
Abstract
Heart failure (HF) remains one of the major causes of morbidity and mortality worldwide. Recent studies have shown that stem cells (SCs) including bone marrow mesenchymal stem (BMSC), embryonic bodies (EB), embryonic stem (ESC), human induced pluripotent stem (hiPSC)-derived cardiac cells generation, and transplantation treated myocardial infarction (MI) in vivo and in human. However, the immature phenotypes compromise their clinical application requiring immediate intervention to improve stem-derived cardiac cell (S-CCs) maturation. Recently, an unbiased multi-omic analysis involving genomics, transcriptomics, epigenomics, proteomics, and metabolomics identified specific strategies for the generation of matured S-CCs that may enhance patients’ recovery processes upon transplantation. However, these strategies still remain undisclosed. Here, we summarize the recently discovered strategies for the matured S-CC generation. In addition, cardiac patch formation and transplantation that accelerated HF recuperation in clinical trials are discussed. A better understanding of this work may lead to efficient generation of matured S-CCs for regenerative medicine.

Graphical abstract
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Heart failure (HF) remains one of the major causes of morbidity and mortality worldwide. In spite of effective contemporary treatment strategies such as chemotherapy, catheterization, and surgical interventions, HF recurrence is still high [1]. Recent observations indicate that patients who underwent mitral-valve repair or replacement for severe ischemic mitral valve regurgitation had frequent recurrence with subsequent HF [2]. Approximately, 50% of patients who underwent the left ventricular assist device therapy died after 1 year due to HF recurrence [3]. Pulmonary artery catheters worsened HF and to some extent, HF reoccurred in patients who recovered from such treatment [4]. Lack of replacing or repairing damaged cardiac cells as a result of aging explains this phenomenon [5]. The higher HF mortality and its recurrence have intensified the necessity to explore novel strategies for the replacement of damaged cardiac cells to improve cardiac function.
Preclinical studies showed that stem cells such as bone marrow mesenchymal stem (BMSC), embryonic stem (EB); embryonic stem (ESC), human-induced pluripotent stem (hiPSC)-derived cardiac cells including endothelial cells (ECs), cardiac conducting system such as sinoatrial nodes (SANs) and atrioventricular nodes (AVNs), cardiomyocytes (CMs), cardiac fibroblasts (CFs), and epicardial cells (EpCs) treated myocardial infarction (MI) in vivo and in human [6]. Nonetheless, the immature phenotype of stem-derived cardiac cells (S-CCs) compromises their clinical application for MI and HF treatment [7].
Recently, an unbiased multi-omic analysis involving genomics, transcriptomics, epigenomics, proteomics, and metabolomics identified specific strategies for the generation of matured S-CCs that may enhance patients’ recovery processes upon transplantation. However, these strategies still remain undisclosed. Here, we summarize the recently discovered strategies for the matured S-CC generation. In addition, matured cardiac patch formation and transplantation that treated HF in clinical trials are discussed. A better understanding of this work may lead to effective and efficient generation of matured S-CCs for cardiac regenerative medicine.
Matured Stem-Cardiac Cell Generation
Cardiac Conducting System
The SANs, AVNs, and bundles of His and Purkinje fibers transmit electrical impulse to initiate heartbeat. Continuous damage of the cardiac tissues causes arrhythmia, HF, and frailty in elderly people [8]. The strategies for generation, transplantation, and replacement of the damaged conducting system are necessary for the prevention of arrhythmia, HF, and death in aging individuals. Recently, a 2-component cardiac organoid model, a 3-dimentional (3D) model that mimics the adult cardiac cells’ physiological environment to bridge ex vivo and in vitro techniques, has been developed. This 3D model utilized EB to engineer heart tissue (EHT). The EB-EHT cultured with fibrinogen and thrombin expressed SAN transcription factors (TFs) such as hyperpolarization-activated cyclic nucleotide-gated channel 4 and T-box-3, 18 (Tbx3, 18) which further exhibited specific matured EBs-SAN genes, including α-actinin, myosin light chain (MLC-2a), and MLC-2v with spontaneous beats. The generated EB-SAN further expressed matured gene including connexin 43 (Cx43) with accurate calcium signaling, diastolic depolarization period, action potential (AP), and autonomous beating provening the generation of pacemaker cells [9, 10]. It was observed that wingless 3a (Wnt3a) ligand triggered TFs including the short stature homeobox 2 (Shox2) expression and repressed pan-cardiac genes such as cardiac troponin I,T (cTNT) leading to matured-like spontaneous beating with AP similar to native SA pacemaker cells in mouse ESCs. The generated ES pacemaker cells further exhibited matured features such as appropriate Ca2+-clock mechanisms, maintained rapid automaticity and short AP duration [11]. In addition, EBs with activated exogenous Shox2 induced endogenous Shox2 expression which stimulated pacemaker-specific genes including gap junction alpha-7 protein (GJA7) and Cx45 leading to EB pacemaker cell generation. Observations showed that generated EB pacemaker cells had elevated Na+-Ca2+ exchanger coupling, spontaneous intracellular Ca2+ release, and high voltage oscillations [12]. Moreover, contactin-2 activation, a cell adhesion molecule critical for neuronal patterning and ion channel clustering, activation in EB-generated cells expressing Cx40 with cardiac pore-forming sodium channel subunits such as Nav1.5/Scn5a led to spontaneous electrical oscillations causing early after depolarizations and prolong AP which progressed to EB-Purkinje fiber network formation in vivo [13, 14]. It was identified that cyclic adenosine monophosphate (cAMP) activation by sodium nitroprusside upregulated generated Purkinje fiber maturity by triggering Cx30.2, 45, and contactin-2 [15], showing that treatment with fibrinogen and thrombin and activation of Wnt3a, Shox2, and contactin-2 promote cardiac conducting system maturation both in vitro and in vivo. Therefore, targeting these novel genes may enable the generation of matured cardiac conducting system capable of treating arrythmia and HF resulting from damaged SANs, AVNs, and Purkinje fibers.
S-ECs
ECs line the inner layer of the vasculature and the endocardium to prevent clot formation and thrombosis. A massive loss of ECs in the coronary microvasculature promotes the development of MI and HF requiring immediate EC replacement strategy to prevent HF in patients. More recent studies have shown that genomic editing strategies such as gene insertion and deletion generated matured S-CCs. For instance, zinc finger nuclease technology allowed the insertion of reporter genes into the safe harbor gene locus (protein phosphatase 1 regulatory subunit 12C (PPP1R12C) or adeno-associated virus integration site 1 (AAVS1)) in the hESC and hiPSC. The application of zinc finger nuclease technology led to the generation of immature hiPS-ECs [16]. In addition, type II clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) (CRISPR/Cas9) technique inserted factor VIII gene into hemophilia A, a coagulation abnormality disease caused by the deletion of the blood coagulation factor VIII (FVIII) gene, patient-derived iPSCs which generated immature hiPS-ECs evidenced by the expression of pan-cardiac cell genes such as octamer-binding transcription factor 4 (Oct4), SRY-box transcription factor 2 (Sox2), and Lin28 [17]. Additionally, DNA promoter methylation increases immature EC generation by augmenting proteins and TFs such as ETS variant transcription factor-2, nuclear receptor 2 subfamily 2, and Gata2 in hiPSCs, EBs, and ESCs [18, 19]. Furthermore, elevated glucose uptake increases the stimulation of proteins that induce iPSC differentiation, including vascular epithelial growth factor (VEGF-A) leading to EC TF activation and immature hiPS-EC generation [20, 21], indicating that zinc finger nuclease technology, CRISPR/Cas9, and DNA methylation provide a platform for the generation of immature S-CCs.
The identification that stepping up S-EC generation techniques to produce mature S-ECs is important to cardiac regenerative therapy obtained particular attention. This led to development of other strategies such as the adherent feeder-free differentiation protocol for matured S-EC production. The adherent feeder-free differentiation protocol successfully generated mature hiPS-ECs that continued to tube formation by activating endothelial nitric oxide synthase (eNOS), von Willebrand factor (vWF), and Weibel-Palade bodies (WPBs) in type 1 diabetes mellitus patient-derived hiPSCs [22, 23]. The matured hiPS-ECs transplanted into rat peri-infarct area secreted VEGF-A, increased neovascularization, improved left ventricular ejection fraction (LVEF), and prevented the occurrence of ischemic HF [24]. It was reported that transplanted hiPS-ECs induce structural maturation in pre-existing endocardial cells via the activation of TnI1,3, myosin-heavy chain 6,7 (MYH6,7), myosin-light chain (MYL2,7), and specific gap junction including Cx43 and zonula occludens-1 (ZO-1). In addition, transplanted hiPS-ECs had higher expression of extracellular matrix, collagen I, III, and fibronectin which enhanced hiPS-EC-hiPS-EC communications and signal transduction restoring Duchenne muscular dystrophy (DMD), a lethal degenerative muscle disease caused by over 3000 different mutations in the X-linked dystrophin gene clustered, mice cardiac function [25]. In addition, Cx43 facilitated the native ECs-hiPS-EC electrophysiological communication [25]. Additionally, higher expression of ZO-1, vascular endothelial cadherin (Ve-cad), cluster of differentiation 34 (CD34), and vWF increased native ECs-hiPS-EC tight junction formation and prevented permeability [25, 26]. However, apolipoprotein E4 gene overexpression increased proinflammatory and prothrombotic states repressing tight junction protein function which increased permeability and hiPS-EC dysfunction [26], suggesting that tight junction protein activation improves hiPS-EC maturation, native EC-hiPS-EC, and hiPS-EC-hiPS-EC junction communication. Therefore, strategies to maintain progressive matured genes and tight junction protein expression to enhance continuous hiPS-EC-native EC communication may accelerate the recuperation rate in patients.
Recent reports have demonstrated that hiPSCs treated with CHIR-99021 glycogen synthase kinase 3-β inhibitors, followed by bone morphogenetic protein 4 (BMP4) and a brief period of VEGF-A and Forskolin activation, maintained Ve-Cad and matured gene expression and matured EC generation [27]. It was identified that hESCs treated with Wnt3a and BMP-2 generated immature atrioventricular and endocardium cells. The later further differentiated to pre-valvular cells by triggering Smad6, Sox9, cadhrin-11, N-cadhrin, periostin, and endoglin [28]. However, treatment with VEGF-A and BMP-4 synergistically represses immature genes including enhancer of zeste homolog-2, increased matured hiPS-EC generation which continued to tube formation in vitro, and angiogenesis in vivo [29, 30]. An RNA-seq analysis demonstrated that ESCs with friend leukemia integration 1(FLI1) and protein kinase C (PKC) co-activation differentiated to immature ECs [31], while VEGF-A and BMP-4 activation increased matured gene expression that progressed to hiPS-EC network formation [32]. Collectively, these data indicate that genome editing techniques and DNA methylation generated immature hiPS-ECs. However, an optimized and adherent feeder-free differentiation procedure generated matured hiPS-ECs and improved adult ECs-hipS-EC junction communication. These suggest that utilizing the optimized and adherent feeder-free differentiation procedure to generate matured hiPS-EC may improve the function of transplanted hiPS-ECs and prevent HF.
