
Abstract
Proper atrioventricular canal (AVC) patterning and subsequent valvulogenesis is a complex process, and defects can result in disease or early death. The zebrafish Danio rerio has become a useful model system for studying AVC development, and much progress has been made in dissecting out the critical steps. Here, we review the recent advances in the field and highlight the cellular and molecular changes observed during zebrafish AVC development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proper atrioventricular canal (AVC) development is essential for dividing the heart into the appropriate chambers and setting up the morphological milieu that allows for normal heart valve formation. Defects in AVC development can lead to early death or result in valvular diseases that affect the unidirectional flow of blood through the heart. There is some inherent difficulty to studying AVC development in higher vertebrates, primarily due to the early lethality that results from essential gene knockouts.
AVC development can be studied quite easily in the zebrafish Danio rerio. The early heart is a simple tube that is divided into a ventricle and atrium by the AV canal. The heart develops rapidly over a 4-day period, and during this time, the zebrafish embryo is transparent, allowing observation of development without the detriment of invasive techniques. A further advantage lies in the small size of the embryo. So small in fact, that oxygen can diffuse directly into the embryo for the first 4 days, rendering the need for intact circulation irrelevant. This allows the process of cardiac development to be probed using chemical and genetic disruption techniques without the embryonic lethality seen in higher organisms.
An array of markers has been used to determine the role of numerous components in zebrafish AVC development. Transgenic fluorescence expression, in situ hybridization techniques, and antibody staining in conjunction with confocal microscopy can be used to great advantage in zebrafish, as embryo transparency allows for simple whole-mount observation.
Recent zebrafish research has shed light on atrioventricular canal development. Chemical and genetic knockdowns, along with overexpression studies, have resulted in the discovery and characterization of multiple genes and signaling pathways that are essential for AVC and subsequent valve formation. In this review, we will highlight many of these discoveries by examining how the AVC forms at the organ, cellular, and molecular level over time.
Atrioventricular Canal Formation
General AVC Formation in Vertebrates
Work in chick and mouse has delineated the basic process by which the AVC develops, matures, and forms valves [1]. In the region between the developing atrium and ventricle, endothelial cells detach from their initial cell layer and migrate into the extracellular matrix (ECM) separating the inner endocardium from the outer myocardial cells. The ECM is rich in collagen and long-chain sugars attached to core proteins, termed proteoglycans. Migration of the endothelial cells into the ECM is followed by transdifferentiation into mesenchymal cells, a process known as the endothelial to mesenchyme transition (EMT). The cellularized ECM creates a cardiac cushion, which then pushes out the endothelial layer and eventually provides the substrate for development of the mature valve. While there are some differences in this process in zebrafish, we can break this development into three general steps: (1) early AV canal patterning, (2) endocardial cushion formation, and (3) valve maturation.
Early AV Canal Patterning (22–48 h Post-fertilization)
Cellular and Organ Development
The primitive heart in zebrafish is formed by cardiac progenitor cells that produce two bilateral tubular cardiac primordia which fuse to form the definitive heart tube by 24 h post-fertilization (hpf) [2] (Fig. 1). The heart is actively beating at this time, and circulation of the blood has started. Although structurally still a tube, the future atrium can be delineated from the ventricle using antibody staining [3]. The heart tube is composed of a single layer of myocardial cells surrounding a layer of endocardial cells. Surprisingly, the endocardial layer is not absolutely required for this early stage of organogenesis. The cloche mutant is missing the endocardium; yet, the heart tube still forms and beats allowing blood to circulate [4].

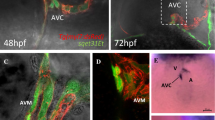
Morphological changes in the zebrafish atrioventricular canal between 24 and 96 h post-fertilization. Dark red ventricular myocardium, light red ventricular endocardium, green atrial myocardium, light green atrial endocardium, rectangles cuboidal cells, ovals squamous or trapezoidal cells, grayish blue Dm-grasp expression, sky blue chondroitin sulfate expression, dark blue Z01 expression, A atrium, V ventricle
Between 30 and 36 hpf, the heart tube loops, and a clear constriction separates the atrium from the ventricle. Cellular changes in the pre-valve AVC endocardium begin to occur. Early endocardial cells appear to be squamous in shape, but at 36 h, cells at the atrioventricular border start adopting a cuboidal shape [5]. Furthermore, these cuboidal endocardial cells start expressing Dm-grasp (a cell surface adhesion molecule) on their lateral surfaces. Squamous endocardial cells do not express this molecule. These changes have been suggested to be the first signs of endocardial transdifferentiation within the developing AVC [5].
