
Abstract
Elongation and pathological thickening of the mitral valve (MV) is commonly seen in hypertrophic cardiomyopathy (HCM), and its pathogenic basis is poorly understood. Associated features include mal-positioning of the papillary muscles and MV, as well as systolic anterior motion (SAM) of the MV leaflets, which can worsen the turbulence and dynamic left ventricular outflow tract (LVOT) gradient. Coaptation of the MV leaflets depends on both anterior and posterior leaflet length and position, and failure of either to optimally adapt in this setting can result in mitral regurgitation or worsened LVOT obstruction. The cause of MV enlargement in HCM is not currently understood, and several different hypotheses may be relevant. The lack of correlation between MV size and the severity of left ventricular hypertrophy, as well as the early findings in genetically predisposed individuals with sarcomere mutations, suggest that it may be an intrinsic aspect of HCM in certain individuals. Other evidence points to a reactive process in the setting of excess production of paracrine growth factors in diseased myocardium that may influence valve overgrowth. Improved understanding of the responsible adaptive mechanisms will pave the way for studies targeted on the prevention and treatment of MV disease in HCM.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM) is a genetic disorder typically caused by heterozygous mutations in a gene encoding an element of the cardiac sarcomere [1]. Unexplained thickening of the left ventricular (LV) walls is present among 1:500 individuals [2]. Phenotypic presentations range from asymptomatic to early sudden death or heart failure. Impaired diastolic LV filling and left ventricular outflow tract (LVOT) obstruction contribute to symptoms in HCM. LVOT obstruction is dynamic and multifactorial, including narrowing of the LVOT due to septal hypertrophy as well as further obstruction frequently mediated by systolic anterior motion (SAM) of the anterior and/or posterior leaflet of the mitral valve (MV) [3]. Optimal treatment strategies for these different aspects of HCM require attention to their individual cause and impact. Most human and murine studies to date have focused on the LV wall with particular attention to the subaortic septum, yet abnormal MV morphology and function are widely prevalent in HCM [4, 5]. The largest pathology-based investigation of MV disease in HCM investigated 94 MV from people with HCM and 45 MV from people without structural heart disease [4]. Structural MV abnormalities were present in 66% of those with HCM, as compared with only 2% of the normal controls [4]. Although limited data have been published to address the specificity of MV enlargement in HCM versus other causes of LV hypertrophy, one study compared MV size among people with aortic valve stenosis or regurgitation, with or without LV dilation, to MV size in HCM and controls [6]. In this study, MV size was increased in the setting of LV dilation, but not when aortic stenosis occurred without LV dilation despite LVH [6].
The Mitral Valve Contributes to Obstruction and Symptoms
Catheterization, echo-Doppler, and simultaneous Doppler-catheterization evaluations have demonstrated that the gradient observed in HCM represents an obstacle to systolic ventricular flow [7–9]. This acceleration of the systolic flow within the LVOT is generally due to SAM of the MV towards the upper septum. The combination of septal hypertrophy and SAM may produce an intraventricular pressure gradient measured non-invasively by continuous wave Doppler echocardiography.
The hemodynamic consequences of SAM include not only creation of a dynamic LVOT obstruction (defined as a peak instantaneous LVOT pressure gradient ≥30 mmHg), but also prolongation of ejection time and a reduction in stroke volume, while systolic coaptation of the mitral leaflets may be disrupted resulting in frequent mitral regurgitation (MR). The presence of such a resting obstruction is an important cause of symptoms and disease progression, but it also has prognostic significance in HCM as a predictor of progression to heart failure and sudden cardiac death [10]. Recently, the presence of structural MV abnormalities has been highlighted in HCM, and its importance in generating SAM and obstruction has been emphasized, while the pathogenesis of such changes begins to be elucidated. Observations regarding the functional and structural characteristics of the MV in HCM are presented in the next sections, and they are summarized in Table 1.
SAM Is a Functional Abnormality
Abnormalities of the MV in HCM have long been thought to be limited to SAM, which was first described as a diagnostic feature of obstructive HCM, but has now also been demonstrated in the absence of HCM [11]. Roughly 30 to 60% of patients with HCM demonstrate SAM, with approximately one-third demonstrating LVOT obstruction at rest [12]. Moreover, around one-third of patients with symptomatic non-obstructive HCM at rest have latent LVOT obstruction revealed by echocardiographic examination during or in the minutes following upright cardiopulmonary exercise testing; the resulting pressure gradient is highly variable and strongly influenced by central blood volume and contractile state [13]. All symptomatic patients without evidence of a resting gradient should be investigated for dynamic LVOT obstruction using exercise or the Valsalva maneuver if the patient is unable to perform an adequate exercise test. Thus, SAM with obstruction is present in approximately two-thirds of all patients with HCM, either at rest or exercise, and there is a consensus to state that in HCM, a rough correlation exists between heart failure symptoms (dyspnea, asthenia) and the presence and importance of obstruction, and that its reduction improves functional status [14].