S-CMs
A genomic analysis showed that gene insertion at AAVS1 locus by the transcription activator-like effector nucleases (TALEN) technology in hiPSCs produced matured hiPS-CMs that mimicked native CM functionalities [33]. In addition, point mutations efficiently restored dystrophin protein function promoting appropriate contractility and the generation of matured hiPS-CMs in hiPSCs derived from DMD patients [34]. A whole transcriptome sequencing shows that inhibition of heart break long noncoding RNA 1 and miR-1 allowed α/β-myosin heavy chain α/β (α/β-MHC) activation and increased mature hiPS-CM generation [35]. The transplantation of 1 × 106 matured hiSP-CMs exhibiting higher Cx43, N-cadherin, α/β-MHC, rapid contraction, and relaxation rate which significantly reduced MI progression and improved LVEF in rats [36, 37]. In addition, bioinformatic analyses of miRs and TF regulatory programs demonstrated that miR-200c inhibition escalated hESC differentiation TFs such as CACNA1C, KCNJ2, and SCN5A (Ca2+, K+, and Na+ ion channel genes) increased matured hiPS-CM generation [38]. Moreover, deep RNA sequencing analysis of cardiac biopsy from dilated cardiomyopathy patient shows that circular RNAs such as circ-MYOD, -SLC8A1, -ATXN7. and -PHF21A interaction with argonaute-2 protein complexes attenuated matured hiPS-CM generation. However, circ-SLC8A1, -CACNA1D, -SPHKAP. and -ALPK2 upregulation accelerated matured hiPS-CM generation [39], suggesting that gene insertions and point mutations generate matured hiPS-CMs. Moreover, whole transcriptome sequencing and bioinformatic analyses provide accurate strategies for the identification of specific TFs and miRs that promote hiPS-CM generation. The application of these protocols to generate mature hiPS-CMs that perpetually maintain matured gene expression may decline the HF progression providing an avenue for cardiac generative medicine.
Epigenetic modification has recently been implicated into matured S-CC generation and cardiac regenerative medicine. The application of the rank correlation method identified that spalt-like transcription factor-3 (SALL3), a TF that interacts with DNA methyltransferases-3A and 3B at CpG methylation sites to activate the CM stemness gene, overexpression increased ectoderm differentiation propensity, and decreased mesoderm and endoderm differentiation propensity. The attenuation of the SALL3 expression declined ectoderm, mesoderm, endoderm propensity gene such as nestin, Sox1, GATA-binding factor 4 (Gata4), and kinase insert domain receptor (KDR), forkhead box-A2, and alpha-fetoprotein, respectively, in hiPSCs. It was observed that treatment of endoderm with CHIR99021 followed by IWP4, a Wnt inhibitor, led to the generation of immature hiPS-CMs [40]. Moreover, class I and II histone methylation regulatory elements have been found to play an important role in the generation of mature hiPS-CMs. Class I histone methylation regulatory elements (ubiquitous enhancer (H3K4me1) and promoter (H3K4me3) promote pan-CC generation. However, class II histone methylation regulatory elements H3K4me1 and promoter H3K4me3 activation accelerated matured hiPS-CM generation [18]. In addition, bone marrow progenitor cells treated with trichostatin A, a histone deacetylase inhibitor, and 5-aza-2`-deoxycytidine, a DNA methylation inhibitor, activated matured genes such as cardiotroponin I, α-sarcomeric actinin, α-MHC and elevated matured hiPS-CM generation. The intramyocardial injection of 5 × 105 hiPS-CMs into the infarct area declined MI, improved LVEF, and prevented MI-associated HF [41].
Furthermore, transcriptomic analysis showed that mouse embryos expressing insulin gene enhancer protein 1 (Isl1), a TF that can augment maturation, differentiated into cells expressing both MLC2a and MLC2v [42]. However, activin A and BMP4 activation specified cell into ventricular CMs with immature TFs including Tbx5ILKNkx2–5+, Tbx5+Nkx2–5−, Tbx5−Nkx2–5+, and Tbx5−Nkx2–5− [42,43,44,45]. Interestingly, BMP-2 activation suppressed the immature TFs, including Nanog, Sox2, Oct-4, GATA-binding factor 6 (Gata6), hyaluronan, and proteoglycan link protein 1 (HAND1) and increased the expression of matured TFs such as MYL kinase 7, 9, tropomyosin1, and MYH9. The purification and transplantation of >15 million hiPS-CMs into rats and non-human primates’ myocardium significantly treated MI [46]. It was observed that 5 × 105 transplanted hiPS-CMs secreted exosomes/microvesicles containing miRNAs such as Nanog-regulated miR-21 and hypoxia inducible factor-1α-regulated miR-210 which prevented MI and the HF progression in mice [47]. In addition, hMSC-secreted soluble factors increased the expression of tight junction proteins including Cx43, N-cadherin and myofibrils A-, H-, and I-bands alignment, structural framework, electric pacing and cell-cell interactions. The hMSC-derived soluble factors elevated hiPSC-CM metabolic activities which accelerated adenosine triphosphate (ATP) production in hiPSC-CM and hMSC co-culture [37]. The tight junction proteins increased native CMs-hiPS-CM structural communication at post-transplantation [37], indicating the metabolic role in mature hiPSC-CM generation.
The failing heart undergoes a metabolic switch from fatty acid oxidation (FAO) to glycolysis resulting in low ATP synthesis. In addition, depleted ATP production declined 5′adenosin monophosphate kinase α2 (AMPKα2) and increased AMPKα1 (AMPKα2 to AMPKα1 switch) and promotes endocardial inflammation and triggers CM dysfunction. The metabolic AMPKα2 to AMPKα1 switches accelerate HF development in patients with failing heart [48,49,50,51]. Studies have demonstrated that generated matured hiPS-CMs effectively regulate ketogenesis, ketolysis, and methylglyoxal-related metabolism with depletion of oxidation stress-associated hypertrophic cardiomyopathy (HCM) via activation of angiogenic proteomes [52]. In addition, abundance of mitochondrial proteins including ATP synthase subunit γ, succinyl-CoA:3-ketoacid coenzyme A transferase 1, aldehyde dehydrogenase, 3-ketoacyl-CoA thiolase, and isocitrate dehydrogenase subunit γ upregulated mitochondrial mass and mtDNA content which increased FAO, ATP production, and the matured hiPS-CM generation [53, 54], indicating that higher FAO, AMPKα2 activation, and mitochondria biogenesis accelerate maturation of hiPS-CMs. FAO elevated native ECs-hiPS-CM communication by escalating cluster of differentiation 31 (CD31) and MYL3,4, respectively. In addition, vimentin and periostin enhanced fibroblast-hiPS-CM communication [53, 55]. Collectively, these show that employing genomic editing, transcriptomic and specific metabolomic for mature hiPS-CM generation and transplantation may prevent HF development.
S-CFs
Epicardial tissue constitutes approximately 80% of cardiac fibroblasts (CFs) in the adult heart [56]. Various studies have shown that CFs contribute to hypertrophic cardiomyopathy and HF development [57]. However, recent studies have demonstrated cardioprotective effect of hiPS-CFs [47]. For instance, in vitro, scaffold-free 3D microtissue approach first differentiated hiPSCs into EPCs which further transitioned to hiPS-CFs by suppressing WT1, Tbx18, and upregulating matured hiPS-CF genes such as gap junction alpha-1 protein (GJA1), integrin subunit alpha 4, collagen type alpha1, and periostin following basic fibroblast growth factor (bFGF2) activation [58]. Matured hiPS-CFs induced matured phenotypic traits such as electrophysiological maturity via KCND3 and KCNA4, typical transient outward potassium current (Ito) genes, increased AP amplitudes, and prolonged duration at 90% of repolarization in hiPS-CMs and hiPS-CFs co-cultured [58]. In addition, matured hiPS-CFs elevated structural maturity by escalating cardiac sarcomeric genes, desmin, titin-cap, and accelerated well-organized sarcomere formation with regular Z-lines, I-bands, H-zones, M-lines, and T-tubule-like structures in hiPS-CMs and hiPS-CF co-culture [58]. Additionally, the matured hiPS-CFs elevated hiPS-CM sarcoplasmic reticulum Ca2+ handling potential via calsequestrin, calmodulin 2, phospholamban, and triadin, Ca2+-handling proteins activation which led to appropriate negative and positive inotropic responses to pharmacological agents such as verapamil (L-type calcium channel antagonist) and K-8644 (L-type calcium channel agonist) [58]. These data demonstrate that utilizing the scaffold-free 3D microtissue approach for mature hiPS-CF generation further induce hiPS-CM generation. In addition, this imply that matured hiPS-CFs have higher potential of induce native CM generation to improve injured heart recovery. The transplantation of matured hiPS-CFs may facilitate appropriate contraction, relaxation, electrophysiology, and drug responses to prevent HF serving as generative therapeutic strategy.
S-EpCs
The epicardium forms a monolayer that surround the heart surface to protect the endocardium and supports CM proliferation [59, 60]. The 2D monolayer-based direct differentiation approach utilizes well-defined, growth factor- and xeno-free system for the generation of EpCs from hiPSCs. This protocol activated EpC TFs such as WT1, transcription factor 21 (TCF21), Tbx18, and α-actinin both in vivo and in vitro upon BMP-4 activation in the hESC culture [60]. In addition, EBs treated with activin A and BMP-4 produce EpCs with higher expression of ZO-1, indicating matured hiPS-EpCs [43, 60]. Altogether, these data indicate that the activin A and BMP-4 overactivation promote matured EpC generation providing a promising avenue for replacement of dead EpCs in MI for the prevention of MI-associated HF.
Formation of Cardiac Patches
Researchers have employed different approaches for the generation of cardiac patch (Table 1).
For instance, Ong et al. reported that a hiPSC-CM, human umbilical vein EC (HUVEC), and adult ventricular CF co-culture formed cardiospheres expressing the matured markers of individual cells such as cTNT, CD31, and vimentin, respectively. Recently, biomaterial-free cardiac patch using a 3D bioprinter has been developed for the generation of cardiac patch. Applying the 3D design software of the 3D bioprinter, the cardiospheres were identified, isolated with vacuum suction, transferred, and loaded onto a needle array in a sterile environment. The cardiospheres were bioprinted and allowed to mature. The needle array was removed to obtain the cardiac patch [61]. The 3D bioprinting techniques have been utilized to generate 3D myocardium with synchronous macroscopic beating. In addition, the hybrid technology combining guided self-assembly and 3D bioprinting created ECs with higher CD31 expression, tight junctions with adjacent cells, self-assembly, vessel-like conduits, and cardiac patch formation [71]. The cardiac patch engraftment onto rat myocardium promoted vascularization and maturation in pre-existing CMs indicating significant step toward generation of stem cell-based HF treatment [61]. Similarly, Gao et al.’s generated cardiac trilineage cell patch consisting of hiPSC-CMs, -ECs, and -smooth muscle cells. An epicardially engrafted mature 4 cm × 2 cm × 1.25 mm cardiac patch, a clinically relevant dimensions, released exosomes that reduced MI and hypertrophy and significantly improved LVEF in swine [6].