Cellular changes also occur to the AVC myocardium. By 40 hpf, atrial myocardial cells display a squamous appearance, while ventricular myocardial cells are cuboidal. In contrast, AVC cells have a trapezoid morphology, due to a widened basolateral surface, and an extended apical surface [6]. Much like the endocardium, strong expression of Dm-grasp is observed in the AVC myocardium, although weaker expression is observed in myocardial cells of the atrium and ventricle [5]. The functional relevance behind the appearance of Dm-grasp in the AVC has yet to be determined.
These changes in cellular morphology are in part controlled by an array of T-box transcription factors. Foxn4 and Tbx5 switch on Tbx2 in early myocardial cells of the AVC, and knockdown of foxn4 or tbx2 results in myocardial cells that do not change into a trapezoid morphology and endocardial cells that remain squamous [6]. Similarly, knockdown of pdlim7, which interacts with tbx5, results in endocardial cells remaining squamous and failing to express Dm-grasp [7].
Thus, the earliest features of AVC patterning are heralded by morphologic changes in both endothelial and myocardial AVC cells. These alterations in cellular morphology likely reflect changes in cell differentiation or fate and are dependent on T-box transcription factors including Tbx2 and Tbx5.
Formation of the Cardiac Jelly
By 33 hpf, the cardiac jelly has formed between the endocardial and myocardial layers of the heart tube. The cardiac jelly in other vertebrates has been shown to be a complex mixture of extracellular matrix components, including proteoglycans, hyaluronic acid, and collagen [8]. Proteoglycans are complex molecules consisting of a long-chain sugar, such as chondroitin sulfate (CS), covalently attached to a core protein. There are several core proteins known to be important in AVC and valve development, including versican and aggrecan, but the role of the attached sugar polymer has only recently been addressed. Using an antibody against chondroitin sulfate proteoglycans (CSPGs) it has been demonstrated that this complex molecule is detected between the endocardial and myocardial layers of the zebrafish heart tube at approximately 33 hpf [9]. Chondroitin expression is initially observed throughout the heart tube, although heaviest in the ventricle. By 36 hpf, CS becomes more restricted to the developing atrioventricular canal and outflow tract. CS is always covalently attached to a core protein, and the gene versican may encode the relevant core protein. In situ hybridization reveals that versican is initially expressed throughout the myocardium of the heart tube, but at 37 hpf becomes restricted to myocardial cells in the same region as CS [10]. If versican were the cognate core protein for CS, this would imply that this CSPG is initially expressed by cardiomyocytes before being secreted into the ECM.
Another component of the cardiac jelly is the long-chain sugar hyaluronic acid (HA). Although no report has yet to demonstrate the presence of HA in the zebrafish heart, has-2, the gene encoding the enzyme that produces HA, is expressed within the AVC endocardium at 48 hpf [11].
Bidirectional Endocardial/Myocardial Signaling in the AVC
The myocardial and endocardial cells in the developing zebrafish AVC appear to communicate to each other across the cardiac jelly, specifically through the bmp and notch signaling pathways. Early in development, bmp4 is expressed throughout the myocardium, but at 37 hpf becomes restricted to myocardial cells in the AVC [10]. BMPs are part of the TGF-β superfamily of signaling molecules, which are known to play important roles in valve development in other vertebrates [1]. Multiple BMP ligand and receptor pairs can interact at the cell surface, which ultimately results in activation of cytoplasmic SMAD proteins that regulate gene expression [12]. SMAD activation can be observed using a transgenic zebrafish that has a SMAD-responsive element driving a GFP reporter [13]. Using this line, it was demonstrated that SMADs are activated, possibly through BMP, in the endocardium of the primitive heart tube by 24 hpf, but later in myocardial cells of the AVC by 48 hpf. The early endocardial activation in this line is interesting, as bmp4 expression is myocardial at this time, suggesting that the transgene is activated in the endothelium by BMP4 signals from the myocardium.