For years, it has been recognized that the simple attraction of the MV towards the interventricular septum due to high systolic velocities in the vicinity of the leaflets contributes to SAM (Venturi effect). These increased velocities are created by the increase in left ventricular pump function in a tight outflow tract (due to predominant upper septal hypertrophy), explaining the triggering role of exercise in obstruction and its relief or reduction by negative inotropic drugs or procedures (surgical and nonsurgical) that reduce the thickness the subaortic septum. While the Venturi effect likely contributes to the propagation and worsening of SAM, it is not adequate to initiate SAM.
SAM Is a Structural Abnormality
In vivo echocardiographic observations that the beginning of SAM occurs before outflow tract acceleration at normal LVOT velocity have highlighted complementary mechanisms centered on structural MV abnormalities [15].
Experimental Data
Experimental in vitro studies as computational works have indeed demonstrated that isolated anterior and internal displacement of the papillary muscles (PM) combined with leaflet elongation (especially of the posterior leaflet) is able to create SAM, the degree of which is related to leaflet length, even in the absence of septal hypertrophy [15–18]. Such a displacement creates a long distal residual leaflet portion interposed in the pathway of outflow. Chordal and leaflet slack is further facilitated by the presence of enlarged MV leaflets, which allows interposition of the valve within the outflow tract flow at the beginning of ejection, a prerequisite for SAM, while secondary systolic acceleration further increases obstruction due to the Venturi effect. These findings indicate that drag, the pushing force of flow, contributes to SAM, which is generated only if structural abnormalities of the MV are present [19]. Leaflet elongation promotes the development of SAM in response to PM displacement by creating long overlapping residual leaflets capable of moving anteriorly, as demonstrated by prospectively altering leaflet length. Posterior leaflet elongation also promotes SAM by shifting leaflet coaptation anteriorly, with progressive increases in SAM. Basal and mid-anterior leaflet elongation causes SAM with prolapse, while distal anterior leaflet elongation creates SAM with a mobile flap. Leaflet elongation without PM displacement creates prolapse. Residual leaflet length correlates well with total leaflet length, and the degree of SAM in turn correlates well with residual leaflet length [20].
Imaging Data
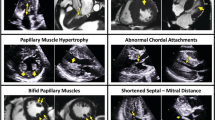
A large diversity of structural abnormalities of the MV apparatus has been demonstrated non-invasively by echocardiography in HCM patients with or without obstruction. These include: (1) malposition with anterior displacement of the MV occurring with anterior and internal displacement of the PM (Fig. 1) [21]; (2) redundant elongation of MV leaflet(s) occurring with hypertrophied PM (Fig. 2) [20]; (3) direct insertion of the anterior PM onto the anterior leaflet without interposition of chordae or accessory MV tissue (Fig. 3) [22, 23]; and (4) signs of degeneration with increased leaflet and/or chordal thicknesses, ruptured chordae, and prolapsed MV [24, 25]. These associated findings do not prove causation, but there is a recurrent theme of abnormal PM or chordae occurring in HCM in these reports.

Typical two-dimensional echocardiographic views in a normal (A) and HCM (B–F) heart; parasternal long-axis (A, B) and parasternal short-axis views (C–F). Note in B the anterior position within the LV of both papillary muscles and of the coaptation point (solid arrow) of the mitral valve; the dotted line connects the annular hinge points. Thus, at beginning of systole, the MV leaflets and chordae are interposed within ejection flow (B, transparent arrow), which then may push the leaflets and facilitate SAM at relatively low velocities. a Anterolateral papillary muscle, b posterior papillary muscle, AO aortic valve, LA left atrium, LV left ventricle, RV right ventricle. Note the anterior malposition of the anterior (APM) and posterior (PPM) papillary muscles (white arrows, C) as well as of the mitral valve in diastole (D), all essentials to the genesis of SAM of the anterior leaflet in early systole (E, F)

Two-dimensional echocardiographic apical long-axis views of the left ventricle in an HCM patient. Note the markedly elongated posterior leaflet (PL) and moderately elongated anterior leaflet (AL) on the end-diastolic frame (a). The posterior leaflet causes SAM (b) and outflow tract obstruction (OTO) seen using the color Doppler (c)

Parasternal long-axis echocardiographic view of an HCM patient with marked structural mitral valve apparatus abnormalities. Note the elongated antero-lateral papillary muscle and its direct insertion (arrow) into the AL of the MV. LA left atrium, LV left ventricle, AO aorta, RV right ventricle
In a pioneering study, structural abnormalities unique to HCM patients with SAM were demonstrated, including anterior and inward displacement of the PM and anterior displacement with elongation of the mitral leaflets, which were on average 1.5–1.7 cm longer compared to that in other subjects [21]. This suggests a mechanism for SAM, which combines the effects of LVOT narrowing with those of PM displacement. Thus, displacement of the PM alters the effectiveness of chordal support so that the central leaflet portions become relatively slack and more readily displaced anteriorly. Altered distribution of chordal tension orients the distal leaflets upward into the LVOT at the onset of systole, prior to aortic valve opening, so that ventricular ejection will actually drag the interposed leaflets anteriorly. The resolution of SAM can be understood in terms of a reverse Venturi effect created by MR, as well as continued traction of the centrally displaced PM on the lateral leaflet margins. Intraoperative transesophageal Doppler echocardiography in a small group of HCM patients undergoing myectomy found similarly longer anterior and posterior leaflets as compared to controls (31 ± 4 vs. 22 ± 3 mm and 20 ± 2 vs. 15 ± 3 mm, respectively) [26]. Transthoracic echocardiography was also able to demonstrate frequently such elongated leaflets in HCM with or without SAM and obstruction [27]. Mitral redundancy in HCM may also lead to MV prolapse instead of SAM as demonstrated by echocardiographic coexistence of prolapse and HCM in a few patients [25]. The MV opening area on a parasternal diastolic short-axis view of the left ventricle seems the best single predictor of actual mitral leaflet area measured from specimen [28].