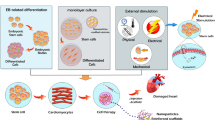
Furthermore, laser-induced-forward-transfer (LIFT) bioprinting technology used to create EBs from mouse ESCs further directed cardiogenesis [72]. The LIFT bioprinting technique prints HUVEC and hMSC onto a cardiac patch to enhance angiogenesis. LIFT-generated cardiac patch transplantation attenuated the progressive hypertrophy and cardiac fibrosis 8 weeks post-MI [73]. Additionally, tissue printing (TP) technology printed human cardiac-derived CM progenitor cells in an alginate hydrogel scaffold with higher matured transcription factor expression, cardiac lineage commitment, and viability [74]. Moreover, the automated bio-3D printer created cardiac spheroid from induced pluripotent SCs (iCells), ECs, and fibroblasts which formed tubular cardiac construct which had spontaneous beating with higher amplitude [75]. A follow-up study indicates that 0.2 × 0.2 cm epicall patching retained the CM phenotype by expressing TnI, cardiac actinin, and Cx43 and decreased mice MI progression [76]. In additon, Peti et al.’s printed heart tissue constructs using extrusion bioprinting by utilizing decellularized extracellular matrix bioink extracted from heart tissue and laden with myoblasts [77]. The transplantation of bioink generated cardiac patch comprising extracellular matrix hydrogel (cECM) and human hCPC showed high cell viability, vascularization, and improved cardiac function in MI mice [78]. Additionally, cardiac patch influences regeneration, proliferation, and maturation of host CMs [62, 63]. It has been confirmed that implanted cardiac patch form CM networks induced native heart tissue proliferation and vascularization with evidence of appropriate electrical coupling and declined the MI progression in guinea pig [64, 65]. The engrafted cardiac patch recapitulated short QT syndrome, an inherited arrhythmogenic syndrome characterized by an abnormal ion channel function, by improving the IKr current density in vitro, prevented MI and HF in rats [66]. In additon, the transplanted cardiac patch continued to express angiogenic growth factors such as angiopoietin 1, VEGF-A, insulin growth factor-1, and CM structural proteins including Cx43 and β-MHC which declined chronic HF progression in rats [67]. The cardiac patch implantation maximized LVEF while decreasing LV end diastolic pressure to prevent chronic HF in rats (Figure 1) [68], suggesting that cardiac patch implantation prevents arrythmias, MI, and HF in vivo. The cardiac patch generation and transplantation have demonstrate high effectiveness and safety in animal models which suggest its application in clinical trials.

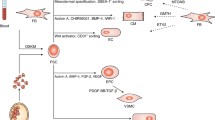
Diagrammatic representation of hiPS-cardiac cell generation, maturation, and transplantation as heart failure therapy. The treatment of hiPSCs with activin A, BMP4, and Wnt3A/5A activates transcription factors such as Sox7,17, Pax6, and Gata4,6, leading to the expression of immature ECs, while the activation of Ve-cad, CD34, vWF, and p-selectin promotes the generation of matured ECs. In addition, Hc4, Tbx3,18, and Shox2 promote the generation of immature cardiac conducting system but the progressive stimulation of MLC-2a,2v leads to the generation of the cardiac conducting system similar to native cells. Similarly, Gata4, MESP1, FoxC1, and Octa3/4 expression retain the pluripotent phenotype of CMs with immature characteristics. However, troponin, tropomyosin, MYH6,7, and MYL2,7 enhance matured CM generation with appropriate contraction and relaxation periods. Moreover, Tbx5,18 and WT1 expression indicate immature cardiac fibroblasts (CFs) while GAJ1, COL1A1,2, TNNT2, and MYL3,4 progressively produce CFs that secrete exosomes that accelerate cardiac cell replication and increase injury recovery. Furthermore, the activation of KDR, PDGFRA, WT1, and TCF21 represses immature genes such as Tbx5,18 and elevates the generation of EpCs. The cardiopatch technology generates a cardiac sheet consisting of all the S-CCs and omental flap. The omental flap which is rich in vasculature promotes angiogenesis, cardiac cell proliferation, and nutrition and creates an anti-ischemic environment to allow native cardiac cell replication and replacement of lost or damaged cells leading to the prevention of heart failure
Clinical Trials
Heart Failure
The S-CC generation and transplantation have gained adequate clinical attention. Recent reports have shown that surgical transplantation of 4 million hiPS-CMs that have lost immature markers, including Sox-2, Nanog, but expressing Isl-1 as matured marker into the infarct area of a 68-year-old patient with severe HF improved LVEF after 3 months of treatment. Upon frequent follow-up, the patient reported no complications but demonstrated the feasibility of hiPS-CM-based therapy [79]. 8.2 million hESC-CMs embedded in a cardiac patch which was epicardially delivered during a coronary artery bypass procedure improved LVEF and terminated HF after 12 months [80]. The CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction) reported that infusion of 12.5 to 25 × 106 cardiosphere-derived cells expressing matured markers improved LVEF and prevented HF within 12 months [81]. In addition, the congestive HF cardiopoietic regenerative therapy (CHART-1) trial showed that retention-enhanced intramyocardial injection catheter implantation of 600 × 106 bone marrow-derived and lineage-directed autologous cardiopoietic SCs elevated LVEF and prevented HF progression in chronic ischemic HF patients [82]. Additionally, intracoronary transplantation of autologous bone marrow mesenchymal SCs enhanced LV systolic function and reduced acute HF within 4 months in patients who underwent percutaneous coronary intervention [83]. Moreover, retention-enhanced catheter endomyocardial delivery of 24 million cardiopoietic cells terminated HF in symptomatic ischemic HF after 39 weeks with high safety and efficacy (Table 2) [84, 85].
In brief, data presented here support the fact that S-CC generation and transplantation prevent the progress of MI and HF in vitro and in vivo. It is clear that transplanted S-CCs proliferate and replace damaged cells via the secretion of exosomes that have the potential. In addition, implanted S-CCs promote the proliferation of pre-existing cells for rapid lost cell replacement and reversal of damaged cardiac tissues. These data prove that targeting matured S-CC generation and transplantation may serve as a unique strategy for the treatment of HF [81].
Advantages, Challenges, and Strategic Mitigation in Mature Cardiac Patch Generation
The CRISPR/Cas9 and TALEN techniques provide efficient approach for gene deletion and insertion for S-CC generation. However, CRISPR/Cas9 has a high risk of off-target mutations in human cells [86]. Chih-Che et al.’s synthetic switch protocol allows CRISPR/Cas9 self-regulation in both transcription and translation step to prevent off-target mutations. The approach also enables simultaneous transcriptional and translational suppression, increasing on-target indel and minimizing off-target mutation leading to “hit and run” genome editing in vivo [87]. Although CRISPR/Cas9 has been shown to be more efficient than the TALEN technique, however, the TALEN technique application indicates significantly lower DNA mutation in S-CC generation [88]. For instance, TALEN faithful gene knockout by selectively targeting the knockout of TNNT2 p.R173W pathogenic mutation in patient hiPSC-CMs. TALEN knockout of pathogenic mutation ameliorated dilated cardiomyopathy phenotype in vitro [89]. The genome editing technologies provide a powerful framework for S-CCs and mitigate mutation that may be detrimental in the cardiac regenerative medicine.
Studies have shown that the human heart consists of approximately 109 cells in which CMs form one third of the total cells. The ability to engineer such a large number of S-CCs still remains a challenge. Recently, highly efficient differentiation protocols generated approximately 107 cells in a single dish but scaling up to 109 cells required higher expertise and labor intensiveness. However, the active gas ventilation, a scalable 2D culture system using multi-layer culture plates, generated 109 with 90% CPCs, over 60% purity and higher feasibility [90, 91]. In addition, the 3D suspension differentiation platforms such as microcarriers generated approximately 109 hiPS-CMs with 80–90% purity, more space-efficient and cost-effective manner [92, 93]. The next step toward engineering clinically relevant myocardial tissues would include ECs, fibroblasts, atrial, ventricular, and conducting system to enhance easy integration of transplanted myocardial tissues into the host myocardium [94,95,96].
Another major constrain is the maintenance of the generated and transplanted S-CC survival. Studies have shown that transplanted tissue integration into the host myocardium and survival are not well sustained in a deprived vascular network heart. It has been reported that activation of angiogenic molecules and increasing cell-cell interactions to improve implanted S-CC perfusion may enhance the survival rate [97]. It has been demonstrated that hiPSC-CMs, ECs, or EPCs co-culture formed primitive vessel-like structures with higher potential for in vivo anastomosis which may increase transplanted tissue perfusion rate and enhance survival [97, 98]. The 3D bioprinting, micropatterning, and microfluidic systems provide a greater strategy for controlling vessel architecture, anastomosis, and tissue integration [99,100,101]. Moreover, 3D bioprinting provides higher promising strategy for maintaining transplanted tissue’s survival. However, the application of 3D bioprinting is currently limited due to the inadequate bioinks and multi-material bioprinting modalities necessary for 3D vascular constructs to maintain tissue-mimetic stiffness, cell density, and function. The next expectation is the creation of vascularized 3D engineered myocardial constructs with higher perfusion rate in vitro and in vivo to enhance transplanted tissues maturity, survival, and function [99, 102].
More recent studies have shown that vascular supply to transplanted cardiac patch contributes to the survival and ability to induce cellular proliferation in pre-existing cells [69]. Omentum has been found to be a good source of blood supply which increased the survival rate of the transplanted cardiac patch. The omental flap patching on EBs-cardiac sheet increased perfusion, nutrient diffusion, angiogenetic factor secretion, and angiogenesis [69]. The omental flap further created an anti-ischemic environment which promoted maturation, prolonged the survival of transplanted S-CCs, and improved cardiac function in mini pigs [69, 70]. These indicate that omental flap covering enhanced angiogenesis and promoted S-CCs’ maturation and survival and provided longer therapeutic effects at post-transplantation in a porcine ischemic cardiomyopathy model [69]. It is clear that strategies to improve transplanted tissue vascularization may improve cardiac stem cell therapy. Recently, the aeronautics and space administration (NASA) and the new organ alliance initiated a competition to overcome the vascularization bottledneck with an awarding of a $500 K to the teams that can successfully generate functionally vascularized cardiac tissues [103], but the results are yet to be reported.
Moreover, appropriate integration of the transplanted myocardial tissue into the host myocardium is another hurdle. The transplanted cell heterogeneity accounts for low engraftment and poor electromechanical communication-associated reentry arrhythmias. A wave block of electrical transmission that occurs as signal is transmitted via fibrotic interface sandwiched in transplanted cardiac patch and pre-existing cardiac tissue that causes abnormal AP duration or excitability [104]. However, the conductive scaffolds approach improves electrical transmission between transplanted tissue and the host myocardium by activating Cx43 [105,106,107,108]. In addition, transplanted patch are physically detach from the pericardium which impairs electrical coupling and repair processes. Nonetheless, bioactive peptides such as poly (glycerol sebacate)-poly(ε-caprolactone) activation prevented the physical separation of the transplanted tissue from the host tissue and increased asculogenesis and myogenesis without altering electrical transmission and structural protein expression [109, 110], serving as a promising approach to improve transplanted tissue integration into host myocardium. These features also demonstrate the ability of the cardiac patch to maintain continuous matured phenotypes to enhance cardiac stem cell therapy.
