improved LV function and attenuated diabetic cardiac hypertrophy, and suppression of IRF1 had a negatively feedback effect on the accumulation of macrophages and the secretion of inflammatory cytokines in the diabetic heart. Moreover, overexpression of IRF1 in AIM2-/- mice markedly reversed the protective effect of AIM2 deficiency on HFD/STZ-induced diabetic cardiomyopathy, accumulation of cardiac infiltrating CD68+ macrophages, increased NLRC4 inflammasome assembly in cardiac tissues, and enhanced hypertrophic response were observed in AIM2-/- mice after activating IRF1.
Recently, the cardioprotective effect of AIM2 deficiency on DCM has also been observed in a type 2 diabetic rat model [31]. This is a very similar study to ours. In this article, the author highlighted the importance of increased cardiac ROS production and pyrotosis activation in AIM2-mediated diabetic cardiomyopathy. However, in our study we mainly focus on how macrophage polarization is regulated by AIM2, because M1 polarization of macrophage plays an important role in the development of DCM. Consistently, we both observed increased cell death, enhanced fibrosis and aggravated cardiac dysfunction under HFD-induced diabetic conditions, and AIM2 deficiency benefits the heart from diabetic remodeling and dysfunction. The difference is that, in addition to NLRP3-mediated pyrotosis activation, we also found that NLRC4-mediated IRF1/Nf-kB signaling pathway and cardiac inflammation contribute significantly to AIM2-induced pathological hypertrophy and DCM. Our research adds to the theory of cardiomyopathy mediated by AIM2 inflammasome and provides novel targets for the treatment of related diseases. In summary, our findings demonstrate that AIM2 plays a key role in the regulation of macrophage polarization and accumulation in the heart, leading to the occurrence of DCM. Inhibition of AIM2 impairs the proinflammatory state of macrophages, which obstructs the NLRC4 inflammasome assembly, reduces cardiac inflammation and protected STZ-mediated diabetic hypertrophy and dysfunction. We believe that this study will open up a possible new therapeutical target for the prevention of DCM linking to macrophage-mediated cardiac innate immune response.
Study Limitations
Our study has several limitations. Firstly, our findings indicate that AIM2 plays a functional role in regulating the polarization and infiltration of cardiac macrophages, and inhibition of AIM2 has been demonstrated to significantly abolish M1 macrophage-mediated cardiac inflammation, remodeling and dysfunction in HFD/STZ-induced mice. However, the present data have been obtained using mouse animal models or mouse-derived cell lines, which limit the generalizability of corresponding anti-inflammatory therapies by targeting AIM2 in patients with diabetic cardiomyopathy. It is postulated that the related methods and results may be similar to those predicted in human diabetic cardiomyopathy, but the pathogenesis of human diabetic cardiomyopathy may be more complex than previously thought. Given the diversity of downstream inflammasomes activated by AIM2, it is possible that the specific inflammasome involved in this process may differ between humans and mice. Consequently, the supplementary files contain data indicating that impaired NLRC4 inflammasome assembly is also observed in human-derived AC16 cardiomyocytes following AIM2 knockdown. However, these findings must be further validated in human diabetic models to confirm the potential role of AIM2 in cardiomyopathy.
Secondly, the duration of the present experiments may not be sufficiently long to observe a longer-term effect of AIM2 deficiency on HFD/STZ-induced diabetic cardiomyopathy. The development of diabetic cardiomyopathy is a relatively lengthy process. A high-fat diet induces a reduction in insulin sensitivity, which subsequently results in impairment of glucose tolerance. This, in turn, leads to the development of insulin resistance and cardiac remodeling. Consequently, it can be anticipated that the development of AIM2-targeted therapies should also have a relatively long-lasting effect. Due to the constraints of the experimental design, the observation period for the endpoint was limited to 12 weeks following STZ/HFZ stimulation. This was because symptoms such as cardiac hypertrophy, fibrosis and LV dysfunction were observed during this period. The knockout of AIM2 resulted in the inhibition of cardiac infiltrating macrophages, thereby improving cardiac inflammation and delaying the occurrence of diabetic cardiomyopathy. Nevertheless, it cannot be guaranteed that the application of AIM2 antagonists will be effective for an extended period of time. Consequently, further research is required to assess the long-term therapeutic efficacy of targeting the AIM2 signaling pathway.
Last but not the least, the potential negative effects of AIM2 deletion or inhibition of the AIM2/NLRC4/IRF1 signaling axis used in the study on organs other than the cardiac system have not been extensively explored. In the previous reports, AIM2 inflammasome has been shown to serve as a guardian of cellular integrity in modulating chronic inflammatory diseases, carcinoma and infection [32, 33]. Therefore, AIM2 deficiency might promote tumor progression and weaken the anti-tumor immunotherapy. Nevertheless, the genetic inactivation of AIM2 has been demonstrated to reduce liver damage and hepatocellular carcinoma development in mice. In our study, no pathological changes or liver toxicity were observed in AIM2-/- mice after HFD/STZ for 12 weeks (Supplementary Fig. 2-3). Anyway, further research is required to confirm these findings in the future.