The spectrum of morphologic variations seen echocardiographically in patients relates similarly to MV structure and, for example, posterior leaflet SAM can be related to a long scallop of the posterior leaflet moving anteriorly between the chordal connections to the anterior leaflet. These findings are consistent with recent observations that reducing leaflet redundancy and posterior leaflet height can reduce obstructive SAM following MV repair in patients with MV prolapse and can help to relieve obstruction in patients with HCM and enlarged leaflets [20]. Similarly, clinical examples abound in which obstructive SAM accompanies comparatively modest septal hypertrophy due to excessive or segmental leaflet elongation and PM malposition that fails in its basic design to restrain the leaflets posteriorly. Finally, relief of obstruction using ventricular septal ablation may not eliminate SAM of the MV and MR in patients with HCM due to anterior PM displacement and malcoaptation of MV leaflets [29].
Great inter-individual differences in MR can occur for comparable degrees of SAM. These differences relate to variations in posterior leaflet length and mobility, restricting its ability to follow the anterior leaflet and to coapt effectively. As shown in vitro and in patients studied by intraoperative transesophageal echocardiography, MR correlates inversely with the length over which the leaflets coapt. The most severe regurgitation occurs when there is a visible gap. Regurgitation worsens with increasing mismatch of anterior to posterior leaflet length and reduced posterior leaflet mobility. Thus, SAM produces greater MR if the posterior leaflet is limited in its ability to move anteriorly to coapt effectively [3].
In a large cohort of HCM patients who underwent surgery with concomitant MV procedures (48% with repair and 42% with replacement), detailed analysis of the intra- and post-operatively echocardiograms showed that valve abnormalities as assessed by echocardiography or the surgeon evoked diverse MV alterations: degenerative (31%), myxomatous (20%), PM (20%), restrictive chordal (19%), restrictive leaflet (70%) and long leaflet (56%) [30]. However, no systematic histologic examination was available.
Recently, cardiac magnetic resonance (CMR) imaging of the MV in HCM was published, and the findings parallel data acquired from prior echocardiographic imaging reports [31, 32]. A cohort of 172 individuals with HCM was compared to 172 controls, and both the AML and PML were longer in HCM (26 ± 5 vs. 19 ± 5 mm for AML, 14 ± 4 vs. 10 ± 3 mm for PML; P < 0.001 for each) [31]. There was no relationship between leaflet length and LV wall thickness or LVOT gradient.
Anatomical Data
Anatomic examination of the MV has clearly demonstrated the high frequency of such intrinsic abnormalities of the MV. In a group of highly symptomatic HCM patients who underwent surgery for relief of severe obstruction, 66% presented with significant structural MV abnormalities: increased valve area (12.9 ± 3.7 and >12 cm2 in 58% of the cases vs. 8.7 ± 2 cm2 and 1% in control subjects, respectively) largely caused by an increase in anterior leaflet length (2.2 ± .5 vs. 1.8 ± 3 cm in controls), increased scallop number of the posterior leaflet (36%), increased leaflet thickness and valve weight (around twice of the controls), and direct insertion of one PM onto the leaflet without interposition of chordae (10%) [4]. In some instances, either both leaflets or only a portion of posterior leaflet were enlarged. These abnormalities were not directly linked to left ventricular size, degree of obstruction or importance of hypertrophy [4].
Histological Data
Influence of histological changes on leaflet motion is underlined by sequential ultrasonic/histologic examinations in HCM patients [28]. As compared with normal-sized MV, the enlarged valves are situated more posteriorly in a larger LVOT and also had greater systolic excursion of the anterior leaflet with typical SAM. This pattern of SAM is possible because the central and distal portions of the leaflet are relatively free of fibrous thickening. In contrast, normal-sized MV are situated more anteriorally in a smaller LVOT and frequently show a different mechanism of SAM and LVOT obstruction with relatively limited leaflet motion, absence of a sharp bend, and septal contact involving more substantial portions of the anterior leaflet and contiguous chordae [28]. Mitral–septal apposition is facilitated by posterior ventricular septal motion, and this pattern of SAM is associated with a diffuse pattern of fibrous thickening [28].
