
Abstract
Inflammation has emerged as a critical biological process contributing to nearly all aspects of cardiovascular diseases including heart failure. Heart failure represents the final consequence of a diverse set of initial insults to the myocardium, among which myocardial infarction (MI) is the most common cause. After MI, the lack of perfusion often leads to the death of cardiomyocytes. The necrotic cells trigger a cascade of inflammatory pathways that work to clear the dead cells and matrix debris, as well as to repair and heal damaged tissues. For the heart, an organ with limited regeneration capacity, the consequence of MI (termed post-MI remodeling) comprises a series of structural and functional changes, including scar formation at the infarct zone, reactive hypertrophy of the remaining cardiomyocytes at the noninfarct area, ventricular chamber dilatation, and molecular changes marked by fetal gene up-regulation, all of which have been linked to the activation of the inflammatory pathways. Inadequate or excessive inflammatory response may lead to improper cellular repair, tissue damage, and dysfunction. Herein, we summarize the current understanding of the role of inflammation in cardiac injury and repair and put forth the hypothesis that temporally regulated activation and suppression of inflammation may be critical for achieving effective cardiac repair and regeneration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cardiovascular disease remains the number one cause of death in the United States, with myocardial infarction (MI) and other cardiovascular insults often leading to the subsequent development of heart failure [1]. The post-MI remodeling as well as morbidity and mortality are highly dependent on the number of cardiomyocytes lost during ischemic injury or the infarct size. Until recently, the heart was thought to be a terminally differentiated organ without regenerative capacity. Emerging data, however, now provide convincing evidence that the heart does, in fact, possess a regenerative capacity from both endogenous and exogenous sources [2–8]. Although such capacity is limited, the notion of regenerating lost heart tissue has fueled tremendous hope of one day being able to achieve complete cardiac repair and regeneration.
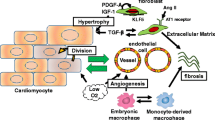
Since the first description of cardiomyogenic differentiation of stem cell populations approximately a decade ago, scientists worldwide have worked to understand this process, with a goal of controlling this process and ultimately attaining effective cardiac regeneration. Cell-based therapy offers a unique and novel therapeutic strategy for the treatment of a multitude of heart diseases. Although initial animal and human studies have been rather encouraging, true physiologically relevant cardiac regeneration remains an unachieved goal. The overarching roadmap to a successful cell therapy requires improving the long-term survival and engraftment and proper differentiation of implanted stem cells. Most positive effects observed with cell-based therapy reported thus far have been attributed to paracrine effects rather than direct differentiation of engrafted cells. It may be somewhat discouraging, at first, that cell therapy to date has not yielded new stem cell-derived cardiac tissue; however, cell therapy has been suggested to protect remaining cardiac cells from dying, lessen inflammation, induce angiogenesis, promote endogenous repair with de novel myocyte formation, and attenuate fibrosis, all of which are beneficial. It seems almost as if cell-based therapy may actually activate the heart’s endogenous healing mechanisms and provide the organ a second chance to repair cellular damage. The important lessons we learn from the so termed paracrine effects may provide critical clues as how to achieve cardiac repair/regeneration either with or even without cell implantation. Inflammation has been implicated in tissue repair and/or wound healing in many organs, although its significance has gained less attention in cardiac tissue regeneration. The current review highlights the role of inflammation and potential impact of modulating inflammatory pathways in cardiac repair and regeneration.
Post-MI Cardiac Tissue Repair and Remodeling
Under normal immune function, the inflammatory response occurs immediately after tissue injury. The injury-triggered inflammatory signaling cascade is a programmed process to remove dead cells and matrix debris; repair damaged tissues through the following one or, most often, both processes; scar formation by fibrogenesis and scarless healing via tissue regeneration; and ultimately, restore a given organ's original structure and function [9, 10]. The repair and healing process is likely far more challenging in a continuously beating organ such as the heart relative to other static tissues. This is not only attributed to the exceedingly low proliferative capacity of existing cardiomyocytes but also to the fact that the heart cannot cease contraction during the repair process and, critically, the space vacuum created by dead cells needs to be occupied immediately, even if by scar tissue, to prevent ventricular rupture. As such, after MI or ischemia–reperfusion (I/R) injury, necrotic cells activate a series of inflammatory pathways using chemokines and cytokines to recruit leukocytes, including neutrophils and monocytes/macrophages among other cells, to clear the infarct zone of dead cells and matrix debris. This inflammatory chain reaction serves to repair and heal damage tissues; however, for myocardium, the infarct healing results in profound molecular and structural remodeling, with scar formation to replace lost cardiomyocytes in the infarct zone and reactive hypertrophy of the noninfarct area accompanied by chamber dilation and up-regulation of fetal gene expression [11]. The subsequent chronic elevation in tissue inflammation results in continued fibrosis and cardiomyocyte loss and contributes to adverse cardiac remodeling and ultimately the development of heart failure.