Data Availability
The raw data are available and can be obtained from the corresponding author upon reasonable request.
References
Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. 2016;12:144–53.
Lorenzo-Almorós A, Tuñón J, Orejas M, Cortés M, Egido J, Lorenzo Ó. Diagnostic approaches for diabetic cardiomyopathy. Cardiovasc Diabetol. 2017;16(1):28.
Yu Q, Vazquez R, Khojeini EV, Patel C, Venkataramani R, Larson DF. IL-18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol. 2009;297:H76–85.
Nakamura K, Miyoshi T, Yoshida M, Akagi S, Saito Y, Ejiri K, Matsuo N, Ichikawa K, Iwasaki K, Naito T, Namba Y, Sugiyama H, Ito H. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int J Mol Sci. 2022;23(7):3587.
Durga Devi T, Babu M, Mäkinen P, Kaikkonen MU, Heinaniemi M, Laakso H, Ylä-Herttuala E, Rieppo L, Liimatainen T, Naumenko N, Tavi P, Ylä-Herttuala S. Aggravated Postinfarct Heart Failure in Type 2 Diabetes Is Associated with Impaired Mitophagy and Exaggerated Inflammasome Activation. Am J Pathol. 2017;187:2659–73.
Yang N, Wang M, Lin K, Xu D, Han X, Zhao X, Wang Y, Wu G, Luo W, Liang G, Shan P. Dectin-1 deficiency alleviates diabetic cardiomyopathy by attenuating macrophage-mediated inflammatory response. Biochim Biophys Acta Mol basis Dis. 2023;1869:166710. https://doi.org/10.1016/j.bbadis.2023.166710.
Ridker PM, Devalaraja M, Baeres FMM, Engelmann MDM, Hovingh GK, Ivkovic M, Lo L, Kling D, Pergola P, Raj D, Libby P, Davidson M. IL-6 inhibition with ziltivekimab in patients at high atherosclerotic risk (RESCUE): a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397:2060–9.
Broch K, Anstensrud AK, Woxholt S, Sharma K, Tøllefsen IM, Bendz B, Aakhus S, Ueland T, Amundsen BH, Damås JK, Berg ES, Bjørkelund E, Bendz C, Hopp E, Kleveland O, Stensæth KH, Opdahl A, Kløw NE, Seljeflot I, et al. Randomized Trial of Interleukin-6 Receptor Inhibition in Patients With Acute ST-Segment Elevation Myocardial Infarction. J Am Coll Cardiol. 2021;77:1845–55.
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249:158–75.
Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597:415–9.
Fidler TP, Xue C, Yalcinkaya M, Hardaway B, Abramowicz S, Xiao T, Liu W, Thomas DG, Hajebrahimi MA, Pircher J, Silvestre-Roig C, Kotini AG, Luchsinger LL, Wei Y, Westerterp M, Snoeck HW, Papapetrou EP, Schulz C, Massberg S, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592:296–301.
Paulin N, Viola JR, Maas SL, de Jong R, Fernandes-Alnemri T, Weber C, Drechsler M, Döring Y, Soehnlein O. Double-Strand DNA Sensing Aim2 Inflammasome Regulates Atherosclerotic Plaque Vulnerability. Circulation. 2018;138:321–3.
Ciążyńska M, Olejniczak-Staruch I, Sobolewska-Sztychny D, Narbutt J, Skibińska M, Lesiak A. The Role of NLRP1, NLRP3, and AIM2 Inflammasomes in Psoriasis: Review. Int J Mol Sci. 2021;22(11):5898. https://doi.org/10.3390/ijms22115898.
Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell. 2017;171:1057–1071.e1011.
Simpson DS, Pang J, Weir A, Kong IY, Fritsch M, Rashidi M, Cooney JP, Davidson KC, Speir M, Djajawi TM, Hughes S, Mackiewicz L, Dayton M, Anderton H, Doerflinger M, Deng Y, Huang AS, Conos SA, Tye H, et al. Interferon-γ primes macrophages for pathogen ligand-induced killing via a caspase-8 and mitochondrial cell death pathway. Immunity. 2022;55:423–441.e429.
Li H, Li Y, Song C, Hu Y, Dai M, Liu B, Pan P. Neutrophil Extracellular Traps Augmented Alveolar Macrophage Pyroptosis via AIM2 Inflammasome Activation in LPS-Induced ALI/ARDS. J Inflamm Res. 2021;14:4839–58.
Pan J, Han L, Guo J, Wang X, Liu D, Tian J, Zhang M, An F. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem Biophys Res Commun. 2018;498:487–94.
Rosain J, Neehus AL, Manry J, Yang R, Le Pen J, Daher W, Liu Z, Chan YH, Tahuil N, Türel Ö, Bourgey M, Ogishi M, Doisne JM, Izquierdo HM, Shirasaki T, Le Voyer T, Guérin A, Bastard P, Moncada-Vélez M, et al. Human IRF1 governs macrophagic IFN-γ immunity to mycobacteria. Cell. 2023;186:621–645.e633.