Previous reports showed that highly efficient protocols only generated immature cardiac tissues with low survival suggesting need for the discovering of new protocol that maintain the continuous maturity. Recent studies show that mechanical factors such as substrates and physiological stiffness at 6–10 kPa imparting physiological shapes, 2000-μm (2) rectangles with length:width aspect ratios at 5:1–7:1, and inducing mature alignment of myofibrils. The mechanical factors continued to improve electrophysiology by increasing calcium handling causing appropriate generated cardiac tissue contractility [111]. In addition, the application of Young’s modulus-induced stress and strain in generated cardiac tissue at 15 μm in 15 s intervals immediately increased tissue force between 0.4 and 0.5 mN. This passive stretch further increased maturation by activating calcium and potassium ion channels, β-adrenergic receptors, t-tubule protein, and caveolin-3 [7, 112]. These data indicate the significance of mechanical factors in enhancing cardiac tissue maturity and function to prevent rejection.
Furthermore, acute and chronic immune rejections at post-transplantation remaining major roadblock in animal models compelling researchers to immediately develop immune tolerance S-CC generation protocols. The human leukocyte antigen (HLA)-matched tissues indicate immune rejection which require immunosuppression. The HLA-matched cardiac tissue banks have the potential to serve as a valuable source of tissues with effective and safer way to deliver cell therapy to a large number of patients. The generation and transplantation of HLA haplotype homozygous hiPSC in allogeneic settings showed favorable engraftment with minimal acute graft-versus-host disease implying the merit of banking hiPSC and transplantation [113]. However, the unexplained complexities in genetics and ethnic diversity are still a drawback [114]. According to Suji et al., comprehensive genomic hybridization-based single nucleotide polymorphism and copy number variation showed that only 10 HLA-homozygous hiPSC lines matched 41.07% of the Korean population. This study served as a useful indicator for the development of new methods for hiPSC generation and quality control. Pierre-Antoine et al.’s probabilistic models show that hiPSC bank comprising 100 hiPSC lines generated most frequent HLA haplotypes that matched only 22% European Americans, 37% Asians, 48% Hispanics, and 55% African Americans [115, 116]. This notion indicates that allogeneic hiPSC bank development may be feasible for ethnically homogenous countries but may be challenging for heterozygote or diverse countries. Interestingly, Riolobos et al. (2013), beta-2 microglobulin gene homozygote deficient ESCs that do not express HLA indicated immune tolerance [117]. These indicate the feasibility of developing off-the-shelf S-CCs with immune tolerance. Tobias et al. (2019) have reported that inactivation of histocompatibility complex (MHC) class I and II genes and overexpression of CD47 in both mouse and hiPSCs demonstrated immune tolerance in fully MHC-mismatched allogeneic recipients without immunosuppression (Table 3) [118]. More importantly, the HLA- and MHC I/II-null application could be applied to engineer a hypo-immunogenic cardiac patch as an off-the-shelf product for universal cardiac repair. In addition, improving the presented protocol may lead to the generation of S-CCs and cardiac patch that escape immune rejection for therapeutic transplantation.
Safety
Recently, there were serious safety-related concerns for S-CC-based therapy due to recorded genetic and epigenetic abnormalities, tumorigenicity, and immunosuppression that occur at post-transplantation [121]. National Institutes for Food and Drug Control (NIFDC) extensively reviewed the biological safety of S-CC-based therapy and reported that it was safe for clinical trials or therapies [121]. In addition, recent preclinical and clinical data reported the S-CC-based therapy efficacy without the occurrence of teratoma [122]. For instance, non-ischemic dilated cardiomyopathy mice receiving hiPS-CMs exhibited no acute or chronic adverse effect at post-transplantation [123]. Furthermore, immunosuppression at pre-transplantation and post-transplantation was another major concern in S-CC-based therapy [118]. However, recent studies have deduced that S-CCs generated from HF animals and transplanted into the donor neither required immunosuppression nor demonstrated immune rejection [124, 125]. Mini pigs receiving omental flaps and hiPS-CM sheets indicated higher cardiogenesis and angiogenesis without short- or long-term effects [122]. Moreover, S-CC transplantation that induced cardiac repair and prevented HF in swine was rapid, safe, and efficacious without teratoma formation or immunosuppression (Table 4) [6, 126]. Altogether, this data proves that S-CCs or cardiac patch transplantation for the HF treatment is safe and effective. In addition, extracting patient-derived S-CC generation and transplantation into donor may avoid the need for immunosuppression at pre-transplantation and tissue rejection at post-transplantation.
In summary, the immature traits of S-CCs limit their clinical application which made SC therapy seem like a myth. However, omic studies have eliminated all barriers that compromise S-CC clinical application for HF treatment. This confirms that cardiac regeretive medicine represents a potential therapeutic strategy for the treatment of HF.
Conclusion
Cardiomyocytes are cell types with slow proliferation rate, and aging further reduces their renewal and replacement resulting in HF recurrence and sudden death. The generation and transplantation of S-CCs to replace damaged and lost cells present a promising strategy for the treatment of HF. However, the immature phenotype of S-CCs limits their clinical application. Recent omic studies such as genome editing, epigenetic, metabolomics, and other approaches including 3D bioprint and omental flap studies have generated matured S-CCs for the treatment of HF in clinical trials which eliminates the limitations in their clinical application. Recently, Su-Yi Tsai et al. (2020) created MYH6:mCherry hES-CM reporters that effectively monitored drug-induced cardiotoxicity which can be used to monitor transplanted S-CC safety [127]. In addition, deletion of HLA and MCH-I/II genes promotes immune tolerance and increases cardiac repair with higher efficacy and safety. This study shows that applying the listed protocol for the generation of matured S-CC and increasing immune tolerance may enhance their clinical application for the treatment of HF as SC-based therapy.
Perspectives
Currently, the hiPSCs and cardiac patch engineering for cardiac regenerative medicine have greatly advanced due to the utilization of biocompatible materials and mechanical properties modulation. However, bottlenecks such as 3D cryopreservation, full myocardium engineering to attain a full functional cardiac tissue in vitro, require further attention. In addition, native cell-S-CC interactions and signal transmissions need further consideration. In future, we envision the maximum microphysiological system utilization for the generation of S-CCs and cardiac patch with the full adult cardiac tissue composition, optimal paracrine factors secretion, and survival. This approach aims to scale down the number of reagents needed to optimize engineered individual myocardial cells and intensity. To enhance clinical application, the MHC-I/II and HLA-null approach can be improved to be amenable to cryopreservation which may allow a true off-the-shelf product.
The Japanese pharmaceuticals, medical devices, and other therapeutic product acts categorized regenerative medical products as independent from conventional pharmaceuticals and medical approaches. The act approves hiPSC-based clinical studies for safety data and allowed up to 7 years for researchers to ascertain further evidence of safety and efficacy [119]. The regulatory act minimizes cost and provides adequate time for regenerative medical studies. In addition, the Japanese ministry of health, labour, and welfare (MHLW) initiated the “Project for Enhanced Practical Application of Innovative Drugs, Medical Devices and Regenerative Medical Products” to support the personnel exchange and cooperation in writing of guidelines regarding gene and cellular therapy [120]. The guideline facilitates the discovery of specific mature genes, number of hiPSCs required for myocardium generation, improves host cell-hiPSC communication, appropriate transplantation strategy, and protects the patient’s right. Finally, we envision a future in which heart failure patients can simply receive cryopreserved, immunotolerant S-CCs and myocardium as approved therapy. It is expected that a similar framework may be applied for the treatment of cerebrovascular and renovascular diseases and abdominal aortic aneurysms. Although cardiac regenerative medicine has proven feasible, improving engineering protocols is labor intensive, time consuming, and costly which requires collaboration of local and large international research community to achieve the goal.
Clinical Knowledge
-
Omic studies have provided an avenue for the generation of matured S-CCs and cardiac patch to improve the cardiac regenerative medicine.
-
Transplanted S-CCs provide conducive microenvironment for native CM replication.
-
Transplanted S-CCs replaced damaged CMs and increase recuperation.
-
Cardiac patch transplantation increases CM perforation.
-
Cardiac patch creates anti-ischemic microenvironment which prevents MI and HF.
-
Transplanted S-CCs and cardiac patch increase LVEF and prevent heart failure.
References
Tae-Oh Kim, M.-S. K., Kim, J.-J., & Song, J.-K. (2014). Efficacy and optimal duration of heart failure medications in preventing the recurrence of stress-induced cardiomyopathy. Circulation, 130, A17862.
Goldstein, D., Moskowitz, A. J., Gelijns, A. C., Ailawadi, G., Parides, M. K., Perrault, L. P., Hung, J. W., Voisine, P., Dagenais, F., Gillinov, A. M., Thourani, V., Argenziano, M., Gammie, J. S., Mack, M., Demers, P., Atluri, P., Rose, E. A., O'Sullivan, K., Williams, D. L., Bagiella, E., Michler, R. E., Weisel, R. D., Miller, M. A., Geller, N. L., Taddei-Peters, W. C., Smith, P. K., Moquete, E., Overbey, J. R., Kron, I. L., O'Gara, P. T., & Acker, M. A. (2016). Ctsn, Two-year outcomes of surgical treatment of severe ischemic mitral regurgitation. The New England Journal of Medicine, 374(4), 344–353.
Tomohiro Saito, M. D., Weng, Y., Potapov, E., & Krabatsch, T. (2018). Left ventricular assist device reimplantation for recurrent heart failure after myocardial recovery and weaning. Circulation, 130, A12417.
Binanay, C., Califf, R. M., Hasselblad, V., O'Connor, C. M., Shah, M. R., Sopko, G., Stevenson, L. W., Francis, G. S., Leier, C. V., Miller, L. W., Investigators, E., & Coordinators, E. S. (2005). Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA, 294(13), 1625–1633.
Parmar, K. R., Xiu, P. Y., Chowdhury, M. R., Patel, E., & Cohen, M. (2015). In-hospital treatment and outcomes of heart failure in specialist and non-specialist services: a retrospective cohort study in the elderly. Open Heart, 2(1), e000095.
Gao, L., Gregorich, Z. R., Zhu, W., Mattapally, S., Oduk, Y., Lou, X., Kannappan, R., Borovjagin, A. V., Walcott, G. P., Pollard, A. E., Fast, V. G., Hu, X., Lloyd, S. G., Ge, Y., & Zhang, J. (2018). Large cardiac muscle patches engineered from human induced-pluripotent stem cell-derived cardiac cells improve recovery from myocardial infarction in swine. Circulation, 137(16), 1712–1730.
Ronaldson-Bouchard, K., Ma, S. P., Yeager, K., Chen, T., Song, L., Sirabella, D., Morikawa, K., Teles, D., Yazawa, M., & Vunjak-Novakovic, G. (2018). Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature, 556(7700), 239–243.