Similar signaling between the cell layers is observed for notch1b. Notch signaling is an evolutionarily conserved pathway that is critically important for cell fate decisions and communication between adjacent cells [14]. Initially, notch1b is expressed throughout the endocardial layer of the developing heart, but by 45 hpf becomes primarily restricted to the endocardial valve forming cells of the AVC [10, 15]. Notch knockdown in zebrafish results in loss of myocardial differentiation into the slowly conducting phenotype of the mature AV ring [16]. Thus, loss of endocardial notch expression results in a myocardial AVC phenotype providing another example of signaling between the endocardial and myocardial layers.
Regulation of Endothelial Division via VEGF/Calcineurin/NFAT Signaling
VEGF signaling plays an important dual role during AVC development. Early hints of this role were observed in a zebrafish transgenic line that used the murine tie-2 promoter to drive GFP [17]. Tie-2 is the VEGF receptor expressed in vascular endothelial tissue. This GFP fusion resulted in surprisingly specific upregulation of GFP in endocardial valve precursor cells starting around 43 hpf [10, 17]. While the mechanism behind this specific expression pattern has yet to be determined, the implication is that VEGF must be important in AVC and valve formation. Indeed, there appears to be an essential early role for the VEGF signaling pathway during AVC development, which is controlled by the calcineurin/nuclear factor of activated T cells (NFAT) pathway. Chemical inhibition of VEGF signaling at 17–19 hpf results in the failure of the AVC to properly develop [18]. Thus, VEGF is essential for AVC formation at 17–19 hpf; however repression of VEGF by calcineurin/NFAT is needed from 20–33 hpf. Using either cyclosporine A or FK506 to block calcineurin/NFAT signaling between 22 and 30 hpf in zebrafish results in the failure of the AVC to mature and form valves [19]. Endocardial cells do undergo the change in cell shape from squamous to cuboidal, but Dm-grasp is not expressed [5]. These calcineurin signaling defects could be rescued by adding a soluble protein inhibitor of the VEGF pathway [19]. It is hypothesized that VEGF causes endothelial cell marker changes and division [18, 19], which may be needed early on, but could be disruptive to proper AV canal formation at the later stage.
Summary
As the zebrafish heart changes from a linear tube to a looped structure with specified chambers, the AVC becomes an important region that separates the atrium from the ventricle. An interesting theme that emerges is the change in expression of multiple genes from heart-wide to specific cells within the AVC. Chondroitin sulfate, versican, has-2, bmp4, and notch1b all undergo such changes. Furthermore, the changes in cellular structure and morphology support a model where AVC cells are patterned to a specific type of tissue that differs from both atrium and ventricle.
Endocardial Cushion Formation (48–96 hpf)
Organ Development
By 72 hpf, the heart has finished looping, and defined atrial and ventricular chambers exist, along with an inflow chamber before the atrium (the sinus venosus) and an outflow chamber after the ventricle (the bulbous arteriosus). The myocardial and endocardial layers are primarily one-cell thick while the ventricular myocardium begins to thicken after 96 hpf [20]. During this time, the heart is contracting, and circulation has become vigorous throughout the zebrafish. Although discrete valve leaflets have not fully developed, blood is kept flowing unidirectionally through the apposition of the cardiac cushions within the AVC [2, 20].
Endocardial Cushions
In other vertebrate model systems, the endocardial cushions (EC) are defined as the structures that appear after endothelial cells have migrated away from the endothelial layer, entered the cardiac jelly, and transdifferentiated into mesenchyme cells (EMT). In zebrafish, however, there has been little definitive evidence that the EMT process occurs. Using the terminology that has evolved in the zebrafish field, we will therefore define the zebrafish cardiac cushion as the thickened extracellular matrix and endothelial cell layer that appears in the AV canal, including the pre-valve endothelium.
