
Abstract
The Mediterranean Sea (MS) is a large oligotrophic sea whose productivity is sensitive to riverine nutrient inputs. More specifically, phosphorus (P) river supply is crucial for the MS, with an important role of the estuarine/deltaic filter especially for the storage and recycling in sediments. A benthic dataset from the Rhône River prodelta was used to derive P budgets, by means of an early diagenetic model including the benthic P cycle. The model was fitted to pore water profiles of oxygen, nitrate, sulfate, dissolved inorganic carbon, ammonium, oxygen demand units, dissolved inorganic phosphorus (DIP) and solid data (organic carbon (OC), Fe-bound P, Ca-bound P and organic P). Results indicated that the intensity of biogeochemical processes occurring below the sediment–water interface decreased from the river mouth to the adjacent continental shelf with decreasing integrated rates of OC mineralization (160–10 mmol m−2 day−1). The organic P mineralization was intense near the river mouth and decreased offshore (1196–80 μmol m−2 day−1). Its contribution to DIP release was large (> 90%). Fe-bound P had a key role in transferring P to deeper layers. These deltaic sediments played an important role as a source of regenerated DIP. A significant part of DIP was recycled to the overlying waters (72–94%), representing 25% of the riverine DIP discharge. Simultaneously, 6–28% of DIP produced in sediments was buried as Ca-bound P. Overall, this study highlighted the importance of deltaic sediments as an additional source of DIP to the coastal sea, and a minor but permanent sink of phosphorus as solid P burial.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Phosphorus is a key macronutrient for all life forms (Ruttenberg 1992; Tyrrell 1999). It is a typical limiting element of primary production in marine areas (Benitez-Nelson 2000), such as the Mediterranean Sea, with a prominent role in the Eastern part (Krom et al. 1991). Overall, particulate phosphorus (P) is partitioned as P associated to organic matter, Fe oxyhydroxides (henceforth called Fe oxides), aluminum oxides, vivianite and apatite (Lebo 1991). Meybeck (1982) showed on a global scale that rivers deliver globally 21 × 1012 g P year−1 to the sea. However, only 10% of this flux is available to the marine biota, because more than 90% of the P load is bound to particles and, therefore, settles rapidly to the seafloor in the coastal zones.
Although eutrophication has existed throughout the geological times (Nixon 1995; Cloern 2001), the second half of the twentieth century has witnessed the dramatic increase of anthropogenic activities (i.e. agricultural practice, industrial and urban waste, dam building and perturbation of hydrological cycles and sediment loads) which degraded deltaic and coastal ecosystems and altered biogeochemical cycles of carbon (C), nitrogen (N) and P in these vulnerable areas (Howarth et al. 1996; Vitousek et al. 1997; Jackson et al. 2001; Diaz and Rosenberg 2008; Wilkinson 2017). These anthropogenic changes led also to large organic matter inputs (McKee et al. 2004; Canfield et al. 2005; Bianchi and Allison 2009; Syvitski et al. 2009) and intense nutrient (N and P) loads to coastal margins compared to the middle of the twentieth century (Downing et al. 1999; Cloern 2001; Ludwig et al. 2009). The nutrient enrichment stimulates primary production and phytoplankton growth in adjacent coastal waters (Milliman and Meade 1983; Cloern 2001), which leads to phytoplankton proliferation, alteration in species compositions, hypoxia and deterioration of water quality (Smith et al. 1999).
River-dominated Ocean Margins (RiOMar) are important interfaces, connecting approximatively 87% of land surfaces to the oceans (Ludwig and Probst 1998; McKee et al. 2004). They support a large human population and provide ecological, economical and societal development due to their richness (Batker et al. 2014). They are important burial sites and preserve more than 40% of organic matter in the world ocean (Muller-Karger et al. 2005; Burdige 2007). Their sediments are characterized by high spatio-temporal variability of riverine discharge to the seafloor and strong biogeochemical gradients offshore (McKee et al. 2004; Lansard et al. 2009; Rassmann et al. 2016).
The Mediterranean Sea is a quasi-enclosed marginal sea (0.8% of the world ocean surface; Powley et al. 2017), often characterized as a replicate of an open ocean (Antoine and Morel 1995). This regional sea is oligotrophic, with lower nutrient concentrations (N and P) compared to oceanic areas (Dugdale and Wilkerson 1988), especially for dissolved inorganic phosphorus. A west–east gradient of decreasing P concentrations (Moutin et al. 2012) leads to higher primary production in the western Mediterranean Basin (Van Den Broeck and Moutin 2002; Moutin and Raimbault 2002). Overall, it has been shown that the P availability plays a key role as a limiting nutrient in the entire Mediterranean Sea (Krom et al. 1991; Diaz et al. 2001).