Initiation of Post-MI Inflammatory Response
It is recognized that the heart expresses all components of the innate immune system, including effectors and pattern recognition receptors. More than a decade ago, a danger model proposed by Matzinger and colleagues suggested that the presence of potentially infectious contents alone is not sufficient to activate the immune system without an appropriate alarm signal from stressed or damaged tissue/cells [12]. Under the danger model, tissue or cells sense danger from stressors or damaging agents, and to exert self-protection, certain molecules are released by the necrotic or dying cells. These released molecules serve as alarm signals that then initiate an inflammatory response by interacting with pattern recognition receptors such as Toll-like receptors (TLRs) [13–15] and nucleotide-binding oligomerization domain (NOD) receptors [16–21].
TLRs are a group of pattern recognition receptors well known to recognize structurally conserved molecules derived from microbial origin, referred to as pathogen-associated molecular patterns. To date, 11 human and 13 mouse TLRs have been identified [13], among which TLR2, 3, 4, and 6 are expressed in the heart. Importantly, accumulating evidence suggest that TLRs also play critical roles in initiating inflammation by recognizing constitute damage-associated molecular patterns [14, 22–25]. For example, necrotic or dying cells can release high-mobility group box protein 1 (HMGB1), a nuclear nonhistone protein ubiquitously and highly expressed in most cell types [26]. HMGB1 can initiate inflammatory response in surrounding cells through TLR4 signaling [22, 27–29]. Fragmented hyaluronan and heparin sulfate, two components released from the extracellular matrix during tissue injury, are also considered as danger signals [23, 30–34]. Both of these have been demonstrated to initiate innate immune response through interaction with TLR4 [31–33]. TLR4 in response to the danger signals can activate nuclear factor (NF)-κB, an essential transcription factor that regulates the expression of numerous inflammatory genes such as adhesion molecules, chemokines, and cytokines. NF-κB is critical for triggering inflammation and is involved in the cardioprotection and healing process post-MI, though the effects of activation appear to be cell type dependent.
The family of NOD-like receptors (NLR) similar to TLRs, although not fully characterized, are thought to also interact with danger signals to initiate an inflammatory response. Approximately 20 mammalian NLR proteins have been identified and include two major subfamilies, NLRC and NLRP. The C of NLRC symbolizes a caspase recruitment domain (CARD), which exists at the N-terminus of the protein, whereas the P of NLRP stands for a pyrin domain [18, 35]. Both CARD and pyrin domains are members of the death-domain-fold family and resemble six antiparallel α-helices with a similar three-dimensional fold [36, 37]. These domains mediate protein–protein interactions and are important components in the formation of the inflammasome complex. NLRP3 (also known as cryopyrin), for example, upon interaction with danger signals, induces assembly of the cellular inflammasome complex followed by activation of caspase-1 (also called IL-1β-converting enzyme or ICE) [16, 18, 19, 38]. Activated caspase-1 processes the maturation and secretion of interleukin (IL)-1β and IL-18 [18–20]. If caspase-1 activity is high enough, it can induce pyroptosis, a programmed cell death characterized by release of intracellular contents, which is similar to necrosis but distinct from apoptosis, and may amplify the inflammatory response [18, 19].
Inflammatory Cytokines in Cell Death and Regeneration
An array of cytokines has been implicated in the activation of inflammatory response as well as in the healing and repair processes. Here, we highlight several examples.
TNFα
TNFα (tumor necrosis factor-α) is one of most thoroughly studied cytokines in cardiac physiology and pathology. Overexression of TNFα in mouse model or infusion of recombinant TNFα faithfully results in cardiac dysfunction with many symptoms mimicking heart failure in humans [39–41]. More importantly, circulating levels of TNFα have been shown to be up-regulated in patients with heart failure and correlate with mortality [42]. A protective role for TNFα, however, has also been reported [43–45]. This seemingly dichotomous function for TNFα has been explained by its distinct receptors, TNFR1 and TNFR2. Recent data suggest that TNFα signaling through TNFR1 results in deleterious effects, whereas TNFR2 mediates protective signaling [46, 47]. Depending on the cell type and cellular microenvironment, however, activation of TNFR1 alone can promote either cell survival or cell death [48, 49]. In this regard, recent findings indicate that protein kinases of the receptor interacting protein (RIP) family play critical roles in the activated TNFR1 complex, directing either survival or cell death [48–51]. RIP1, for example, may either activate prosurvival signalings such as mitogen-activated protein kinases and NF-κB or trigger cell apoptotic or necrotic signaling. Such a switch role depends on the ubiquitination state of RIP1, which can be regulated by multiple factors including inhibitors of apoptosis proteins that promote polyubiquitination [52, 53] and the deubiquitinases cylindromatosis (CYLD) and A20 [54–57]. Taken together, these findings serve to highlight the complexity of the cytokine signaling and may provide explanations for the lack of clear results in clinical studies of cytokine-target therapies.