By 55 hpf, a single layer of about 20 cuboidal endothelial cells have developed on each side of the AV canal, forming the early endocardial cushion [5]. It is proposed that the EC at the inner curvature of the developing heart be termed the superior EC, while that at the outer curvature be the inferior EC. The cuboidal cells express the surface adhesion marker Dm-grasp on their lateral sides. All the endocardial cells express ZO1, a marker of tight junctions between the cells. By 60 hpf, specific endocardial cells of the superior endocardial cushion have lost ZO1 expression (perhaps indicative of EMT) and have sent cellular extensions into the cardiac jelly. The inferior EC follows suit at 80 hpf. Both zebrafish cushions have formed by 96 hpf. The differential development of these cushions is interesting to note. In other vertebrate species, the major endocardial cushions form first, followed by two other mural or lateral cushions. While the cushions observed in zebrafish have been assumed to both be major cushions, another hypothesis is that the earlier developing superior EC is the major cushion, while the inferior EC is a lateral structure.
Myocardial cells of the AVC also have a specific cell shape at this time that is reliant on the cyclooxygenase (Cox) pathway. The myocardial cells overlying the superior EC are large and elongated, but when treated with Cox2 inhibitors, become smaller and rounder [21]. Treatment of zebrafish embryos with the widely prescribed Cox2 inhibitor celecoxib results in failure of the AVC to mature [22].
The Role of the Cardiac Jelly and Extracellular Matrix
Genetic clues about AVC formation in zebrafish were first gained in a large-scale mutagenesis screen [23]. One of the mutants isolated was jekyll, which lacks an AV valve, causing the blood to toggle back and forth between the atrium and the ventricle. This results in lack of proper circulation and eventual death. The mutated locus encodes the gene UDP-glucose dehydrogenase (ugdh). This enzyme is needed to synthesize three important glycosaminoglycans (GAGs): chondroitin sulfate, heparan sulfate, and hyaluronic acid. Presumably, the jekyll phenotype is due to the lack of one or more of these GAGs. Interestingly, mice with mutations in the chondroitin sulfate core protein versican have a similar AV canal phenotype, as do mice defective in hyaluronic acid synthesis [24, 25].
As a start in determining the relevant GAG, CS was experimentally downregulated by both morpholino knockdown of the chondroitin synthase-1 gene (chys-1) and the CS chemical inhibitor DX [9]. Normally at 48 hpf, chondroitin sulfate is localized primarily to the cardiac jelly in AVC and outflow tract. Both chemical and genetic downregulation of CS lead to a jekyll-like phenotype. We note that CS disruption does not completely phenocopy jekyll, however, suggesting that each GAG has unique functions and that loss of all GAGs in jekyll mutants affect multiple pathways in a complex manner. This hypothesis is strengthened by the differences in signaling pathway expression observed in jekyll and CS-depleted zebrafish embryos. For example, notch1b loses AVC restriction and is expressed throughout the heart in jekyll mutants, but completely disappears during CS downregulation. Both the jekyll and CS results imply that GAGs are responsible for setting up the boundary between the atrium and ventricle and patterning the developing AV canal.
There have been some clues on how normal formation of the cardiac jelly is controlled. Micro-RNAs (miRs) have recently been shown to be important molecular modulators for numerous signaling pathways and processes [26]. Binding of miRs to the 3′ end of target mRNA usually results in downregulation or prevention of protein translation. Overexpression of miR-138 causes AVC defects comparable to jekyll mutants, and examination of predicted target genes has revealed that versican, the gene that encodes the putative core protein for chondroitin in the cardiac jelly, has a miR-138 binding site [27]. Prevention of miR production should cause upregulation of the target gene, and indeed, morpholino knockdown of miR-138 causes expansion of versican throughout the ventricle (instead of the normal AVC restriction). The retinoic acid pathway has also been implicated in this process, as the gene encoding retinoic acid dehydrogenase (raldh1a2) has a miR-138 binding site, and treatment of fish with RA also causes versican expansion throughout the ventricle [27]. It appears that miR-138 normally downregulates both raldh1a2 and versican within the ventricle in order to keep both gene products localized to the AVC, while RA production through Aldh1a2 upregulates versican.