Moghtadaei, M., Jansen, H. J., Mackasey, M., Rafferty, S. A., Bogachev, O., Sapp, J. L., Howlett, S. E., & Rose, R. A. (2016). The impacts of age and frailty on heart rate and sinoatrial node function. The Journal of Physiology, 594(23), 7105–7126.
Gorabi, A. M., Hajighasemi, S., Tafti, H. A., & Atashi, A. (2019). TBX18 transcription factor overexpression in human-induced pluripotent stem cells increases their differentiation into pacemaker-like cells. Journal of Cellular Physiology, 234, 1534–1546.
Schulze, M. L., Lemoine, M., & Fischer, A. W. (2019). Dissecting hiPSC-CM pacemaker function in a cardiac organoid model. Biomaterials., 206, 133–145.
Liang, W., Han, P., Kim, E. H., Mak, J., Zhang, R., Torrente, A. G., Goldhaber, J. I., Marban, E., & Cho, H. C. (2019). Canonical Wnt signaling promotes pacemaker cell specification of cardiac mesodermal cells derived from mouse and human embryonic stem cells. Stem Cells, 38(3), 352–368.
Ionta, V., Liang, W., Kim, E. H., Rafie, R., Giacomello, A., Marban, E., & Cho, H. C. (2015). SHOX2 overexpression favors differentiation of embryonic stem cells into cardiac pacemaker cells, improving biological pacing ability. Stem Cell Reports, 4(1), 129–142.
Tsai, S. Y., Chen, S., & Evans, T. (2017). Efficient generation of cardiac Purkinje-like cells from embryonic stem cells by activating cAMP signaling. Current Protocols in Stem Cell Biology, 40, 1F 16 1-1F 16 13.
Pallante, B. A., Giovannone, S., Fang-Yu, L., Zhang, J., Liu, N., Kang, G., Dun, W., Boyden, P. A., & Fishman, G. I. (2010). Contactin-2 expression in the cardiac Purkinje fiber network. Circulation. Arrhythmia and Electrophysiology, 3(2), 186–194.
Tsai, S. Y., Maass, K., Lu, J., Fishman, G. I., Chen, S., & Evans, T. (2015). Efficient generation of cardiac Purkinje cells from ESCs by activating cAMP signaling. Stem Cell Reports, 4(6), 1089–1102.
Wang, Y., Zhang, W. Y., Hu, S., Lan, F., Lee, A. S., Huber, B., Lisowski, L., Liang, P., Huang, M., de Almeida, P. E., Won, J. H., Sun, N., Robbins, R. C., Kay, M. A., Urnov, F. D., & Wu, J. C. (2012). Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circulation Research, 111(12), 1494–1503.
Park, C. Y., Sung, J. J., Cho, S. R., Kim, J., & Kim, D. W. (2019). Universal correction of blood coagulation factor VIII in patient-derived induced pluripotent stem cells using CRISPR/Cas9. Stem Cell Reports, 12(6), 1242–1249.
Zhao, M. T., Shao, N. Y., Hu, S., Ma, N., Srinivasan, R., Jahanbani, F., Lee, J., Zhang, S. L., Snyder, M. P., & Wu, J. C. (2017). Cell type-specific chromatin signatures underline regulatory DNA elements in human induced pluripotent stem cells and somatic cells. Circulation Research, 121(11), 1237–1250.
Tanaka, T., Izawa, K., Maniwa, Y., Okamura, M., Okada, A., Yamaguchi, T., Shirakura, K., Maekawa, N., Matsui, H., Ishimoto, K., Hino, N., Nakagawa, O., Aird, W. C., Mizuguchi, H., Kawabata, K., Doi, T., & Okada, Y. (2018). ETV2-TET1/TET2 complexes induce endothelial cell-specific Robo4 expression via promoter demethylation. Scientific Reports, 8(1), 5653.
Panopoulos, A. D., Yanes, O., Ruiz, S., Kida, Y. S., Diep, D., Tautenhahn, R., Herrerias, A., Batchelder, E. M., Plongthongkum, N., Lutz, M., Berggren, W. T., Zhang, K., Evans, R. M., Siuzdak, G., & Izpisua Belmonte, J. C. (2012). The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Research, 22(1), 168–177.
Tiemeier, G. L., de Koning, R., Wang, G., Kostidis, S., Rietjens, R. G. J., Sol, W., Dumas, S. J., Giera, M., van den Berg, C. W., Eikenboom, J. C. J., van den Berg, B. M., Carmeliet, P., & Rabelink, T. J. (2020). Lowering the increased intracellular pH of human-induced pluripotent stem cell-derived endothelial cells induces formation of mature Weibel-Palade bodies. Stem Cells Translational Medicine, 9(7), 758–772.
Hellen, N., Pinto Ricardo, C., Vauchez, K., Whiting, G., Wheeler, J. X., & Harding, S. E. (2019). Proteomic analysis reveals temporal changes in protein expression in human induced pluripotent stem cell-derived cardiomyocytes in vitro. Stem Cells and Development, 28(9), 565–578.
Chan, X. Y., Black, R., Dickerman, K., Federico, J., Levesque, M., Mumm, J., & Gerecht, S. (2015). Three-dimensional vascular network assembly from diabetic patient-derived induced pluripotent stem cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(12), 2677–2685.
Alrefai, M. T., Tarola, C. L., Raagas, R., Ridwan, K., Shalal, M., Lomis, N., Paul, A., Alrefai, M. D., Prakash, S., Schwertani, A., & Shum-Tim, D. (2019). Functional assessment of pluripotent and mesenchymal stem cell derived secretome in heart disease. Ann Stem Cell Res, 2(1), 29–36.
Al-Rewashdy, H., Ljubicic, V., Lin, W., Renaud, J. M., & Jasmin, B. J. (2015). Utrophin a is essential in mediating the functional adaptations of mdx mouse muscle following chronic AMPK activation. Human Molecular Genetics, 24(5), 1243–1255.
Rieker, C., Migliavacca, E., Vaucher, A., Baud, G., Marquis, J., Charpagne, A., Hegde, N., Guignard, L., McLachlan, M., & Pooler, A. M. (2019). Apolipoprotein E4 expression causes gain of toxic function in isogenic human induced pluripotent stem cell-derived endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 39(9), e195–e207.
Patsch, C., Challet-Meylan, L., Thoma, E. C., Urich, E., Heckel, T., O'Sullivan, J. F., Grainger, S. J., Kapp, F. G., Sun, L., Christensen, K., Xia, Y., Florido, M. H., He, W., Pan, W., Prummer, M., Warren, C. R., Jakob-Roetne, R., Certa, U., Jagasia, R., Freskgard, P. O., Adatto, I., Kling, D., Huang, P., Zon, L. I., Chaikof, E. L., Gerszten, R. E., Graf, M., Iacone, R., & Cowan, C. A. (2015). Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nature Cell Biology, 17(8), 994–1003.
Neri, T., Hiriart, E., van Vliet, P. P., Faure, E., Norris, R. A., Farhat, B., Jagla, B., Lefrancois, J., Sugi, Y., Moore-Morris, T., Zaffran, S., Faustino, R. S., Zambon, A. C., Desvignes, J. P., Salgado, D., Levine, R. A., de la Pompa, J. L., Terzic, A., Evans, S. M., Markwald, R., & Puceat, M. (2019). Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nature Communications, 10(1), 1929.
Harding, A., Cortez-Toledo, E., Magner, N. L., Beegle, J. R., Coleal-Bergum, D. P., Hao, D., Wang, A., Nolta, J. A., & Zhou, P. (2017). Highly efficient differentiation of endothelial cells from pluripotent stem cells requires the MAPK and the PI3K pathways. Stem Cells, 35(4), 909–919.
Descamps, B., Saif, J., Benest, A. V., Biglino, G., Bates, D. O., Chamorro-Jorganes, A., & Emanueli, C. (2018). BDNF (brain-derived neurotrophic factor) promotes embryonic stem cells differentiation to endothelial cells via a molecular pathway, including microRNA-214, EZH2 (enhancer of Zeste homolog 2), and eNOS (endothelial nitric oxide synthase). Arteriosclerosis, Thrombosis, and Vascular Biology, 38(9), 2117–2125.
Zhao, H., Zhao, Y., Li, Z., Ouyang, Q., Sun, Y., Zhou, D., Xie, P., Zeng, S., Dong, L., Wen, H., Lu, G., Lin, G., & Hu, L. (2018). FLI1 and PKC co-activation promote highly efficient differentiation of human embryonic stem cells into endothelial-like cells. Cell Death & Disease, 9(2), 131.
Olmer, R., Engels, L., Usman, A., Menke, S., Malik, M. N. H., Pessler, F., Gohring, G., Bornhorst, D., Bolten, S., Abdelilah-Seyfried, S., Scheper, T., Kempf, H., Zweigerdt, R., & Martin, U. (2018). Differentiation of human pluripotent stem cells into functional endothelial cells in scalable suspension culture. Stem Cell Reports, 10(5), 1657–1672.
Luo, Y., Liu, C., Cerbini, T., San, H., Lin, Y., Chen, G., Rao, M. S., & Zou, J. (2014). Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by AAVS1 transcription activator-like effector nucleases. Stem Cells Translational Medicine, 3(7), 821–835.
Long, C., Li, H., Tiburcy, M., Rodriguez-Caycedo, C., Kyrychenko, V., Zhou, H., Zhang, Y., Min, Y. L., Shelton, J. M., Mammen, P. P. A., Liaw, N. Y., Zimmermann, W. H., Bassel-Duby, R., Schneider, J. W., & Olson, E. N. (2018). Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Science Advances, 4(1), eaap9004.
Liu, J., Li, Y., Lin, B., Sheng, Y., & Yang, L. (2017). HBL1 is a human long noncoding RNA that modulates cardiomyocyte development from pluripotent stem cells by counteracting MIR1. Developmental Cell, 42(4), 333–348 e5.
Khan, M., Nickoloff, E., Abramova, T., Johnson, J., Verma, S. K., Krishnamurthy, P., Mackie, A. R., Vaughan, E., Garikipati, V. N., Benedict, C., Ramirez, V., Lambers, E., Ito, A., Gao, E., Misener, S., Luongo, T., Elrod, J., Qin, G., Houser, S. R., Koch, W. J., & Kishore, R. (2015). Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circulation Research, 117(1), 52–64.
Yoshida, S., Miyagawa, S., Fukushima, S., Kawamura, T., Kashiyama, N., Ohashi, F., Toyofuku, T., Toda, K., & Sawa, Y. (2018). Maturation of human induced pluripotent stem cell-derived cardiomyocytes by soluble factors from human mesenchymal stem cells. Molecular Therapy, 26(11), 2681–2695.
Poon, E. N.-y., Hao, B., et al. (2018). Integrated transcriptomic and regulatory network analyses identify microRNA-200c as a novel repressor of human pluripotent stem cell-derived cardiomyocyte differentiation and maturation. Cardiovascular Research, 114, 894–906.
Siede, D., Rapti, K., Gorska, A. A., Katus, H. A., Altmuller, J., Boeckel, J. N., Meder, B., Maack, C., Volkers, M., Muller, O. J., Backs, J., & Dieterich, C. (2017). Identification of circular RNAs with host gene-independent expression in human model systems for cardiac differentiation and disease. Journal of Molecular and Cellular Cardiology, 109, 48–56.