IL-1
IL-1 plays a central role in programming the inflammatory process [15, 58, 59]. Increased IL-1 in either an autocrine or paracrine manner through IL-1RI/Myd88-mediated activation of NF-κB is essential in establishing a concentration gradient of adhesion molecules and chemoattractants around the injury site and guiding the movement of neutrophils, monocytes/macrophages, and likely also surrounding or circulating repair cells to damaged tissues [60–62]. Transmigration or infiltration of neutrophils into the injury site followed by the recruitment of monocytes and/or macrophages is required to remove dead cells and matrix debris [63, 64]. Excess accumulation and activation of leukocytes, however, particularly neutrophils, may lead to further tissue damage, potentially through the release of large amounts of toxic or destructive materials such as reactive oxygen species and proteases [65]. Toward this, blocking IL-1 signaling by knockout of MyD88 in mouse has been shown to prevent the development of cardiac fibrosis and heart failure [66]. Interestingly, an IL-1R antagonist (IL-1Ra) has been found to be expressed in the injury site to potentially attenuate IL-1 effects after acute MI [67]. These data suggest that an endogenous anti-inflammatory mechanism may be coactivated to balance the degree of tissue inflammatory response. Consistent with this notion, recent studies show that administration of recombinant IL-1Ra or overexpression of IL-1Ra is able to reduce cardiomyocyte apoptosis in mouse or rat models [68, 69].
IL-6
IL-6 plays an important role in regulating cell survival, growth, and differentiation in various cell types [70]. A recent study has suggested that mice with IL-6 deficiency have increased accumulation of interstitial collagen and severe cardiac dilatation, associated with a marked increase in the cardiac fibroblast population and a concomitant decrease in other cardiac cell populations [71]. Furthermore, it has been suggested that both IL-6 and TNF-α may exert a cardioprotective role mediated via late preconditioning after I/R [45, 72].
TWEAK
Recently, Engel et al. reported that a novel cytokine, TWEAK (TNF-related weak inducer of apoptosis), induces proliferation of neonatal rat cardiomyocyte, and this effect is mediated via up-regulation of cyclin D2 and Ki67 and down-regulation of the cell cycle inhibitor, p27KIP1 [73]. Interestingly, our laboratory has found that TWEAK, although it did not induce proliferation of adult rat cardiomyocytes, stimulated adult rat cardiomyocytes to undergo dedifferentiation (Shi, Jiang, and Liao, unpublished data). Chronic overexpression of TWEAK in vivo, however, leads to the development of dilated cardiomyopathy associated with cardiomyocyte elongation and cardiac fibrosis [74].
HMGB1
HMGB1, which is released from necrotic cells and initiates tissue inflammatory response, is reported to stimulate angiogenesis [75] and promote cardiac stem cell growth and differentiation [76, 77]. Surprisingly, the effect of HMGB1 is associated with increased expression of numerous inflammatory genes including VEGF, PlGF, Mip-1α, GM-CSF, IL-1β, IL-1ra, IL-4, IL-9, IL-10, TNFα, and IFNγ in cardiac fibroblasts [78].
These findings may suggest a pivotal role for cytokines in not only regulating the inflammatory response but also the critical balance among fibrogenesis, tissue regeneration, and cell death (apoptosis and/or necrosis), which may be a major determinants for the final outcome of tissue remodeling.
Targeting the Inflammatory Pathways to Optimize Cardiac Repair and Regeneration
It is well documented that growth factors (tyrosine kinase receptor ligands), such as HGF, FGF2, IGF1, EGF, and VEGF, regulate angiogenesis, progenitor cell survival, proliferation, and differentiation [64]. These growth factors may act in conjunction with other chemokines and cytokines to regulate tissue regeneration. In contrast, TGFβ1, another cellular cytokine, plays a critical role in regulating cardiac tissue fibrogenesis [79, 80]. TGFβ1 binds to a serine/threonine kinase receptor and activates downstream SMAD signaling pathways. Vasoactive agents such as Ang II, endothelin-1, and adrenergic receptor agonists have been reported to stimulate TGFβ expression and thereby contribute to fibrogenesis [81–83].