Moreover, histological description of the MV in HCM is scarce and undeveloped. Large studies with systematic examination of excised MV in HCM patients have not been previously reported. In the study by Klues et al. [4], only 22 MV out of 94 were submitted to histological analysis. Surprisingly, they reported no myxomatous changes in the spongiosa of the MV or fibrous thickening of the fibrosa.
A major gross pathologic feature of MV disease in HCM is the change in MV geometry: the length and the surface area of both anterior and posterior leaflets are increased [4, 30]. Contrasting with this homogeneous pattern of gross pathologic abnormalities, the histological changes are highly variable in their descriptions, including plain fibrosis [4], myxomatous valves, chordae tendinae elongation or rupture leading to MV prolapse [30].
Pathogenesis of MV Disease in HCM
Several theories exist regarding the pathogenesis of MV disease in HCM, and very little experimental data have been published to address this topic. These hypotheses are outlined in the sections below.
The Genetic Hypothesis: Elongation Depends on the Gene Defect
Within the same families, posterior leaflet length is increased in healthy carriers of a morbid mutation (>14 mm in 37% of patients with left ventricular hypertrophy, 9.5% of patients without hypertrophy and less than 2% of normal relatives without the mutation) [32]. Similarly, in an MRI study, the AML was longer among 15 individuals with a sarcomere gene mutation but without LVH (genotype-positive, phenotype-negative) [31]. The presence of such an elongated valve in HCM patients without hypertrophy and obstruction suggests that this phenomenon is not adaptive to changes in ventricular geometry or consecutive to leaflet stress. Finally, observations that MYBPC3 knock-out mice, which model HCM, express MV elongation despite the absence of obstruction also support that hypothesis [33].
How a mutation in a sarcomere gene can lead to pathologic changes in the growth and structure of MV is unclear as such proteins are not abundant within the MV apparatus (except for the PMs). Cultured valvular interstitial cells express certain sarcomere genes, and their role in valvular formation, development, homeostasis, and function is currently not well understood [34]. Thus, alternative mechanisms for the observed leaflet and chordal abnormalities have been hypothesized.
The Adaptative Hypothesis: Elongation is due to a Stretch Effect
The original observations of enlarged but non-myxomatous leaflets led to the suggestion that leaflet stretching through repeated episodes of SAM was responsible. Experimentally in animals, MV area increases with mechanical stretch created by PM displacement through cell activation [35]. Thus, mechanical stresses imposed by PM tethering increase over time MV leaflet area and thickness, with cellular changes suggesting reactivated embryonic developmental pathways with endothelial–mesenchymal transdifferentiation (EMT) [35]. α-Smooth muscle actin-positive cells appear in the atrial endothelium, penetrating into the interstitium, with increased collagen deposition. Thickened chordae showed endothelial and subendothelial α-smooth muscle actin. EMT capacity was also demonstrated in cultured MV endothelial cells [35].
However, many of the patients with leaflet enlargement do not have SAM, presumably lacking other preconditions of septal and PM geometry and furthermore, anatomic studies failed to demonstrate a correlation between the degree of SAM and obstruction and the existence or importance of leaflet elongation [28]. Thus, even if present, this hypothesis fails to explain alone the changes observed.
The Concurrent Disease Hypothesis: Elongation Is a Separate Disorder
Another possibility is that MV disease concomitantly occurs in some people with HCM, resulting in abnormal MV leaflets that arise independently from the HCM. The most common MV abnormality in population studies is prolapse, and its prevalence has been variably assessed in echocardiographic studies, but recognition of the saddle shape of the MV annulus has led to more accurate diagnosis and improved ascertainment of MV disease prevalence [36]. Echocardiograms in participants in the Framingham studies indicate that the prevalence of MV prolapse is approximately 2% in controls [37]. The largest study addressing MV prolapse in HCM found it in 3% of 528 people with HCM, which might suggest that HCM and MVP are two separate concurrent diseases in some cases [25]. Yet the incidence of other MV disease is much higher in HCM, as high as 66% in one study [4]. This suggests that the MV abnormalities in HCM are intrinsic to HCM, either as a primary trait or a secondary consequence of hypertrophy and its physiologic adaptation.
The Developmental Hypothesis: Elongation is due to Paracrine Effects from the Myocardium
Valvulogenesis involves a series of cellular transformations under the influence of a family of molecular signals generated by the valve itself as well as by the surrounding myocardium [38]. That some of these same signals can affect adult valves is indicated by reactivation of EMT in adult valve endocardial cells during degenerative (myxomatous) diseases and when the valve leaflets are stretched using a surgical model of MR due to leaflet stretch and tethering [35, 39]. Thus, valve plasticity persists in adult life and it can be postulated that in HCM, the mutated myocardium may provide abnormal developmental, biochemical, and mechanical clues that alter both MV biology and PM position.