Kuroda, T., Yasuda, S., Tachi, S., Matsuyama, S., Kusakawa, S., Tano, K., Miura, T., Matsuyama, A., & Sato, Y. (2019). SALL3 expression balance underlies lineage biases in human induced pluripotent stem cell differentiation. Nature Communications, 10(1), 2175.
Rajasingh, J., Thangavel, J., Siddiqui, M. R., Gomes, I., Gao, X. P., Kishore, R., & Malik, A. B. (2011). Improvement of cardiac function in mouse myocardial infarction after transplantation of epigenetically-modified bone marrow progenitor cells. PLoS One, 6(7), e22550.
Cai, C. L., Liang, X., Shi, Y., Chu, P. H., Pfaff, S. L., Chen, J., & Evans, S. (2003). Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Developmental Cell, 5(6), 877–889.
Zhang, J. Z., Termglinchan, V., Shao, N. Y., Itzhaki, I., Liu, C., Ma, N., Tian, L., Wang, V. Y., Chang, A. C. Y., Guo, H., Kitani, T., Wu, H., Lam, C. K., Kodo, K., Sayed, N., Blau, H. M., & Wu, J. C. (2019). A human iPSC double-reporter system enables purification of cardiac lineage subpopulations with distinct function and drug response profiles. Cell Stem Cell, 24(5), 802–811 e5.
Lee, J. H., Protze, S. I., Laksman, Z., Backx, P. H., & Keller, G. M. (2017). Human pluripotent stem cell-derived atrial and ventricular cardiomyocytes develop from distinct mesoderm populations. Cell Stem Cell, 21(2), 179–194 e4.
Spater, D., Abramczuk, M. K., Buac, K., Zangi, L., Stachel, M. W., Clarke, J., Sahara, M., Ludwig, A., & Chien, K. R. (2013). A HCN4+ cardiomyogenic progenitor derived from the first heart field and human pluripotent stem cells. Nature Cell Biology, 15(9), 1098–1106.
Menasche, P., Vanneaux, V., Fabreguettes, J. R., Bel, A., Tosca, L., Garcia, S., Bellamy, V., Farouz, Y., Pouly, J., Damour, O., Perier, M. C., Desnos, M., Hagege, A., Agbulut, O., Bruneval, P., Tachdjian, G., Trouvin, J. H., & Larghero, J. (2015). Towards a clinical use of human embryonic stem cell-derived cardiac progenitors: a translational experience. European Heart Journal, 36(12), 743–750.
Wang, Y., Zhang, L., Li, Y., Chen, L., Wang, X., Guo, W., Zhang, X., Qin, G., He, S. H., Zimmerman, A., Liu, Y., Kim, I. M., Weintraub, N. L., & Tang, Y. (2015). Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. International Journal of Cardiology, 192, 61–69.
Bedi Jr., K. C., Snyder, N. W., Brandimarto, J., Aziz, M., Mesaros, C., Worth, A. J., Wang, L. L., Javaheri, A., Blair, I. A., Margulies, K. B., & Rame, J. E. (2016). Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation, 133(8), 706–716.
Schick, R., Mekies, L. N., Shemer, Y., Eisen, B., Hallas, T., Ben Jehuda, R., Ben-Ari, M., Szantai, A., Willi, L., Shulman, R., Gramlich, M., Pane, L. S., My, I., Freimark, D., Murgia, M., Santamaria, G., Gherghiceanu, M., Arad, M., Moretti, A., & Binah, O. (2018). Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS One, 13(10), e0205719.
Wang, B., Nie, J., Wu, L., Hu, Y., Wen, Z., Dong, L., Zou, M. H., Chen, C., & Wang, D. W. (2018). AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circulation Research, 122(5), 712–729.
Lai, L., Reineke, E., Hamilton, D. J., & Cooke, J. P. (2019). Glycolytic switch is required for transdifferentiation to endothelial lineage. Circulation, 139(1), 119–133.
Zhao, X., Chen, H., Xiao, D., Yang, H., Itzhaki, I., Qin, X., Chour, T., Aguirre, A., Lehmann, K., Kim, Y., Shukla, P., Holmstrom, A., Zhang, J. Z., Zhuge, Y., Ndoye, B. C., Zhao, M., Neofytou, E., Zimmermann, W. H., Jain, M., & Wu, J. C. (2018). Comparison of non-human primate versus human induced pluripotent stem cell-derived cardiomyocytes for treatment of myocardial infarction. Stem Cell Reports, 10(2), 422–435.
Ulmer, B. M., Stoehr, A., Schulze, M. L., Patel, S., Gucek, M., Mannhardt, I., Funcke, S., Murphy, E., Eschenhagen, T., & Hansen, A. (2018). Contractile work contributes to maturation of energy metabolism in hiPSC-derived cardiomyocytes. Stem Cell Reports, 10(3), 834–847.
Ramachandra, C. J. A., Mehta, A., Wong, P., Ja, K., Fritsche-Danielson, R., Bhat, R. V., Hausenloy, D. J., Kovalik, J. P., & Shim, W. (2018). Fatty acid metabolism driven mitochondrial bioenergetics promotes advanced developmental phenotypes in human induced pluripotent stem cell derived cardiomyocytes. International Journal of Cardiology, 272, 288–297.
Horikoshi, Y., Yan, Y., Terashvili, M., Wells, C., Horikoshi, H., Fujita, S., Bosnjak, Z. J., & Bai, X. (2019). Fatty acid-treated induced pluripotent stem cell-derived human c0ardiomyocytes exhibit adult cardiomyocyte-like energy metabolism phenotypes. Cells, 8(9), 1095.
Tallquist, M. D., & Molkentin, J. D. (2017). Redefining the identity of cardiac fibroblasts. Nature Reviews. Cardiology, 14(8), 484–491.
Chen, G., Bracamonte-Baran, W., Diny, N. L., Hou, X., Talor, M. V., Fu, K., Liu, Y., Davogustto, G., Vasquez, H., Taegtmeyer, H., Frazier, O. H., Waisman, A., Conway, S. J., Wan, F., & Cihakova, D. (2018). Sca-1(+) cardiac fibroblasts promote development of heart failure. European Journal of Immunology, 48(9), 1522–1538.
Giacomelli, E., Meraviglia, V., Campostrini, G., Cochrane, A., Cao, X., van Helden, R. W. J., Garcia, A. K., Mircea, M., Kostidis, S., Davis, R. P., van Meer, B. J., Jost, C. R., Koster, A. J., Mei, H., Miguez, D. G., Mulder, A. A., Ledesma-Terron, M., Pompilio, G., Sala, L., Salvatori, D. C. F., Slieker, R. C., Sommariva, E., de Vries, A. A. F., Giera, M., Semrau, S., Tertoolen, L. G. J., Orlova, V. V., Bellin, M., & Mummery, C. L. (2020). Human-iPSC-derived cardiac stromal cells enhance maturation in 3D cardiac microtissues and reveal non-cardiomyocyte contributions to heart disease. Cell Stem Cell, 26(6), 862–879 e11.
Bao, X., Lian, X., Qian, T., Bhute, V. J., Han, T., & Palecek, S. P. (2017). Directed differentiation and long-term maintenance of epicardial cells derived from human pluripotent stem cells under fully defined conditions. Nature Protocols, 12(9), 1890–1900.
Guadix, J. A., Orlova, V. V., Giacomelli, E., Bellin, M., Ribeiro, M. C., Mummery, C. L., Perez-Pomares, J. M., & Passier, R. (2017). Human pluripotent stem cell differentiation into functional epicardial progenitor cells. Stem Cell Reports, 9(6), 1754–1764.
Ong, C. S., Fukunishi, T., Zhang, H., Huang, C. Y., Nashed, A., Blazeski, A., DiSilvestre, D., Vricella, L., Conte, J., Tung, L., Tomaselli, G. F., & Hibino, N. (2017). Biomaterial-free three-dimensional bioprinting of cardiac tissue using human induced pluripotent stem cell derived cardiomyocytes. Scientific Reports, 7(1), 4566.
Sugiura, T., Hibino, N., Breuer, C. K., & Shinoka, T. (2016). Tissue-engineered cardiac patch seeded with human induced pluripotent stem cell derived cardiomyocytes promoted the regeneration of host cardiomyocytes in a rat model. Journal of Cardiothoracic Surgery, 11(1), 163.
Khan, M., Xu, Y., Hua, S., Johnson, J., Belevych, A., Janssen, P. M., Gyorke, S., Guan, J., & Angelos, M. G. (2015). Evaluation of changes in morphology and function of human induced pluripotent stem cell derived cardiomyocytes (HiPSC-CMs) cultured on an aligned-nanofiber cardiac patch. PLoS One, 10(5), e0126338.
Castro, L., Geertz, B., Reinsch, M., Aksehirlioglu, B., Hansen, A., Eschenhagen, T., Reichenspurner, H., Weinberger, F., & Pecha, S. (2019). Implantation of hiPSC-derived cardiac-muscle patches after myocardial injury in a Guinea pig model. Journal of Visualized Experiments, 18, 145.
Ma, J., Guo, L., Fiene, S. J., Anson, B. D., Thomson, J. A., Kamp, T. J., Kolaja, K. L., Swanson, B. J., & January, C. T. (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. American Journal of Physiology. Heart and Circulatory Physiology, 301(5), H2006–H2017.
Shinnawi, R., Shaheen, N., Huber, I., Shiti, A., Arbel, G., Gepstein, A., Ballan, N., Setter, N., Tijsen, A. J., Borggrefe, M., & Gepstein, L. (2019). Modeling reentry in the short QT syndrome with human-induced pluripotent stem cell-derived cardiac cell sheets. Journal of the American College of Cardiology, 73(18), 2310–2324.
Lancaster, J. J., Sanchez, P., Repetti, G. G., Juneman, E., Pandey, A. C., Chinyere, I. R., Moukabary, T., LaHood, N., Daugherty, S. L., & Goldman, S. (2019). Human induced pluripotent stem cell-derived cardiomyocyte patch in rats with heart failure. The Annals of Thoracic Surgery, 108(4), 1169–1177.
Lancaster, J. J., Juneman, E., Arnce, S. A., Johnson, N. M., Qin, Y., Witte, R., Thai, H., Kellar, R. S., Ek Vitorin, J., Burt, J., Gaballa, M. A., Bahl, J. J., & Goldman, S. (2014). An electrically coupled tissue-engineered cardiomyocyte scaffold improves cardiac function in rats with chronic heart failure. The Journal of Heart and Lung Transplantation, 33(4), 438–445.
Kawamura, M., Miyagawa, S., Fukushima, S., Saito, A., Miki, K., Funakoshi, S., Yoshida, Y., Yamanaka, S., Shimizu, T., Okano, T., Daimon, T., Toda, K., & Sawa, Y. (2017). Enhanced therapeutic effects of human iPS cell derived-cardiomyocyte by combined cell-sheets with omental flap technique in porcine ischemic cardiomyopathy model. Scientific Reports, 7(1), 8824.