The mitogen-activated protein kinases (MAPK), such as extracellular signal-regulated kinase (ERK), p38 MAPK, and c-Jun NH2-terminal kinase (JNK), are involved in relaying extracellular signals to intracellular responses. Activation of ERK is generally associated with the mitogenic response to growth factors and the differentiation of specific cell lineages. Activation of p38 and JNK is mainly associated with the response to cellular stresses. Activation of ERK signaling positively regulates cell proliferation, whereas activation of p38 MAPK may have a negative role in cardiomyocyte proliferation [84, 85]. The regulation of these MAPK signaling pathways by vasoactive agents may have significant impact on tissue inflammatory and repair process. For example, angiotensin II (Ang II) is a well known mediator of hypertension, cardiac hypertrophy, and fibrosis [86–88]. Angiotensin-converting enzyme inhibitors or Ang II type-1 (AT1) receptor antagonists alleviate cardiovascular pathologies in patients and in animal models [88, 89]. Ang II can differentially regulate the expression of multiple NF-κB-responsive genes such as VCAM-1, MCP-1, iNOS, COX-2, TGF-β1, TNF-α, and IL-6 [90–94], thereby modulating the inflammatory process, potentially via modulation of cytokine-activated MAPK and NF-κB signaling pathways [91, 95]. Therefore, it is plausible that Ang II, through its ability of modulating inflammatory response, may be targeted for tissue repair and regeneration.
Taken together, we submit the hypothesis that activation of NF-κB must be temporally well controlled so that subsequent gene expression can complete the healing process and resolve the inflammation process promptly. Because ERK activity is required for cytokines such as IL-1β to induce a persistent, but not transient, activation of NF-κB [91, 95–97], it is entirely possible that factors that inhibit ERK signaling may interfere with the expression of later-phase NF-κB responsive genes. In such a way, the Ang II and/or other-factors-mediated changes in inflammatory gene expression pattern may influence the repair process. Therefore, it is critically important to investigate whether target TGFβ1 and G-protein-coupled receptor agonists or their respective signaling pathways, such as SMAD and MAPKs, may modulate the balance of fibrogenesis and regeneration in injured cardiac tissues.
Closing Remarks
Effective cardiac repair and regeneration are dependent on a seemingly well-orchestrated cellular response, including the initial inflammatory cascade, and the timely activation of specific downstream signaling mediators through every step of the healing processes. Interventions aimed at any specific inflammatory mediator should bring into consideration each individual's innate immune condition, as well as all temporal, cell-type-specific, and geographic parameters. Given that heart failure is a functional consequence of a large variety of diseases and insults, inflammatory responses may differ in a failing heart resulting from genetic cause, ischemic injury, viral infection, or toxin exposure. Understanding the inflammatory and innate immune regulation in a more systematic manner, conditioned on an individual basis, is the key to achieving more effective cardiac regeneration in the days to come.
References
Lloyd-Jones, D., Adams, R., Carnethon, M., De Simone, G., Ferguson, T. B., Flegal, K., et al. (2009). Heart disease and stroke statistics—2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation, 119, 480–486.
Kajstura, J., Leri, A., Finato, N., Di Loreto, C., Beltrami, C. A., & Anversa, P. (1998). Myocyte proliferation in end-stage cardiac failure in humans. Proceedings of the National Academy of Sciences of the United States of America, 95, 8801–8805.
Laugwitz, K. L., Moretti, A., Lam, J., Gruber, P., Chen, Y., Woodard, S., et al. (2005). Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature, 433, 647–653.
Moretti, A., Caron, L., Nakano, A., Lam, J. T., Bernshausen, A., Chen, Y., et al. (2006). Multipotent embryonic isl1+ progenitor cells lead to cardiac, smooth muscle, and endothelial cell diversification. Cell, 127, 1151–1165.
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872.
Hsieh, P. C., Segers, V. F., Davis, M. E., MacGillivray, C., Gannon, J., Molkentin, J. D., et al. (2007). Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Natural Medicines, 13, 970–974.
Zhang, J., Wilson, G. F., Soerens, A. G., Koonce, C. H., Yu, J., Palecek, S. P., et al. (2009). Functional cardiomyocytes derived from human induced pluripotent stem cells. Circulation Research, 104, e30–e41.
Bergmann, O., Bhardwaj, R. D., Bernard, S., Zdunek, S., Barnabe-Heider, F., Walsh, S., et al. (2009). Evidence for cardiomyocyte renewal in humans. Science, 324, 98–102.
Leibovich, S. J., & Ross, R. (1976). A macrophage-dependent factor that stimulates the proliferation of fibroblasts in vitro. The American Journal of Pathology, 84, 501–514.
Eming, S. A., Hammerschmidt, M., Krieg, T., & Roers, A. (2009). Interrelation of immunity and tissue repair or regeneration. Seminars in Cell & Developmental Biology, 20, 517–527.
Jennings, R. B., Steenbergen, C., Jr., & Reimer, K. A. (1995). Myocardial ischemia and reperfusion. Monographs in Pathology, 37, 47–80.