Moreover, to explain the large variety of phenotypic data observed in HCM (involving the myocardium, the MV and PM, the intramural coronary arteries, and the collagen matrix) by mutations on sarcomeric proteins, a unifying hypothesis has been recently proposed that is based on the abnormal differentiation of a subset of extracardiac mesenchymal cells that are derived from coelomic mesothelium and called epicardial-derived cells (EPDCs) [40, 41]. EPDCs migrate from the surface of the heart into the ventricular and atrial walls and normally differentiate into fibroblasts. EPDCs that migrate into the muscular interventricular septum and around the mitral–aortic continuity region, uniquely differentiate into cardiomyocytes, not fibroblasts [42, 43]. One hypothesis posits that a mutation in sarcomeric proteins can affect valve development if EPDCs — rather than becoming hypertrophic like other cardiomyocytes — differentiate or revert into fibroblast-like cells, with consecutively increased levels of periostin production. Consistent with this hypothesis, (1) markedly elevated levels of periostin are indeed expressed in HCM mice [44, 45] and (2) periostin might be part of a nodal molecular switch that determines whether a mesodermal progenitor cell becomes a cardiomyocyte or fibroblast [46]. Specifically, it has been demonstrated that blocking expression of the serum response factor (SRF) transcription factor, which is required for initiating contractile protein synthesis, causes up-regulation of periostin expression, with diminished differentiation of cardiac mesoderm into myocytes [47]. Consistent with this hypothesis, preliminary evidence exists that the pattern of periostin expression in HCM mice exactly correlates with the hypothesis that EPDC-derived cardiomyocytes become fibroblasts when a gene encoding a contractile protein is mutated [48]. One effect of elevated levels of periostin in close proximity to the MV could be to increase production of collagen, which may result in elongation of the leaflets. New valve tissue might also be added to existing valves through reawakening of the embryonic process of EMT. The potential of adult MV endocardium to rekindle its potential for EMT after TGF-β stimulation has in fact been recently shown both in vitro and in vivo [35, 49]. These findings as a whole suggest that adult heart valves can adapt to changes in their environment through either abnormal development (as with EPDCs in mice with sarcomeric protein mutations) or reactivation of embryonic valvulogenic processes (EMT).
Thus, the available evidence suggests at least four theories for MV elongation and thickening in HCM. The associated MV disease may be due to the sarcomere gene mutation, to stretch induced by turbulence in the LVOT, to concomitant familial MV disease, or to paracrine effects arising in the adjacent hypertrophic ventricle. Each of these theories appears inadequate to explain the breadth and prevalence of MV disease in HCM. Since they are not mutually exclusive, several or all of them may simultaneously or additively contribute to the MV leaflet pathology observed in HCM. The timeframe between the development of MV abnormalities and the development of obstructive physiology is also not well characterized, and longitudinal studies in people or animal models of HCM with MV disease would help to define their temporal association.
Consequences for Management
Diagnosis
In familial HCM, leaflet SAM and chordal SAM are more frequent in mutation carriers and elongation of the mitral leaflets is an indication of possible HCM in children without left ventricular hypertrophy [50]. Mitral redundancy by echocardiography is now considered as a minor criterion for HCM according to European recommendations [51].
Therapeutics
Symptomatic LVOT obstruction is often relieved by interventions that reduce the systolic thickness of the upper septum, which decrease leaflet interposition into the outflow stream and cause a posterior realignment of the PM that restrains the leaflets posteriorly. When severe symptoms are refractory to maximal drug therapy and LVOT gradient ≥50 mmHg either at rest or exercise, invasive therapies (and particularly septal reduction) may be proposed to reduce SAM and obstruction [52]. Important structural MV abnormalities (marked elongation, abnormal PM insertion) represent however contraindications to alcohol septal ablation, due to the risk of persistence of SAM and obstruction.
Surgical data suggest that concomitant MV surgery is required in 11% to 20% of patients operated on for HCM [53]. Particularly in patients with prominent leaflet enlargement or MR, adjunctive procedures have been shown to reduce the risk of persistent SAM and MR. Thus, septal myectomy can be performed with one of several different MV procedures: MV replacement, posterior–superior PM realignment, partial excision and mobilization of PM, anterior mitral leaflet plication, or anterior leaflet extension through insertion of a glutaraldehyde-treated pericardial patch (with stiffening of the mid-portion of the leaflet to prevent the bending motion required for SAM to obstruct) [54–57]. An elongated anterior mitral leaflet should influence the surgical procedure in favor of MV repair instead of replacement [30].
Although infective valve endocarditis is uncommon, individuals with HCM are at increased risk of this condition, and the risk is greater among those with LVOT obstruction in HCM [58]. Due to recent changes in the ACC/AHA guidelines for prevention of endocarditis, prophylactic antibiotic use in people with HCM is no longer recommended [59]. However, there is disagreement in the field, and some continue to advocate for the use of antibiotic prophylaxis in HCM, particularly with LVOT obstruction [60].
Conclusions
HCM commonly involves the MV. Excessive leaflet tissue, anterior displacement of the mitral apparatus, and chordal abnormalities play a primary role in obstruction. Furthermore, the MV demonstrates diverse histological features of degenerative disease, and either this or a combination of inherited and acquired factors may lead to the development of significant LVOT obstruction and MR. A thorough assessment of the MV should be performed in all patients, particularly if a septal reduction procedure is being considered. Several mechanisms may contribute to MV disease in HCM, including a primary sarcomere gene abnormality, response to shear stress in a turbulent LVOT, concomitant but unrelated MV disease, and response to paracrine factors arising from adjacent hypertrophic myocardium. Investigations that focus on animal models of HCM, as well as human studies with longitudinal follow-up and comparisons with other forms of cardiomyopathy or pressure-overload hypertrophy, should help to clarify the etiology of MV disease in HCM. Improved understanding of the responsible adaptive mechanisms will pave the way for studies targeted on the prevention and treatment of MV disease in HCM.