Notch Signaling
Restriction of notch1b from the ventricle to specific cells within the AVC after 45 h [10, 15] implies a role for Notch signaling in AVC or cardiac cushion development. As part of a broader study examining EMT in mice, Timmerman et al. increased Notch signaling by injecting a constitutively active form of notch mRNA into zebrafish embryos [28]. This resulted in hypercellularized, thickened cardiac cushions. In order to perform the inverse experiment, the authors added DAPT, an inhibitor of Notch signaling. This prevented cardiac cushions from developing into valves. Beis et al. further demonstrated that Notch knockdown causes ventricular endocardial squamous cells to adopt a cuboidal shape, as well as express Dm-grasp [5]. Recall that these seem to be indicators of endocardial cushion formation. But unlike the Timmerman study, Beis reports that AVC endocardial cushions do form in the presence of a Notch inhibitor, although they are disorganized, and Dm-grasp is downregulated. These results imply that notch is needed at early stages to prevent an AV cushion phenotype in the ventricle, but later on to cause cushion formation in the AVC.
Wnt/β-Catenin Signaling
The Wnt/β-catenin signaling pathway is highly conserved from worms to mammals and is needed for multiple developmental processes [29]. Wnt signaling controls the translocation of β-catenin into the nucleus to allow for expression of target genes. In order to study the putative role of Wnt signaling in cardiac development, a zebrafish line with a truncation in the Apc gene was generated, which causes constitutive activation of the Wnt/β-catenin pathway [11]. This resulted in zebrafish embryos with hearts that failed to loop and a thickening of both the endocardium and cardiac jelly. The endocardial cell layer was considerably thickened in the AV canal at 72 hpf, in the region where valves should eventually form. Conversely, when β-catenin signaling was blocked by overexpression of either full-length Apc or by the Wnt inhibitor Dkk1, hearts failed to loop, and endocardial cushions failed to develop. The Apc deletion and subsequent upregulation of the Wnt/β-catenin pathway led to changes in multiple cardiac markers. The authors note that the Apc phenotype resembles jekyll and hypothesize that jekyll mutants are deficient in valves due to defects in Wnt signaling through two lines of reasoning: (1) the jekyll Drosophila homologue sugarless leads to defects in Wnt signaling, and (2) jekyll mutants and Wnt/β-catenin disruption both lead to a lack of endocardial cushions.
In a similar manner, plakoglobin, a β-catenin homologue in zebrafish, can antagonize Wnt signaling. Morpholino downregulation of plakoglobin results in an increase in Wnt signaling, expansion of bmp4 and notch1b, and a defect in AVC formation [30]. Analysis of plakoglobin morphants at 72 hpf reveals that instead of a sheet of cuboidal endothelial cells, morphology is irregular, with a decrease in the number of cell junctions. Co-injection of the morpholino with the Wnt inhibitor Dkk1 rescued these phenotypes. Thus, both positive and negative regulators of the Wnt pathway are needed for proper AVC development.
Summary
During this period, the heart has matured enough to vigorously pump blood through the body of the animal, the atrium and ventricle have become separate functioning chambers, and the AVC has developed thickened cardiac cushions that function in a valve-like manner. Proteoglycans, retinoic acid, micro-RNAs, along with cyclooxygenase, Notch, and Wnt pathway components all play important roles in the cellular patterning of the AVC at this time.
Maturation of Valve Leaflets (After 4-Day Post-fertilization)
Mature valves start forming from the AVC cardiac cushion region after day 4 post-fertilization. Most of what is known about this process in zebrafish is descriptive in nature, as knockdown effects of morpholinos only persist until day 3.
From day 6 to day 16, the valve leaflets elongate and grow thinner, but not through a process of cell division, as measured by BrdU incorporation [31]. It is proposed that this growth relies on cell migration or reorganization. There is very little extracellular matrix during this growth phase, and the valve leaflets remain two cells thick [5]. However, by day 28, the ECM has expanded, with expression of versican and collagen. There is some evidence that valve cells undergo EMT after day 16, as they lose tight junctions, and some cells start expressing the EMT markers pancytokeratin, vimentin, and focal adhesion kinase [31]. Many aspects of adult valve anatomy and homeostasis have yet to be explored. While the adult zebrafish valve shows some resemblance to a human mitral valve, it is unknown whether the mature leaflets derive from division of the endocardial cushion valve structures, through development of as-yet-undescribed lateral cushion leaflets, or some combination of both. Further, complete analysis of the cell types present and the ECM of adult valves have not been performed. More work is needed to help delineate the process of valve maturation in the developing zebrafish.