Matzinger, P. (1994). Tolerance, danger, and the extended family. Annual Review of Immunology, 12, 991–1045.
Akira, S. (2006). TLR signaling. Current Topics in Microbiology and Immunology, 311, 1–16.
Chao, W. (2009). Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. American Journal of Physiology. Heart and Circulatory Physiology, 296, H1–H12.
Chen, C. J., Kono, H., Golenbock, D., Reed, G., Akira, S., & Rock, K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Natural Medicines, 13, 851–856.
Petrilli, V., Papin, S., Dostert, C., Mayor, A., Martinon, F., & Tschopp, J. (2007). Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death and Differentiation, 14, 1583–1589.
Willingham, S. B., Allen, I. C., Bergstralh, D. T., Brickey, W. J., Huang, M. T., Taxman, D. J., et al. (2009). NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. Journal of Immunology, 183, 2008–2015.
Bergsbaken, T., Fink, S. L., & Cookson, B. T. (2009). Pyroptosis: host cell death and inflammation. Nature Reviews. Microbiology, 7, 99–109.
Franchi, L., Eigenbrod, T., Munoz-Planillo, R., & Nunez, G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nature Immunology, 10, 241–247.
Li, H., Ambade, A., & Re, F. (2009). Cutting edge: necrosis activates the NLRP3 inflammasome. Journal of Immunology, 183, 1528–1532.
Yamasaki, K., Muto, J., Taylor, K. R., Cogen, A. L., Audish, D., Bertin, J., et al. (2009). NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. The Journal of Biological Chemistry, 284, 12762–12771.
Tsung, A., Sahai, R., Tanaka, H., Nakao, A., Fink, M. P., Lotze, M. T., et al. (2005). The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia–reperfusion. The Journal of Experimental Medicine, 201, 1135–1143.
Wu, H., Chen, G., Wyburn, K. R., Yin, J., Bertolino, P., Eris, J. M., et al. (2007). TLR4 activation mediates kidney ischemia/reperfusion injury. Journal of Clinical Investigation, 117, 2847–2859.
Mollen, K. P., Anand, R. J., Tsung, A., Prince, J. M., Levy, R. M., & Billiar, T. R. (2006). Emerging paradigm: toll-like receptor 4-sentinel for the detection of tissue damage. Shock, 26, 430–437.
Takeishi, Y., & Kubota, I. (2009). Role of Toll-like receptor mediated signaling pathway in ischemic heart. Frontiers in Bioscience, 14, 2553–2558.
Rovere-Querini, P., Capobianco, A., Scaffidi, P., Valentinis, B., Catalanotti, F., Giazzon, M., et al. (2004). HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Reports, 5, 825–830.
Kaczorowski, D. J., Nakao, A., Vallabhaneni, R., Mollen, K. P., Sugimoto, R., Kohmoto, J., et al. (2009). Mechanisms of Toll-like receptor 4 (TLR4)-mediated inflammation after cold ischemia/reperfusion in the heart. Transplantation, 87, 1455–1463.
Oozawa, S., Mori, S., Kanke, T., Takahashi, H., Liu, K., Tomono, Y., et al. (2008). Effects of HMGB1 on ischemia–reperfusion injury in the rat heart. Circulation Journal, 72, 1178–1184.
Andrassy, M., Volz, H. C., Igwe, J. C., Funke, B., Eichberger, S. N., Kaya, Z., et al. (2008). High-mobility group box-1 in ischemia–reperfusion injury of the heart. Circulation, 117, 3216–3226.
Decleves, A. E., Caron, N., Nonclercq, D., Legrand, A., Toubeau, G., Kramp, R., et al. (2006). Dynamics of hyaluronan, CD44, and inflammatory cells in the rat kidney after ischemia/reperfusion injury. International Journal of Molecular Medicine, 18, 83–94.
Taylor, K. R., Trowbridge, J. M., Rudisill, J. A., Termeer, C. C., Simon, J. C., & Gallo, R. L. (2004). Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. The Journal of Biological Chemistry, 279, 17079–17084.
Taylor, K. R., Yamasaki, K., Radek, K. A., Di Nardo, A., Goodarzi, H., Golenbock, D., et al. (2007). Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. The Journal of Biological Chemistry, 282, 18265–18275.
Johnson, G. B., Brunn, G. J., Kodaira, Y., & Platt, J. L. (2002). Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. Journal of Immunology, 168, 5233–5239.
Celie, J. W., Rutjes, N. W., Keuning, E. D., Soininen, R., Heljasvaara, R., Pihlajaniemi, T., et al. (2007). Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. The American Journal of Pathology, 170, 1865–1878.