References
Morita, H., Nagai, R., Seidman, J. G., & Seidman, C. E. (2010). Sarcomere gene mutations in hypertrophy and heart failure. Journal of Cardiovascular Translational Research, 3, 297–303.
Maron, B. J., Gardin, J. M., Flack, J. M., Gidding, S. S., Kurosaki, T. T., & Bild, D. E. (1995). Prevalence of hypertrophic cardiomyopathy in a general population of young adults: echocardiographic analysis of 4111 subjects in the CARDIA Study. Circulation, 92, 785–789.
Schwammenthal, E., Nakatani, S., He, S., et al. (1998). Mechanism of mitral regurgitation in hypertrophic cardiomyopathy: mismatch of posterior to anterior leaflet length and mobility. Circulation, 98, 856–865.
Klues, H. G., Maron, B. J., Dollar, A. L., & Roberts, W. C. (1992). Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation, 85, 1651–1660.
Hagege, A., Desnos, M., Komajda, M., et al. (1994). Physiopathology of mitral mechanics in hypertrophic cardiomyopathy. Groupe de travail "Cardiomyopathies et insuffisance cardiaque" de la Societe Francaise de Cardiologie. Archives des Maladies du Coeur et des Vaisseaux, 87, 345–1352.
Mautner, S. L., Klues, H. G., Mautner, G. C., Proschan, M. A., Roberts, W. C., & Maron, B. J. (1993). Comparison of mitral valve dimensions in adults with valvular aortic stenosis, pure aortic regurgitation and hypertrophic cardiomyopathy. The American Journal of Cardiology, 71, 949–953.
Buda, A. J., MacKenzie, G. W., & Wigle, E. D. (1981). Effect of negative intrathoracic pressure on left ventricular outflow tract obstruction in muscular subaortic stenosis. Circulation, 63, 875–881.
Spirito, P., & Maron, B. J. (1984). Patterns of systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy: assessment by two-dimensional echocardiography. The American Journal of Cardiology, 54, 1039–1046.
Sasson, Z., Henderson, M., Wilansky, S., Rakowski, H., & Wigle, E. D. (1989). Causal relation between the pressure gradient and left ventricular ejection time in hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 13, 1275–1279.
Maron, B. J. (2003). Sudden death in young athletes. The New England Journal of Medicine, 349, 1064–1075.
Luckie, M., & Khattar, R. S. (2008). Systolic anterior motion of the mitral valve—beyond hypertrophic cardiomyopathy. Heart, 94, 1383–1385.
Maron, B. J., & Epstein, S. E. (1980). Hypertrophic cardiomyopathy. Recent observations regarding the specificity of three hallmarks of the disease: asymmetric septal hypertrophy, septal disorganization and systolic anterior motion of the anterior mitral leaflet. The American Journal of Cardiology, 45, 141–154.
Shah, J. S., Esteban, M. T., Thaman, R., et al. (2008). Prevalence of exercise-induced left ventricular outflow tract obstruction in symptomatic patients with non-obstructive hypertrophic cardiomyopathy. Heart, 94, 1288–1294.
Maron, M. S., Olivotto, I., Zenovich, A. G., et al. (2006). Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation, 114, 2232–2239.
Lefebvre, X. P., Yoganathan, A. P., & Levine, R. A. (1992). Insights from in-vitro flow visualization into the mechanism of systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy under steady flow conditions. Journal of Biomechanical Engineering, 114, 406–413.
Lefebvre, X. P., He, S., Levine, R. A., & Yoganathan, A. P. (1995). Systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy: an in vitro pulsatile flow study. The Journal of Heart Valve Disease, 4, 422–438.
Yoganathan, A. P., Lemmon, J. D., Jr., Kim, Y. H., Levine, R. A., & Vesier, C. C. (1995). A three-dimensional computational investigation of intraventricular fluid dynamics: examination into the initiation of systolic anterior motion of the mitral valve leaflets. Journal of Biomechanical Engineering, 117, 94–102.
Levine, R. A., Vlahakes, G. J., Lefebvre, X., et al. (1995). Papillary muscle displacement causes systolic anterior motion of the mitral valve. Experimental validation and insights into the mechanism of subaortic obstruction. Circulation, 91, 1189–1195.
Sherrid, M. V., Gunsburg, D. Z., Moldenhauer, S., & Pearle, G. (2000). Systolic anterior motion begins at low left ventricular outflow tract velocity in obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 36, 1344–1354.
He, S., Hopmeyer, J., Lefebvre, X. P., Schwammenthal, E., Yoganathan, A. P., & Levine, R. A. (1997). Importance of leaflet elongation in causing systolic anterior motion of the mitral valve. The Journal of Heart Valve Disease, 6, 149–159.