Summary
The zebrafish atrioventricular canal develops very rapidly and is dependent upon a complex set of molecular components. The AVC divides the atrium from the ventricle and is responsible for formation of the endocardial cushions and eventually the valves that ensure unidirectional blood flow.
One theme that arises during zebrafish AVC development is the early expression of genes throughout the heart and the later restriction of these genes to the AVC in order to set up cushion formation. It seems that the mechanism behind this restriction is not necessarily activation, but repression of these genes throughout the atrium and ventricle. This is underscored by numerous experiments in which specific gene disruption leads to expression of other genes (such as bmp4 or notch1b) throughout the heart tube.
The zebrafish model system lends itself well to these types of studies. Many of the genetic knockdown experiments described above would result in early lethality in other model organisms, but embryonic zebrafish can survive up to 6 days without a functional heart.
Are zebrafish a relevant model system in which to study AVC development? After all, there are structural differences between the zebrafish AVC compared to chick and mouse models, such as the absence of an annulus fibrosus, tendinous chords, or papillary muscles. Further, there remain outstanding questions such as whether EMT occurs. However, most of the signaling pathways discussed above are conserved in other model systems and appear to be activated in the same sequence. So, while zebrafish display differences in AVC and valve morphology, they appear to use the same molecular and cellular mechanisms to form these structures and therefore can be argued to be a highly relevant model system in which to study AVC and valve development.
We have focused primarily on AVC development, but valve development is directly linked to this process. While most reports have implied a valvulogenesis phenotype during genetic and chemical perturbation, it appears more likely that they are causing defects in cardiac cushion development, preventing further valve formation. Weak hypomorphic mutants, lower concentration morphants, more fully defined chemical perturbations, better conditional knockdowns, and lineage studies to define the specific contributions of cellular progenitors are needed to fully understand the complex process that leads from AVC to valve development.
References
Armstrong, E. J., & Bischoff, J. (2004). Heart valve development: endothelial cell signaling and differentiation. Circulation Research, 95, 459–470.
Stainier, D. Y., Lee, R. K., & Fishman, M. C. (1993). Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development, 119, 31–40.
Stainier, D. Y. R., & Fishman, M. C. (1992). Patterning the zebrafish heart tube: acquisition of anteroposterior polarity. Developmental Biology, 153, 91–101.
Stainier, D. Y., Weinstein, B. M., Detrich, H. W., Zon, L. I., & Fishman, M. C. (1995). Cloche, an early acting zebrafish gene, is required by both the endothelial and hematopoietic lineages. Development, 121, 3141–3150.
Beis, D., Bartman, T., Jin, S.-W., Scott, I. C., D'Amico, L. A., Ober, E. A., et al. (2005). Genetic and cellular analyses of zebrafish atrioventricular cushion and valve development. Development, 132, 4193–4204.
Chi, N., Shaw, R., De Val, S., Kang, G., Jan, L., Black, B., et al. (2008). Foxn4 directly regulates tbx2b expression and atrioventricular canal formation. Genes & Development, 22, 706–710.
Camarata, T., Krcmery, J., Snyder, D., Park, S., Topczewski, J., & Simon, H.-G. (2010). Pdlim7 (LMP4) regulation of Tbx5 specifies zebrafish heart atrio-ventricular boundary and valve formation. Developmental Biology, 337, 233–245.
Little, C. D., & Rongish, B. J. (1995). The extracellular matrix during heart development. Cellular and Molecular Life Sciences, 51, 873–882.
Peal, D. S., Burns, C. G., Macrae, C. A., & Milan, D. (2009). Chondroitin sulfate expression is required for cardiac atrioventricular canal formation. Developmental Dynamics, 238, 3103–3110.
Walsh, E. C., & Stainier, D. Y. R. (2001). UDP-Glucose dehydrogenase required for cardiac valve formation in zebrafish. Science, 293, 1670–1673.
Hurlstone, A. F. L., Haramis, A.-P. G., Wienholds, E., Begthel, H., Korving, J., van Eeden, F., et al. (2003). The Wnt/β-catenin pathway regulates cardiac valve formation. Nature, 425, 633–637.
Massague, J., & Wotton, D. (2000). Transcriptional control by the TGF-β/Smad signaling system. EMBO Journal, 19, 1745–1754.