Mills, K. H., & Dunne, A. (2009). Immune modulation: IL-1, master mediator or initiator of inflammation. Natural Medicines, 15, 1363–1364.
Martinon, F., Hofmann, K., & Tschopp, J. (2001). The pyrin domain: a possible member of the death domain-fold family implicated in apoptosis and inflammation. Current Biology, 11, R118–R120.
Fairbrother, W. J., Gordon, N. C., Humke, E. W., O'Rourke, K. M., Starovasnik, M. A., Yin, J. P., et al. (2001). The PYRIN domain: a member of the death domain-fold superfamily. Protein Science, 10, 1911–1918.
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., Jankowski, W., et al. (2007). The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death and Differentiation, 14, 1590–1604.
Kubota, T., McTiernan, C. F., Frye, C. S., Demetris, A. J., & Feldman, A. M. (1997). Cardiac-specific overexpression of tumor necrosis factor-alpha causes lethal myocarditis in transgenic mice. Journal of Cardiac Failure, 3, 117–124.
Bozkurt, B., Kribbs, S. B., Clubb, F. J., Jr., Michael, L. H., Didenko, V. V., Hornsby, P. J., et al. (1998). Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation, 97, 1382–1391.
Yokoyama, T., Vaca, L., Rossen, R. D., Durante, W., Hazarika, P., & Mann, D. L. (1993). Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. Journal of Clinical Investigation, 92, 2303–2312.
Dunlay, S. M., Weston, S. A., Redfield, M. M., Killian, J. M., & Roger, V. L. (2008). Tumor necrosis factor-alpha and mortality in heart failure: a community study. Circulation, 118, 625–631.
Tanno, M., Gorog, D. A., Bellahcene, M., Cao, X., Quinlan, R. A., & Marber, M. S. (2003). Tumor necrosis factor-induced protection of the murine heart is independent of p38-MAPK activation. Journal of Molecular and Cellular Cardiology, 35, 1523–1527.
Yamashita, N., Hoshida, S., Otsu, K., Taniguchi, N., Kuzuya, T., & Hori, M. (2000). The involvement of cytokines in the second window of ischaemic preconditioning. British Journal of Pharmacology, 131, 415–422.
Dawn, B., Guo, Y., Rezazadeh, A., Wang, O. L., Stein, A. B., Hunt, G., et al. (2004). Tumor necrosis factor-alpha does not modulate ischemia/reperfusion injury in naive myocardium but is essential for the development of late preconditioning. Journal of Molecular and Cellular Cardiology, 37, 51–61.
Higuchi, Y., McTiernan, C. F., Frye, C. B., McGowan, B. S., Chan, T. O., & Feldman, A. M. (2004). Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-alpha-induced cardiomyopathy. Circulation, 109, 1892–1897.
Al-Lamki, R. S., Brookes, A. P., Wang, J., Reid, M. J., Parameshwar, J., Goddard, M. J., et al. (2009). TNF receptors differentially signal and are differentially expressed and regulated in the human heart. American Journal of Transplantation, 9, 2679–2696.
Declercq, W., Vanden Berghe, T., & Vandenabeele, P. (2009). RIP kinases at the crossroads of cell death and survival. Cell, 138, 229–232.
Whelan, R. S., Kaplinskiy, V., & Kitsis, R. N. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol, 72, 19-44.
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell, 137, 1100–1111.
Cho, Y. S., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell, 137, 1112–1123.
Mahoney, D. J., Cheung, H. H., Mrad, R. L., Plenchette, S., Simard, C., Enwere, E., et al. (2008). Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proceedings of the National Academy of Sciences of the United States of America, 105, 11778–11783.
Varfolomeev, E., Goncharov, T., Fedorova, A. V., Dynek, J. N., Zobel, K., Deshayes, K., et al. (2008). c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. The Journal of Biological Chemistry, 283, 24295–24299.
Shembade, N., Ma, A., & Harhaj, E. W. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science, 327, 1135-1139.
Wilson, N. S., Dixit, V., & Ashkenazi, A. (2009). Death receptor signal transducers: nodes of coordination in immune signaling networks. Nature Immunology, 10, 348–355.
Wang, L., Du, F., & Wang, X. (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell, 133, 693–703.
Wertz, I. E., O'Rourke, K. M., Zhou, H., Eby, M., Aravind, L., Seshagiri, S., et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature, 430, 694–699.
Dinarello, C. A. (2009). Immunological and inflammatory functions of the interleukin-1 family. Annual Review of Immunology, 27, 519–550.
Chen, C. J., Shi, Y., Hearn, A., Fitzgerald, K., Golenbock, D., Reed, G., et al. (2006). MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. Journal of Clinical Investigation, 116, 2262–2271.