Jiang, L., Levine, R. A., King, M. E., & Weyman, A. E. (1987). An integrated mechanism for systolic anterior motion of the mitral valve in hypertrophic cardiomyopathy based on echocardiographic observations. American Heart Journal, 113, 633–644.
Klues, H. G., Roberts, W. C., & Maron, B. J. (1991). Anomalous insertion of papillary muscle directly into anterior mitral leaflet in hypertrophic cardiomyopathy. Significance in producing left ventricular outflow obstruction. Circulation, 84, 1188–1197.
Musumeci, B., Spirito, P., Parodi, M. I., Assenza, G. E., & Autore, C. (2011). Congenital accessory mitral valve tissue anomaly in a patient with genetically confirmed hypertrophic cardiomyopathy. Journal of the American Society of Echocardiography, 24(592), e5–e6.
Zhu, W. X., Oh, J. K., Kopecky, S. L., Schaff, H. V., & Tajik, A. J. (1992). Mitral regurgitation due to ruptured chordae tendineae in patients with hypertrophic obstructive cardiomyopathy. Journal of the American College of Cardiology, 20, 242–247.
Petrone, R. K., Klues, H. G., Panza, J. A., Peterson, E. E., & Maron, B. J. (1992). Coexistence of mitral valve prolapse in a consecutive group of 528 patients with hypertrophic cardiomyopathy assessed with echocardiography. Journal of the American College of Cardiology, 20, 55–61.
Grigg, L. E., Wigle, E. D., Williams, W. G., Daniel, L. B., & Rakowski, H. (1992). Transesophageal Doppler echocardiography in obstructive hypertrophic cardiomyopathy: clarification of pathophysiology and importance in intraoperative decision making. Journal of the American College of Cardiology, 20, 42–52.
Pai, R. G., Jintapakorn, W., Tanimoto, M., & Shah, P. M. (1995). Role of papillary muscle position and mitral valve structure in systolic anterior motion of the mitral leaflets in hyperdynamic left ventricular function. The American Journal of Cardiology, 76, 623–628.
Klues, H. G., Roberts, W. C., & Maron, B. J. (1993). Morphological determinants of echocardiographic patterns of mitral valve systolic anterior motion in obstructive hypertrophic cardiomyopathy. Circulation, 87, 1570–1579.
Delling, F. N., Sanborn, D. Y., Levine, R. A., et al. (2007). Frequency and mechanism of persistent systolic anterior motion and mitral regurgitation after septal ablation in obstructive hypertrophic cardiomyopathy. The American Journal of Cardiology, 100, 1691–1695.
Kaple, R. K., Murphy, R. T., DiPaola, L. M., et al. (2008). Mitral valve abnormalities in hypertrophic cardiomyopathy: echocardiographic features and surgical outcomes. The Annals of Thoracic Surgery, 85, 1527–1535. 1535 e1–e2.
Maron, M. S., Olivotto, I., Harrigan, C., et al. (2011). Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation, 124, 40–47.
Hagege, A. A., Dubourg, O., Desnos, M., et al. (1998). Familial hypertrophic cardiomyopathy. Cardiac ultrasonic abnormalities in genetically affected subjects without echocardiographic evidence of left ventricular hypertrophy. European Heart Journal, 19, 490–499.
Nematalla, H., Vignier, N., Judge, D. P., et al. (2011). Targeted Mybpc3 knock-out mice with non-obstructive hypertrophic cardiomyopathy exhibit structural mitral valve abnormalities. Journal of the American College of Cardiology, 57(Suppl), E1397.
Brand, N. J., Roy, A., Hoare, G., Chester, A., & Yacoub, M. H. (2006). Cultured interstitial cells from human heart valves express both specific skeletal muscle and non-muscle markers. The International Journal of Biochemistry & Cell Biology, 38, 30–42.
Dal-Bianco, J. P., Aikawa, E., Bischoff, J., et al. (2009). Active adaptation of the tethered mitral valve. insights into a compensatory mechanism for functional mitral regurgitation. Circulation, 334–342.
Levine, R., Triulzi, M., Harrigan, P., & Weyman, A. (1987). The relationship of mitral annular shape to the diagnosis of mitral valve prolapse. Circulation, 75, 756–767.
Freed, L. A., Benjamin, E. J., Levy, D., et al. (2002). Mitral valve prolapse in the general population: the benign nature of echocardiographic features in the Framingham Heart Study. Journal of the American College of Cardiology, 40, 1298–1304.
Butcher, J. T., & Markwald, R. R. (2007). Valvulogenesis: the moving target. Philosophical Transactions of the Royal Society of London B, Biological Sciences, 362, 1489–1503.
Rabkin, E., Aikawa, M., Stone, J. R., Fukumoto, Y., Libby, P., & Schoen, F. J. (2001). Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation, 104, 2525–2532.
Olivotto, I., Cecchi, F., Poggesi, C., & Yacoub, M. H. (2009). Developmental origins of hypertrophic cardiomyopathy phenotypes: a unifying hypothesis. Nature Reviews Cardiology, 6, 317–321.