Laux, D. W., Febbo, J. A., & Roman, B. L. (2011). Dynamic analysis of BMP-responsive smad activity in live zebrafish embryos. Developmental Dynamics, 240, 682–694.
Artavanis-Tsakonas, S., Rand, M. D., & Lake, R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science, 284, 770–776.
Westin, J., & Lardelli, M. (1997). Three novel Notch genes in zebrafish: implications for vertebrate Notch gene evolution and function. Development Genes and Evolution, 207, 51–63.
Milan, D. J., Giokas, A. C., Serluca, F. C., Peterson, R. T., & MacRae, C. A. (2006). Notch1b and neuregulin are required for specification of central cardiac conduction tissue. Development, 133, 1125–1132.
Motoike, T., Loughna, S., Perens, E., Roman, B. L., Liao, W., Chau, T. C., et al. (2000). Universal GFP reporter for the study of vascular development. Genesis, 28, 75–81.
Lee, Y. M., Cope, J. J., Ackermann, G. E., Goishi, K., Armstrong, E. J., Paw, B. H., et al. (2006). Vascular endothelial growth factor receptor signaling is required for cardiac valve formation in zebrafish. Developmental Dynamics, 235, 29–37.
Chang, C.-P., Neilson, J. R., Bayle, J. H., Gestwicki, J. E., Kuo, A., Stankunas, K., et al. (2004). A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell, 118, 649–663.
Hu, N., Sedmera, D., Yost, H. J., & Clark, E. B. (2000). Structure and function of the developing zebrafish heart. The Anatomical Record, 260, 148–157.
Scherz, P. J., Huisken, J., Sahai-Hernandez, P., & Stainier, D. Y. R. (2008). High-speed imaging of developing heart valves reveals interplay of morphogenesis and function. Development, 135, 1179–1187.
Xu, D.-J., Bu, J.-W., Gu, S.-Y., Xia, Y.-M., Du, J.-I., & Wang, Y.-W. (2011). Celecoxib impairs heart development via inhibiting cyclooxygenase-2 activity in zebrafish embryos. Anesthesiology, 114, 391–400.
Stainier, D. Y. R., Fouquet, B., Chen, J. N., Warren, K. S., Weinstein, B. M., Meiler, S. E., et al. (1996). Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development, 123, 285–292.
Mjaatvedt, C. H., Yamamura, H., Capeheart, A. A., Turner, D., & Markwald, R. (1998). The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardiac cushion formation. Developmental Biology, 202, 56–66.
Camenisch, T. D., Spicer, A. P., Brehm-Gibson, T., Biesterfeldt, J., Augustine, M. L., Calabro, A., Jr., et al. (2000). Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. The Journal of Clinical Investigation, 106, 349–360.
Kloosterman, W. P., & Plasterk, R. H. A. (2006). The diverse functions of microRNAs in animal development and disease. Developmental Cell, 11, 441–450.
Morton, S. U., Scherz, P. J., Cordes, K. R., Ivey, K. N., Stainier, D. Y. R., & Srivastava, D. (2008). microRNA-138 modulates cardiac patterning during embryonic development. Proceedings of the National Academy of Sciences, 105, 17830–17835.
Timmerman, L. A., Grego-Bessa, J., Raya, A., Bertran, E., Perez-Pomares, J. M., Diez, J., et al. (2004). Notch promotes epithelial-mesenchymal transition during cadiac development and oncogenic transformation. Genes & Development, 18, 99–115.
Logan, C. Y., & Nusse, R. (2004). The Wnt signaling pathway in development and disease. Annual Review of Cell and Developmental Biology, 20, 781–810.
Martin, E. D., Moriarty, M. A., Byrnes, L., & Grealy, M. (2009). Plakoglobin has both structural and signalling roles in zebrafish development. Developmental Biology, 327, 83–96.
Martin, R. T., & Bartman, T. (2009). Analysis of heart valve development in larval zebrafish. Developmental Dynamics, 238, 1796–1802.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Peal, D.S., Lynch, S.N. & Milan, D.J. Patterning and Development of the Atrioventricular Canal in Zebrafish. J. of Cardiovasc. Trans. Res. 4, 720–726 (2011). https://doi.org/10.1007/s12265-011-9313-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-011-9313-z