Mitchell, M. D., Laird, R. E., Brown, R. D., & Long, C. S. (2007). IL-1beta stimulates rat cardiac fibroblast migration via MAP kinase pathways. American Journal of Physiology. Heart and Circulatory Physiology, 292, H1139–H1147.
Bujak, M., & Frangogiannis, N. G. (2009). The role of IL-1 in the pathogenesis of heart disease. Archivum Immunologiae et Therapiae Experimentalis (Warsz), 57, 165–176.
Tamaru, M., Tomura, K., Sakamoto, S., Tezuka, K., Tamatani, T., & Narumi, S. (1998). Interleukin-1beta induces tissue- and cell type-specific expression of adhesion molecules in vivo. Arteriosclerosis, Thrombosis, and Vascular Biology, 18, 1292–1303.
Frangogiannis, N. G. (2008). The immune system and cardiac repair. Pharmacological Research, 58, 88–111.
Gurtner, G. C., Werner, S., Barrandon, Y., & Longaker, M. T. (2008). Wound repair and regeneration. Nature, 453, 314–321.
Frangogiannis, N. G., Youker, K. A., & Entman, M. L. (1996). The role of the neutrophil in myocardial ischemia and reperfusion. EXS, 76, 263–284.
Blyszczuk, P., Kania, G., Dieterle, T., Marty, R. R., Valaperti, A., Berthonneche, C., et al. (2009). Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circulation Research, 105, 912–920.
Bonetti, A., Marchini, M., & Ortolani, F. (2008). Immunolocalization of interleukin-1 receptor antagonist in healthy and infarcted myocardium. Histology and Histopathology, 23, 1093–1102.
Abbate, A., Salloum, F. N., Vecile, E., Das, A., Hoke, N. N., Straino, S., et al. (2008). Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation, 117, 2670–2683.
Suzuki, K., Murtuza, B., Smolenski, R. T., Sammut, I. A., Suzuki, N., Kaneda, Y., et al. (2001). Overexpression of interleukin-1 receptor antagonist provides cardioprotection against ischemia–reperfusion injury associated with reduction in apoptosis. Circulation, 104, I308–I303.
Kamimura, D., Ishihara, K., & Hirano, T. (2003). IL-6 signal transduction and its physiological roles: the signal orchestration model. Reviews of Physiology Biochemistry and Pharmacology, 149, 1–38.
Banerjee, I., Fuseler, J. W., Intwala, A. R., & Baudino, T. A. (2009). IL-6 loss causes ventricular dysfunction, fibrosis, reduced capillary density, and dramatically alters the cell populations of the developing and adult heart. American Journal of Physiology. Heart and Circulatory Physiology, 296, H1694–H1704.
Dawn, B., Xuan, Y. T., Guo, Y., Rezazadeh, A., Stein, A. B., Hunt, G., et al. (2004). IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovascular Research, 64, 61–71.
Novoyatleva, T., Diehl, F., van Amerongen, M. J., Patra, C., Ferrazzi, F., Bellazzi, R., et al. (2009). TWEAK is a positive regulator of cardiomyocyte proliferation. Cardiovasc Res.
Jain, M., Jakubowski, A., Cui, L., Shi, J., Su, L., Bauer, M., et al. (2009). A novel role for tumor necrosis factor-like weak inducer of apoptosis (TWEAK) in the development of cardiac dysfunction and failure. Circulation, 119, 2058–2068.
Mitola, S., Belleri, M., Urbinati, C., Coltrini, D., Sparatore, B., Pedrazzi, M., et al. (2006). Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. Journal of Immunology, 176, 12–15.
Limana, F., Germani, A., Zacheo, A., Kajstura, J., Di Carlo, A., Borsellino, G., et al. (2005). Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circulation Research, 97, e73–e83.
Germani, A., Limana, F., & Capogrossi, M. C. (2007). Pivotal advances: high-mobility group box 1 protein—a cytokine with a role in cardiac repair. Journal of Leukocyte Biology, 81, 41–45.
Rossini, A., Zacheo, A., Mocini, D., Totta, P., Facchiano, A., Castoldi, R., et al. (2008). HMGB1-stimulated human primary cardiac fibroblasts exert a paracrine action on human and murine cardiac stem cells. Journal of Molecular and Cellular Cardiology, 44, 683–693.
Border, W. A., & Noble, N. A. (1994). Transforming growth factor beta in tissue fibrosis. The New England Journal of Medicine, 331, 1286–1292.
Rosenkranz, S. (2004). TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovascular Research, 63, 423–432.
Briest, W., Homagk, L., Rassler, B., Ziegelhoffer-Mihalovicova, B., Meier, H., Tannapfel, A., et al. (2004). Norepinephrine-induced changes in cardiac transforming growth factor-beta isoform expression pattern of female and male rats. Hypertension, 44, 410–418.