Kawamura, M., Miyagawa, S., Fukushima, S., Saito, A., Miki, K., Ito, E., Sougawa, N., Kawamura, T., Daimon, T., Shimizu, T., Okano, T., Toda, K., & Sawa, Y. (2013). Enhanced survival of transplanted human induced pluripotent stem cell-derived cardiomyocytes by the combination of cell sheets with the pedicled omental flap technique in a porcine heart. Circulation, 128(11 Suppl 1), S87–S94.
Zhang, Y. S., Arneri, A., Bersini, S., Shin, S. R., Zhu, K., Goli-Malekabadi, Z., Aleman, J., Colosi, C., Busignani, F., Dell'Erba, V., Bishop, C., Shupe, T., Demarchi, D., Moretti, M., Rasponi, M., Dokmeci, M. R., Atala, A., & Khademhosseini, A. (2016). Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials, 110, 45–59.
Bauwens, C. L., Peerani, R., Niebruegge, S., Woodhouse, K. A., Kumacheva, E., Husain, M., & Zandstra, P. W. (2008). Control of human embryonic stem cell colony and aggregate size heterogeneity influences differentiation trajectories. Stem Cells, 26(9), 2300–2310.
Gaebel, R., Ma, N., Liu, J., Guan, J., Koch, L., Klopsch, C., Gruene, M., Toelk, A., Wang, W., Mark, P., Wang, F., Chichkov, B., Li, W., & Steinhoff, G. (2011). Patterning human stem cells and endothelial cells with laser printing for cardiac regeneration. Biomaterials, 32(35), 9218–9230.
Gaetani, R., Doevendans, P. A., Metz, C. H., Alblas, J., Messina, E., Giacomello, A., & Sluijter, J. P. (2012). Cardiac tissue engineering using tissue printing technology and human cardiac progenitor cells. Biomaterials, 33(6), 1782–1790.
Arai, K., Murata, D., Verissimo, A. R., Mukae, Y., Itoh, M., Nakamura, A., Morita, S., & Nakayama, K. (2018). Fabrication of scaffold-free tubular cardiac constructs using a bio-3D printer. PLoS One, 13(12), e0209162.
Gaetani, R., Feyen, D. A., Verhage, V., Slaats, R., Messina, E., Christman, K. L., Giacomello, A., Doevendans, P. A., & Sluijter, J. P. (2015). Epicardial application of cardiac progenitor cells in a 3D-printed gelatin/hyaluronic acid patch preserves cardiac function after myocardial infarction. Biomaterials, 61, 339–348.
Pati, F., Jang, J., Ha, D. H., Won Kim, S., Rhie, J. W., Shim, J. H., Kim, D. H., & Cho, D. W. (2014). Printing three-dimensional tissue analogues with decellularized extracellular matrix bioink. Nature Communications, 5, 3935.
Bejleri, D., Streeter, B. W., Nachlas, A. L. Y., Brown, M. E., Gaetani, R., Christman, K. L., & Davis, M. E. (2018). A bioprinted cardiac patch composed of cardiac-specific extracellular matrix and progenitor cells for heart repair. Advanced Healthcare Materials, 7(23), e1800672.
Menasche, P., Vanneaux, V., Hagege, A., Bel, A., Cholley, B., Cacciapuoti, I., Parouchev, A., Benhamouda, N., Tachdjian, G., Tosca, L., Trouvin, J. H., Fabreguettes, J. R., Bellamy, V., Guillemain, R., Suberbielle Boissel, C., Tartour, E., Desnos, M., & Larghero, J. (2015). Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: first clinical case report. European Heart Journal, 36(30), 2011–2017.
Menasche, P., Vanneaux, V., Hagege, A., Bel, A., Cholley, B., Parouchev, A., Cacciapuoti, I., Al-Daccak, R., Benhamouda, N., Blons, H., Agbulut, O., Tosca, L., Trouvin, J. H., Fabreguettes, J. R., Bellamy, V., Charron, D., Tartour, E., Tachdjian, G., Desnos, M., & Larghero, J. (2018). Transplantation of human embryonic stem cell-derived cardiovascular progenitors for severe ischemic left ventricular dysfunction. Journal of the American College of Cardiology, 71(4), 429–438.
Malliaras, K., Makkar, R. R., Smith, R. R., Cheng, K., Wu, E., Bonow, R. O., Marban, L., Mendizabal, A., Cingolani, E., Johnston, P. V., Gerstenblith, G., Schuleri, K. H., Lardo, A. C., & Marban, E. (2014). Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (CArdiosphere-Derived aUtologous stem CElls to reverse ventricUlar dySfunction). Journal of the American College of Cardiology, 63(2), 110–122.
Bartunek, J., Davison, B., Sherman, W., Povsic, T., Henry, T. D., Gersh, B., Metra, M., Filippatos, G., Hajjar, R., Behfar, A., Homsy, C., Cotter, G., Wijns, W., Tendera, M., & Terzic, A. (2016). Congestive heart failure cardiopoietic regenerative therapy (CHART-1) trial design. European Journal of Heart Failure, 18(2), 160–168.
Kim, S. H., Cho, J. H., Lee, Y. H., Lee, J. H., Kim, S. S., Kim, M. Y., Lee, M. G., Kang, W. Y., Lee, K. S., Ahn, Y. K., Jeong, M. H., & Kim, H. S. (2018). Improvement in left ventricular function with intracoronary mesenchymal stem cell therapy in a patient with anterior Wall ST-segment elevation myocardial infarction. Cardiovascular Drugs and Therapy, 32(4), 329–338.
Teerlink, J. R., Metra, M., Filippatos, G. S., Davison, B. A., Bartunek, J., Terzic, A., Gersh, B. J., Povsic, T. J., Henry, T. D., Alexandre, B., Homsy, C., Edwards, C., Seron, A., Wijns, W., Cotter, G., & Investigators, C. (2017). Benefit of cardiopoietic mesenchymal stem cell therapy on left ventricular remodelling: Results from the congestive heart failure cardiopoietic regenerative therapy (CHART-1) study. European Journal of Heart Failure, 19(11), 1520–1529.
Bartunek, J., Terzic, A., Davison, B. A., Filippatos, G. S., Radovanovic, S., Beleslin, B., Merkely, B., Musialek, P., Wojakowski, W., Andreka, P., Horvath, I. G., Katz, A., Dolatabadi, D., El Nakadi, B., Arandjelovic, A., Edes, I., Seferovic, P. M., Obradovic, S., Vanderheyden, M., Jagic, N., Petrov, I., Atar, S., Halabi, M., Gelev, V. L., Shochat, M. K., Kasprzak, J. D., Sanz-Ruiz, R., Heyndrickx, G. R., Nyolczas, N., Legrand, V., Guedes, A., Heyse, A., Moccetti, T., Fernandez-Aviles, F., Jimenez-Quevedo, P., Bayes-Genis, A., Hernandez-Garcia, J. M., Ribichini, F., Gruchala, M., Waldman, S. A., Teerlink, J. R., Gersh, B. J., Povsic, T. J., Henry, T. D., Metra, M., Hajjar, R. J., Tendera, M., Behfar, A., Alexandre, B., Seron, A., Stough, W. G., Sherman, W., Cotter, G., & Wijns, W. (2017). C. Program, Cardiopoietic cell therapy for advanced ischaemic heart failure: results at 39 weeks of the prospective, randomized, double blind, sham-controlled CHART-1 clinical trial. European Heart Journal, 38(9), 648–660.
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., & Sander, J. D. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology, 31(9), 822–826.
Shen, C. C., Hsu, M. N., Chang, C. W., Lin, M. W., Hwu, J. R., Tu, Y., & Hu, Y. C. (2019). Synthetic switch to minimize CRISPR off-target effects by self-restricting Cas9 transcription and translation. Nucleic Acids Research, 47(3), e13.
Yang, Y., Wu, H., Kang, X., Liang, Y., Lan, T., Li, T., Tan, T., Peng, J., Zhang, Q., An, G., Liu, Y., Yu, Q., Ma, Z., Lian, Y., Soh, B. S., Chen, Q., Liu, P., Chen, Y., Sun, X., Li, R., Zhen, X., Liu, P., Yu, Y., Li, X., & Fan, Y. (2018). Targeted elimination of mutant mitochondrial DNA in MELAS-iPSCs by mitoTALENs. Protein & Cell, 9(3), 283–297.
Karakikes, I., Termglinchan, V., Cepeda, D. A., Lee, J., Diecke, S., Hendel, A., Itzhaki, I., Ameen, M., Shrestha, R., Wu, H., Ma, N., Shao, N. Y., Seeger, T., Woo, N., Wilson, K. D., Matsa, E., Porteus, M. H., Sebastiano, V., & Wu, J. C. (2017). A comprehensive TALEN-based knockout library for generating human-induced pluripotent stem cell-based models for cardiovascular diseases. Circulation Research, 120(10), 1561–1571.
Tohyama, S., Fujita, J., Fujita, C., Yamaguchi, M., Kanaami, S., Ohno, R., Sakamoto, K., Kodama, M., Kurokawa, J., Kanazawa, H., Seki, T., Kishino, Y., Okada, M., Nakajima, K., Tanosaki, S., Someya, S., Hirano, A., Kawaguchi, S., Kobayashi, E., & Fukuda, K. (2017). Efficient large-scale 2D culture system for human induced pluripotent stem cells and differentiated cardiomyocytes. Stem Cell Reports, 9(5), 1406–1414.
Hamad, S., Derichsweiler, D., Papadopoulos, S., Nguemo, F., Saric, T., Sachinidis, A., Brockmeier, K., Hescheler, J., Boukens, B. J., & Pfannkuche, K. (2019). Generation of human induced pluripotent stem cell-derived cardiomyocytes in 2D monolayer and scalable 3D suspension bioreactor cultures with reduced batch-to-batch variations. Theranostics, 9(24), 7222–7238.
Chen, V. C., Ye, J., Shukla, P., Hua, G., Chen, D., Lin, Z., Liu, J. C., Chai, J., Gold, J., Wu, J., Hsu, D., & Couture, L. A. (2015). Development of a scalable suspension culture for cardiac differentiation from human pluripotent stem cells. Stem Cell Research, 15(2), 365–375.
Kempf, H., Kropp, C., Olmer, R., Martin, U., & Zweigerdt, R. (2015). Cardiac differentiation of human pluripotent stem cells in scalable suspension culture. Nature Protocols, 10(9), 1345–1361.
Protze, S. I., Liu, J., Nussinovitch, U., Ohana, L., Backx, P. H., Gepstein, L., & Keller, G. M. (2017). Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nature Biotechnology, 35(1), 56–68.
Argenziano, M., Lambers, E., Hong, L., Sridhar, A., Zhang, M., Chalazan, B., Menon, A., Savio-Galimberti, E., Wu, J. C., Rehman, J., & Darbar, D. (2018). Electrophysiologic characterization of calcium handling in human induced pluripotent stem cell-derived atrial cardiomyocytes. Stem Cell Reports, 10(6), 1867–1878.
Maass, K., Shekhar, A., Lu, J., Kang, G., See, F., Kim, E. E., Delgado, C., Shen, S., Cohen, L., & Fishman, G. I. (2015). Isolation and characterization of embryonic stem cell-derived cardiac Purkinje cells. Stem Cells, 33(4), 1102–1112.