Rivers discharge large amounts of sediment and freshwater in the Mediterranean Sea (Ludwig et al. 2009). After the Nile damming, the Rhône River became the most important source of terrestrial organic matter, nutrients and freshwater to the Mediterranean Sea (Antonelli et al. 2008; Ludwig et al. 2009). The average particulate discharge from the Rhône River to the Mediterranean was estimated to be 7.4 Mt year−1 (Pont et al. 2002), with large seasonal and interannual variability. Overall, riverine fluxes at the Mediterranean Sea’s boundaries and the controlling processes at the river–sea connection are a key for understanding P cycling and its control in this oligotrophic sea, especially sediments where particulates from the river rapidly settle and are biogeochemically transformed.
Reactive P reaching the sediment–water interface is mainly associated to organic matter with a molar C/P ratio that is determined by its origin. Additional inputs of P are bound to Fe oxides and as carbonate fluorapatite (CFA; Emsley 1980; Sundby et al. 1992; Ruttenberg and Berner 1993; Slomp et al. 1996; Delaney 1998; Andrieux-Loyer and Aminot 2001; Slomp et al. 2004; Ruttenberg 2014). As organic matter is progressively buried below the sediment–water interface, the non-refractory fraction is degraded via a sequence of biogeochemical processes, releasing dissolved inorganic phosphorus (DIP) to the pore waters (Krom and Berner 1981; Sundby et al. 1992; Joshi et al. 2015). In the oxic zone of sediments, this dissolved phase may diffuse to the overlying waters or be adsorbed onto sediment Fe oxide surfaces reversibly and irreversibly (Fe-bound P; Syers et al. 1973; Froelich 1988; Fox 1990; Balls 1994; Filippelli and Delaney 1996; Anschutz et al. 1998; Slomp et al. 1998; Cade-Menun et al. 2019). In the anoxic layers, DIP concentrations generally show a large increase due to organic P mineralization and P release from Fe oxide dissolution, as the latter are used as a terminal electron acceptor in the mineralization process (Krom and Berner 1981). Therefore, iron reduction can have a significant role in phosphorus cycle as it has been shown for sulfur cycle (Thamdrup et al. 1994). DIP can ultimately be taken up by an authigenic mineral such as CFA through precipitation with Ca2+ (Ruttenberg and Berner 1993) or by vivianite (Emerson and Widmer 1978; Egger et al. 2015; Dijkstra et al. 2016; Lenstra et al. 2018). This “sink-switching” represents the main long-term sink of reactive P, as long as the authigenic mineral acts as an insoluble phase in deep sediments without being reprocessed during the diagenetic processes (Ruttenberg and Berner 1993; Slomp et al. 1998; Kraal et al. 2015). The P regeneration and sink-switching need to be better studied in subaqueous deltaic sediments subjected to high nutrient loads. Considering the large P inputs from the Rhône River, it is necessary to determine the biogeochemical P dynamics and the fate of P input to the deltaic sediments: release versus burial with authigenic minerals.
Biogeochemical modelling by reactive transport models has been largely used to the early diagenesis studies in the aquatic sediments (Berner 1980; Rabouille and Gaillard 1991; Tromp et al. 1995; Boudreau 1996; Soetaert et al. 1996; Wang and Van Cappellen 1996; Boudreau 1997; Wijsman et al. 2002; Berg et al. 2003; Canavan et al. 2006), but the benthic P cycle is rarely incorporated as a function of organic matter mineralization (Canavan and Slomp 2006; Gypens et al. 2008; Lenstra et al. 2018) especially in a highly dynamic system. Here, we analysed spatial variability in the sediment composition, the organic matter transformation and nutrient retention in the Rhône prodelta sediments, in order to quantify the intensity of biogeochemical processes and its effect on the benthic P cycle.
The present paper aims at quantifying the biogeochemical processes that control phosphorus recycling and trapping in these sediments and at determining the spatial impact of riverine organic matter from the river mouth to the adjacent continental shelf. To this end, we used an early diagenetic model (OMEXDIA, Soetaert et al. 1996), with P diagenesis (Ait Ballagh et al. 2020), and applied it to a dataset of sediment and pore water composition in the biogeochemical gradient of the Rhône River prodelta and shelf sediments, including particulate P speciation.
Materials and Methods
Study Site
The Rhône River (RR; Fig. 1) is the largest river in the Mediterranean Sea in terms of sediment loads, nutrient and freshwater fluxes (Antonelli et al. 2008; Ludwig et al. 2009). The RR is 812 km long, characterized by a catchment area of 97,800 km2 originating in the Alps and ending in a delta of 1455 km2 (Pont et al. 2002; Antonelli et al. 2004). Fifty kilometers upstream from the river mouth, the Rhône River splits into two unequal branches: the eastern channel (Grand Rhône, 50 km long, also called Rhône), which discharges 80–90% of the freshwater in the Gulf of Lions, and the western channel (Petit Rhône, 70 km long) that carries 10–20% of total discharge (Ibañez et al. 1997; Sabatier et al. 2006; Provansal et al. 2012). During flood events, 80 to 90% of annual particulate loads are discharged into the Rhône River delta, from the catchment area (Arnau et al. 2004; Ollivier et al. 2010; Eyrolle et al. 2012). The remaining loads originate from the soil erosion in the catchment area, in stream production or resuspension of river sediments from the upstream during lower freshwater discharges (Eyrolle et al. 2012). Overall, the mean water discharge at the Rhône River mouth is 1700 m3 s−1, with a peak discharge of 13,000 m3 s−1 in December 2003 (Lochet and Leveau 1990; Ibañez et al. 1997; Pont et al. 2002; Arnau et al. 2004).