Jain, R., Shaul, P. W., Borok, Z., & Willis, B. C. (2007). Endothelin-1 induces alveolar epithelial–mesenchymal transition through endothelin type A receptor-mediated production of TGF-beta1. American Journal of Respiratory Cell and Molecular Biology, 37, 38–47.
Lee, A. A., Dillmann, W. H., McCulloch, A. D., & Villarreal, F. J. (1995). Angiotensin II stimulates the autocrine production of transforming growth factor-beta 1 in adult rat cardiac fibroblasts. Journal of Molecular and Cellular Cardiology, 27, 2347–2357.
Engel, F. B., Hsieh, P. C., Lee, R. T., & Keating, M. T. (2006). FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proceedings of the National Academy of Sciences of the United States of America, 103, 15546–15551.
Engel, F. B., Schebesta, M., Duong, M. T., Lu, G., Ren, S., Madwed, J. B., et al. (2005). p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes & Development, 19, 1175–1187.
Lips, D. J., deWindt, L. J., van Kraaij, D. J., & Doevendans, P. A. (2003). Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. European Heart Journal, 24, 883–896.
De Angelis, N., Fiordaliso, F., Latini, R., Calvillo, L., Funicello, M., Gobbi, M., et al. (2002). Appraisal of the role of angiotensin II and aldosterone in ventricular myocyte apoptosis in adult normotensive rat. Journal of Molecular and Cellular Cardiology, 34, 1655–1665.
Jessup, M., Abraham, W. T., Casey, D. E., Feldman, A. M., Francis, G. S., Ganiats, T. G., et al. (2009). 2009 focused update: ACCF/AHA Guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation, 119, 1977–2016.
Remme, W. J. (2007). Beta blockers or angiotensin-converting-enzyme inhibitor/angiotensin receptor blocker: what should be first? Cardiol Clin, 25, 581–594. vii.
Ford, C. M., Li, S., & Pickering, J. G. (1999). Angiotensin II stimulates collagen synthesis in human vascular smooth muscle cells. Involvement of the AT(1) receptor, transforming growth factor-beta, and tyrosine phosphorylation. Arteriosclerosis, Thrombosis, and Vascular Biology, 19, 1843–1851.
Jiang, B., Xu, S., Hou, X., Pimentel, D. R., & Cohen, R. A. (2004). Angiotensin II differentially regulates interleukin-1-beta-inducible NO synthase (iNOS) and vascular cell adhesion molecule-1 (VCAM-1) expression: role of p38 MAPK. The Journal of Biological Chemistry, 279, 20363–20368.
Chen, X. L., Tummala, P. E., Olbrych, M. T., Alexander, R. W., & Medford, R. M. (1998). Angiotensin II induces monocyte chemoattractant protein-1 gene expression in rat vascular smooth muscle cells. Circulation Research, 83, 952–959.
Tummala, P. E., Chen, X. L., Sundell, C. L., Laursen, J. B., Hammes, C. P., Alexander, R. W., et al. (1999). Angiotensin II induces vascular cell adhesion molecule-1 expression in rat vasculature: a potential link between the renin–angiotensin system and atherosclerosis. Circulation, 100, 1223–1229.
Nakayama, I., Kawahara, Y., Tsuda, T., Okuda, M., & Yokoyama, M. (1994). Angiotensin II inhibits cytokine-stimulated inducible nitric oxide synthase expression in vascular smooth muscle cells. The Journal of Biological Chemistry, 269, 11628–11633.
Jiang, B., Xu, S., Hou, X., Pimentel, D. R., Brecher, P., & Cohen, R. A. (2004). Temporal control of NF-kappaB activation by ERK differentially regulates interleukin-1beta-induced gene expression. The Journal of Biological Chemistry, 279, 1323–1329.
Jiang, B., Brecher, P., & Cohen, R. A. (2001). Persistent activation of nuclear factor-kappaB by interleukin-1beta and subsequent inducible NO synthase expression requires extracellular signal-regulated kinase. Arteriosclerosis, Thrombosis, and Vascular Biology, 21, 1915–1920.
Jiang, B., Xu, S., Brecher, P., & Cohen, R. A. (2002). Growth factors enhance interleukin-1 beta-induced persistent activation of nuclear factor-kappa B in rat vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 22, 1811–1816.
Acknowledgements
Research is supported by the National Institutes of Health, HL083358 (BJ) and HL088533, HL071775, HL093148 (RL) and American Heart Association grant-in-aid (BJ).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Jiang, B., Liao, R. The Paradoxical Role of Inflammation in Cardiac Repair and Regeneration. J. of Cardiovasc. Trans. Res. 3, 410–416 (2010). https://doi.org/10.1007/s12265-010-9193-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12265-010-9193-7