Yeung, E., Fukunishi, T., Bai, Y., Bedja, D., Pitaktong, I., Mattson, G., Jeyaram, A., Lui, C., Ong, C. S., Inoue, T., Matsushita, H., Abdollahi, S., Jay, S. M., & Hibino, N. (2019). Cardiac regeneration using human-induced pluripotent stem cell-derived biomaterial-free 3D-bioprinted cardiac patch in vivo. Journal of Tissue Engineering and Regenerative Medicine, 13(11), 2031–2039.
Nakane, T., Masumoto, H., Tinney, J. P., Yuan, F., Kowalski, W. J., Ye, F., LeBlanc, A. J., Sakata, R., Yamashita, J. K., & Keller, B. B. (2017). Impact of cell composition and geometry on human induced pluripotent stem cells-derived engineered cardiac tissue. Scientific Reports, 7, 45641.
Kolesky, D. B., Homan, K. A., Skylar-Scott, M. A., & Lewis, J. A. (2016). Three-dimensional bioprinting of thick vascularized tissues. Proceedings of the National Academy of Sciences of the United States of America, 113(12), 3179–3184.
Raghavan, S., Nelson, C. M., Baranski, J. D., Lim, E., & Chen, C. S. (2010). Geometrically controlled endothelial tubulogenesis in micropatterned gels. Tissue Engineering. Part A, 16(7), 2255–2263.
Bettinger, C. J., Weinberg, E. J., Kulig, K. M., Vacanti, J. P., Wang, Y., Borenstein, J. T., & Langer, R. (2005). Three-dimensional microfluidic tissue-engineering scaffolds using a flexible biodegradable polymer. Advanced Materials, 18(2), 165–169.
Jia, W., Gungor-Ozkerim, P. S., Zhang, Y. S., Yue, K., Zhu, K., Liu, W., Pi, Q., Byambaa, B., Dokmeci, M. R., Shin, S. R., & Khademhosseini, A. (2016). Direct 3D bioprinting of perfusable vascular constructs using a blend bioink. Biomaterials, 106, 58–68.
C.C. NASA. STMD, https://www.nasa.gov/directorates/spacetech/centennial_challenges/vascular_tissue/about.html, (retrieved in 2020).
Bursac, N., Loo, Y., Leong, K., & Tung, L. (2007). Novel anisotropic engineered cardiac tissues: Studies of electrical propagation. Biochemical and Biophysical Research Communications, 361(4), 847–853.
Veeraraghavan, R., Lin, J., Hoeker, G. S., Keener, J. P., Gourdie, R. G., & Poelzing, S. (2015). Sodium channels in the Cx43 gap junction perinexus may constitute a cardiac ephapse: An experimental and modeling study. Pflügers Archiv, 467(10), 2093–2105.
Dvir, T., Timko, B. P., Brigham, M. D., Naik, S. R., Karajanagi, S. S., Levy, O., Jin, H., Parker, K. K., Langer, R., & Kohane, D. S. (2011). Nanowired three-dimensional cardiac patches. Nature Nanotechnology, 6(11), 720–725.
Mawad, D., Mansfield, C., Lauto, A., Perbellini, F., Nelson, G. W., Tonkin, J., Bello, S. O., Carrad, D. J., Micolich, A. P., Mahat, M. M., Furman, J., Payne, D., Lyon, A. R., Gooding, J. J., Harding, S. E., Terracciano, C. M., & Stevens, M. M. (2016). A conducting polymer with enhanced electronic stability applied in cardiac models. Science Advances, 2(11), e1601007.
Conant, G., Ahadian, S., Zhao, Y., & Radisic, M. (2017). Kinase inhibitor screening using artificial neural networks and engineered cardiac biowires. Scientific Reports, 7(1), 11807.
Rai, R., Tallawi, M., Frati, C., Falco, A., Gervasi, A., Quaini, F., Roether, J. A., Hochburger, T., Schubert, D. W., Seik, L., Barbani, N., Lazzeri, L., Rosellini, E., & Boccaccini, A. R. (2015). Bioactive electrospun fibers of poly(glycerol sebacate) and poly(epsilon-caprolactone) for cardiac patch application. Advanced Healthcare Materials, 4(13), 2012–2025.
Puig-Sanvicens, V. A., Semino, C. E., & Zur Nieden, N. I. (2015). Cardiac differentiation potential of human induced pluripotent stem cells in a 3D self-assembling peptide scaffold. Differentiation, 90(4–5), 101–110.
Ribeiro, A. J., Ang, Y. S., Fu, J. D., Rivas, R. N., Mohamed, T. M., Higgs, G. C., Srivastava, D., & Pruitt, B. L. (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proceedings of the National Academy of Sciences of the United States of America, 112(41), 12705–12710.
Abilez, O. J., Tzatzalos, E., Yang, H., Zhao, M. T., Jung, G., Zollner, A. M., Tiburcy, M., Riegler, J., Matsa, E., Shukla, P., Zhuge, Y., Chour, T., Chen, V. C., Burridge, P. W., Karakikes, I., Kuhl, E., Bernstein, D., Couture, L. A., Gold, J. D., Zimmermann, W. H., & Wu, J. C. (2018). Passive stretch induces structural and functional maturation of engineered heart muscle as predicted by computational modeling. Stem Cells, 36(2), 265–277.
Morishima, Y., Morishima, S., Murata, M., Arima, N., Uchida, N., Sugio, Y., Takahashi, S., Matsuhashi, Y., Onizuka, M., Eto, T., Nagafuji, K., Onishi, Y., Inoue, M., Atsuta, Y., Fukuda, T., Ichinohe, T., Kato, S., & Kanda, J. (2020). Impact of homozygous conserved extended HLA haplotype on single cord blood transplantation: Lessons for induced pluripotent stem cell banking and transplantation in allogeneic settings. Biology of Blood and Marrow Transplantation, 26(1), 132–138.
Pappas, D. J., Gourraud, P. A., Le Gall, C., Laurent, J., Trounson, A., DeWitt, N., & Talib, S. (2015). Proceedings: human leukocyte antigen haplo-homozygous induced pluripotent stem cell haplobank modeled after the California population: Evaluating matching in a multiethnic and admixed population. Stem Cells Translational Medicine, 4(5), 413–418.
Lee, S., Huh, J. Y., Turner, D. M., Lee, S., Robinson, J., Stein, J. E., Shim, S. H., Hong, C. P., Kang, M. S., Nakagawa, M., Kaneko, S., Nakanishi, M., Rao, M. S., Kurtz, A., Stacey, G. N., Marsh, S. G. E., Turner, M. L., & Song, J. (2018). Repurposing the cord blood Bank for Haplobanking of HLA-homozygous iPSCs and their usefulness to multiple populations. Stem Cells, 36(10), 1552–1566.
Gourraud, P. A., Gilson, L., Girard, M., & Peschanski, M. (2012). The role of human leukocyte antigen matching in the development of multiethnic "haplobank" of induced pluripotent stem cell lines. Stem Cells, 30(2), 180–186.
Riolobos, L., Hirata, R. K., Turtle, C. J., Wang, P. R., Gornalusse, G. G., Zavajlevski, M., Riddell, S. R., & Russell, D. W. (2013). HLA engineering of human pluripotent stem cells. Molecular Therapy, 21(6), 1232–1241.
Deuse, T., Hu, X., Gravina, A., Wang, D., Tediashvili, G., De, C., Thayer, W. O., Wahl, A., Garcia, J. V., Reichenspurner, H., Davis, M. M., Lanier, L. L., & Schrepfer, S. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nature Biotechnology, 37(3), 252–258.
Okada, K., Koike, K., & Sawa, Y. (2015). Consideration of and expectations for the pharmaceuticals, Medical Devices and Other Therapeutic Products Act in Japan. Regenerative Therapy, 1, 80–83.
Nagai, S., & Ozawa, K. (2017). New Japanese regulatory frameworks for clinical research and marketing authorization of gene therapy and cellular therapy products. Current Gene Therapy, 17(1), 17–28.
Okano, H., Nakamura, M., Yoshida, K., Okada, Y., Tsuji, O., Nori, S., Ikeda, E., Yamanaka, S., & Miura, K. (2013). Steps toward safe cell therapy using induced pluripotent stem cells. Circulation Research, 112(3), 523–533.
Kawamura, M., Miyagawa, S., Miki, K., Saito, A., Fukushima, S., Higuchi, T., Kawamura, T., Kuratani, T., Daimon, T., Shimizu, T., Okano, T., & Sawa, Y. (2012). Feasibility, safety, and therapeutic efficacy of human induced pluripotent stem cell-derived cardiomyocyte sheets in a porcine ischemic cardiomyopathy model. Circulation, 126(11 Suppl 1), S29–S37.
Joanne, P., Kitsara, M., Boitard, S. E., Naemetalla, H., Vanneaux, V., Pernot, M., Larghero, J., Forest, P., Chen, Y., Menasche, P., & Agbulut, O. (2016). Nanofibrous clinical-grade collagen scaffolds seeded with human cardiomyocytes induces cardiac remodeling in dilated cardiomyopathy. Biomaterials, 80, 157–168.
Zwi-Dantsis, L., Huber, I., Habib, M., Winterstern, A., Gepstein, A., Arbel, G., & Gepstein, L. (2013). Derivation and cardiomyocyte differentiation of induced pluripotent stem cells from heart failure patients. European Heart Journal, 34(21), 1575–1586.
Zhou, H., Jin, Z., Liu, J., Yu, S., Cui, Q., & Yi, D. (2008). Mesenchymal stem cells might be used to induce tolerance in heart transplantation. Medical Hypotheses, 70(4), 785–787.
Tiburcy, M., Hudson, J. E., Balfanz, P., Schlick, S., Meyer, T., Liao, M. L. C., Levent, E., Raad, F., Zeidler, S., Wingender, E., Riegler, J., Wang, M., Gold, J. D., Kehat, I., Wettwer, E., Ravens, U., Dierickx, P., van Laake, L. W., Goumans, M. J., Khadjeh, S., Toischer, K., Hasenfuss, G., Couture, L. A., Unger, A., Linke, W. A., Araki, T., Neel, B., Keller, G., Gepstein, L., Wu, J. C., & Zimmermann, W. H. (2017). Defined engineered human myocardium with advanced maturation for applications in heart failure modeling and repair. Circulation, 135(19), 1832–1847.
Su-Yi Tsai, Z. G. (2020). Hou-Jun Wang, a human embryonic stem cell reporter line for monitoring chemical-induced cardiotoxicity. Cardiovascular Research, 116, 658–670.
Funding
This research was supported by the Natural Science Foundation of Hunan Province, China (2018JJ2346).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Statement of Informed Consent
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Associate Editor: Junjie Xiao oversaw the review of this article.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jackson, A.O., Rahman, G.A., Yin, K. et al. Enhancing Matured Stem-Cardiac Cell Generation and Transplantation: A Novel Strategy for Heart Failure Therapy. J. of Cardiovasc. Trans. Res. 14, 556–572 (2021). https://doi.org/10.1007/s12265-020-10085-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-020-10085-6