Wang A, Kang X, Wang J, Zhang S. IFIH1/IRF1/STAT1 promotes sepsis associated inflammatory lung injury via activating macrophage M1 polarization. Int Immunopharmacol. 2023;114:109478. https://doi.org/10.1016/j.intimp.2022.109478.
Chai D, Zhang Z, Shi SY, Qiu D, Zhang C, Wang G, Fang L, Li H, Tian H, Zheng J. Absent in melanoma 2-mediating M1 macrophages facilitate tumor rejection in renal carcinoma. Transl Oncol. 2021;14:101018. https://doi.org/10.1016/j.tranon.2021.101018.
Xie M, Yu Y, Kang R, Zhu S, Yang L, Zeng L, Sun X, Yang M, Billiar TR, Wang H, Cao L, Jiang J, Tang D. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat Commun. 2016;7:13280. https://doi.org/10.1038/ncomms13280.
Li YH, Zhang Y, Pan G, Xiang LX, Luo DC, Shao JZ. Occurrences and Functions of Ly6C(hi) and Ly6C(lo) Macrophages in Health and Disease. Front Immunol. 2022;13:901672. https://doi.org/10.3389/fimmu.2022.901672.
Wang J, Li R, Peng Z, Hu B, Rao X, Li J. HMGB1 participates in LPS-induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF-κB signaling pathways. Int J Mol Med. 2020;45:61–80.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–31.
Elmadbouh I, Singla DK. BMP-7 Attenuates Inflammation-Induced Pyroptosis and Improves Cardiac Repair in Diabetic Cardiomyopathy. Cells. 2021;10(10):2640. https://doi.org/10.3390/cells10102640.
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z, Zhang Z, Dai Z. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10:542. https://doi.org/10.1038/s41419-019-1761-4.
Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, Lv J, Li Y, Yu Y, Bai Y, Wang L. Metformin Inhibits the NLRP3 Inflammasome via AMPK/mTOR-dependent Effects in Diabetic Cardiomyopathy. Int J Biol Sci. 2019;15:1010–9.
Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome Treatment Enhances Anti-Inflammatory M2 Macrophages and Reduces Inflammation-Induced Pyroptosis in Doxorubicin-Induced Cardiomyopathy. Cells. 2019;8(10):1224. https://doi.org/10.3390/cells8101224.
Jiang W, Chen G, Pu J. The transcription factor interferon regulatory factor-1 is an endogenous mediator of myocardial ischemia reperfusion injury. Cell Biol Int. 2022;46:63–72.
Jiang DS, Li L, Huang L, Gong J, Xia H, Liu X, Wan N, Wei X, Zhu X, Chen Y, Chen X, Zhang XD, Li H. Interferon regulatory factor 1 is required for cardiac remodeling in response to pressure overload. Hypertension. 2014;64:77–86.
Wang X, Pan J, Liu H, Zhang M, Liu D, Lu L, et al. AIM2 gene silencing attenuates diabetic cardiomyopathy in type 2 diabetic rat model. Life Sci. 2019;2019(221):249–58.
Hu S, Peng L, Kwak YT, Tekippe EM, Pasare C, Malter JS, et al. The DNA Sensor AIM2 Maintains Intestinal Homeostasis via Regulation of Epithelial Antimicrobial Host Defense. Cell Rep. 2015;13:1922–36.
Martínez-Cardona C, Lozano-Ruiz B, Bachiller V, Peiró G, Algaba-Chueca F, Gómez-Hurtado I, et al. AIM2 deficiency reduces the development of hepatocellular carcinoma in mice. Int J Cancer. 2018;2018(143):2997–3007.
Funding
This work was assisted by the Shanghai Natural Science Foundation of China (No. 21ZR1451300) and Natural Science Foundation of China (81870290).
Author information
Authors and Affiliations
Contributions
All the authors listed have made great contributions to this research. Jian-Ping Wu performed the experiments and drafted the manuscript, Cheng Wu contributed to cell culture and molecular experiments, Yuan-Ji Ma performed the animal model and cardiac ultrasound examination, Jian-Bing Zhu analyzed the data, Lei-Lei Ma and Fei-Juan Kong designed and supervised the experiment. All authors have approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. No human studies were carried out by the authors for this article. Disclosure of potential conflicts of interest Research involving Human Participants and/or Animals Informed consent.
Conflict of Interest
All the authors declare that they have no conflict of interest.
Additional information
Associate Editor Guoping Li oversaw the review of this article
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.






Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, JP., Wu, C., Ma, YJ. et al. AIM2 Deficiency Alleviates Cardiac Inflammation and Hypertrophy in HFD/STZ-Induced Diabetic Mice by Inhibiting the NLRC4/IRF1 Signaling Pathway. J. of Cardiovasc. Trans. Res. (2024). https://doi.org/10.1007/s12265-024-10556-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12265-024-10556-0