Gittenberger-de Groot, A. C., Vrancken Peeters, M. P., Mentink, M. M., Gourdie, R. G., & Poelmann, R. E. (1998). Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circulation Research, 82, 1043–1052.
Cai, C. L., Martin, J. C., Sun, Y., et al. (2008). A myocardial lineage derives from Tbx18 epicardial cells. Nature, 454, 104–108.
Zhou, B., Ma, Q., Rajagopal, S., et al. (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature, 454, 109–113.
Rajan, S., Williams, S. S., Jagatheesan, G., et al. (2006). Microarray analysis of gene expression during early stages of mild and severe cardiac hypertrophy. Physiological Genomics, 27, 309–317.
Prabhakar, R., Petrashevskaya, N., Schwartz, A., et al. (2003). A mouse model of familial hypertrophic cardiomyopathy caused by a alpha-tropomyosin mutation. Molecular and Cellular Biochemistry, 251, 33–42.
Norris, R. A., Moreno-Rodriguez, R. A., Sugi, Y., et al. (2008). Periostin regulates atrioventricular valve maturation. Developmental Biology, 316, 200–213.
Niu, Z., Iyer, D., Conway, S. J., et al. (2008). Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proceedings of the National Academy of Sciences of the United States of America, 105, 17824–17829.
Norris, R. A., Kern, C. B., Wessels, A., Moralez, E. I., Markwald, R. R., & Mjaatvedt, C. H. (2004). Identification and detection of the periostin gene in cardiac development. The Anatomical Record. Part A, Discoveries in Molecular, Cellular, and Evolutionary Biology, 281, 1227–1233.
Rabkin-Aikawa, E., Mayer, J. E., Jr., & Schoen, F. J. (2005). Heart valve regeneration. Advances in Biochemical Engineering/Biotechnology, 94, 141–179.
Watkins, H., Conner, D., Thierfelder, L., et al. (1995). Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nature Genetics, 11, 434–437.
McKenna, W. J., Spirito, P., Desnos, M., Dubourg, O., & Komajda, M. (1997). Experience from clinical genetics in hypertrophic cardiomyopathy: proposal for new diagnostic criteria in adult members of affected families. Heart, 77, 130–132.
Maron, B. J., McKenna, W. J., Danielson, G. K., et al. (2003). American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. European Heart Journal, 24, 1965–1991.
Yu, E. H., Omran, A. S., Wigle, E. D., Williams, W. G., Siu, S. C., & Rakowski, H. (2000). Mitral regurgitation in hypertrophic obstructive cardiomyopathy: relationship to obstruction and relief with myectomy. Journal of the American College of Cardiology, 36, 2219–2225.
Reis, R. L., Bolton, M. R., King, J. F., Pugh, D. M., Dunn, M. I., & Mason, D. T. (1974). Anterion–superior displacement of papillary muscles producing obstruction and mitral regurgitation in idiopathic hypertrophic subaortic stenosis. Operative relief by posterior–superior realignment of papillary muscles following ventricular septal myectomy. Circulation, 50, II181–II188.
Schoendube, F. A., Klues, H. G., Reith, S., Flachskampf, F. A., Hanrath, P., & Messmer, B. J. (1995). Long-term clinical and echocardiographic follow-up after surgical correction of hypertrophic obstructive cardiomyopathy with extended myectomy and reconstruction of the subvalvular mitral apparatus. Circulation, 92, II122–II127.
McIntosh, C. L., Maron, B. J., Cannon, R. O., & Klues, H. G. (1992). Initial results of combined anterior mitral leaflet plication and ventricular septal myotomy–myectomy for relief of left ventricular outflow tract obstruction in patients with hypertrophic cardiomyopathy. Circulation, 86, II60–II67.
Kofflard, M. J., van Herwerden, L. A., Waldstein, D. J., et al. (1996). Initial results of combined anterior mitral leaflet extension and myectomy in patients with obstructive hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 28, 197–202.
Spirito, P., Rapezzi, C., Bellone, P., et al. (1999). Infective endocarditis in hypertrophic cardiomyopathy: prevalence, incidence, and indications for antibiotic prophylaxis. Circulation, 99, 2132–2137.
Wilson, W., Taubert, K. A., Gewitz, M., et al. (2007). Prevention of infective endocarditis: guidelines from the American Heart Association. Circulation, 116, 1736–1754.
Maron, B. J., & Lever, H. (2009). In defense of antimicrobial prophylaxis for prevention of infective endocarditis in patients with hypertrophic cardiomyopathy. Journal of the American College of Cardiology, 54, 2339–2340. author reply 2340.
Acknowledgement
This research project was funded by grant 07CVD04 from the Leducq Foundation, Paris, France, for the Leducq MITRAL Transatlantic Network.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hagège, A.A., Bruneval, P., Levine, R.A. et al. The Mitral Valve in Hypertrophic Cardiomyopathy. J. of Cardiovasc. Trans. Res. 4, 757–766 (2011). https://doi.org/10.1007/s12265-011-9319-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-011-9319-6