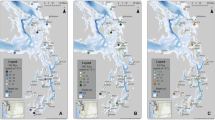
Location of the sampled stations in proximal (stations A, Z and AZ), prodelta (stations AK, K, B and L) and distal (stations C and E) domains of the Rhône River delta
Generally, the Rhône River’s plume extends towards the southwest (SW), as it is oriented by wind regimes close to the Rhône River mouth and by the River flow (Estournel et al. 1997; Naudin et al. 1997; Gatti et al. 2006). Consequently, the delivered loads of particulate material to the delta are mostly transported in the SW direction and some are ultimately driven to the adjacent continental shelf. The largest share of the riverine inputs is deposited at the seafloor near the river mouth, leading to the formation of a deposition fan called prodelta (Got and Aloisi 1990; Lansard et al. 2007). Consequently, the water depth increases from 10 m at the river mouth to 75–90 m on the adjacent continental shelf.
The decrease of sediment loads with distance from the Rhône River mouth is accompanied by a progressive aging of organic particles (Cathalot et al. 2013). The decreasing sediment deposition offshore gives rise to a large biogeochemical gradient from the Rhône River mouth to the continental shelf (Cathalot et al. 2010; Pastor et al. 2011; Rassmann et al. 2016). Concerning the DIP inputs from the Rhône River, Ludwig et al. (2009) showed an increase of DIP loads from 1970 to around 1985, followed by a decrease that may be related to the mitigation of pollution (i.e. the ban of P detergents and/or the upgrading of the waste water treatment plants).
The Rhône prodelta (named because of its prograding properties) is characterized by two regions: the proximal zone located near the river mouth at a water depth of around 20 m and a sedimentation rate of 20–35 cm year−1 depending on the period investigated and the tracer used (Charmasson et al. 1998; Radakovitch et al. 1999; Miralles et al. 2005; Lansard et al. 2007) whereas the prodelta zone is located further at 30–65 m depth and displays sedimentation rates of 1–2 cm year−1. The adjacent continental shelf extends further away from the prodelta and has sedimentation rates of less than 0.1 cm year−1.
Measurements
Sampling Stations
The sampling cruise MissRhoDia was carried out aboard the RV Tethys II in May 2018. Nine stations were sampled between 2 and 18 km from the Rhône River mouth, in the proximal (stations A, Z and AZ), prodelta (stations AK, K, B and L) and distal (stations C and E) domains over a southwest transect, corresponding to the main direction of the river plume. The locations of sampling stations are shown in Fig. 1, and the main characteristics are in Table 1. Sediment cores were collected using an UWITEC® single corer (9.5 cm of inner diameter, 60 cm of length) for pore water and solid-phase analysis. Bottom water samples were taken with a 12-l Niskin® bottle as close as possible to the seafloor (approximately 1 m). Salinity and temperature (°C) were measured using a salinometer with a precision of ± 0.1 and a thermometer with a precision of ± 0.1 °C, respectively (Table 1).
In Situ Microelectrode Measurements
The distribution of O2 and pH at the sediment–water interface was measured using in situ microelectrode with a benthic microprofiler (Unisense®) (Rabouille et al. 2003; Lansard et al. 2008; Cathalot et al. 2010; Rassmann et al. 2016, 2020). The measurements of 4–5 vertical depth O2 profiles per station and 2 pH profiles were achieved with a vertical resolution of 200 μm near the sediment–water interface. The dissolved O2 concentrations were measured by polarographic oxygen microelectrodes that were provided with a built-in reference and internal guard cathode (Revsbech 1989). The microelectrodes were calibrated with a 2-point calibration technique using the bottom water oxygen concentration determined by the Winkler titration (Grasshof et al. 1983) and the anoxic pore waters of the sediment (Cai and Sayles 1996). The pH microelectrodes were calibrated by NBS pH buffers (pH 4.01, 7.00 and 9.00) which provided the slope of the sensor response to pH and which estimated the pH variation. The measurement of seawater pH in bottom waters was carried out by spectrophotometry with unpurified m-cresol purple (Clayton and Byrne 1993; Dickson et al. 2007) and is reported on the total proton scale (pHT). The precision of the dissolved O2 measurement was of ± 0.5 μM, whereas the uncertainty of measured pH was ± 0.01 pH units.
Ex Situ Measurements of Solid and Pore Water Profiles
In order to determine the sediment porosity, a core was sliced with a resolution of 2 mm for 0–10 mm depth, 5 mm for 10–60 mm depth and 10 mm until the bottom of the core. The sediment porosity was calculated using water contents (the difference between the wet and dry weights after 1 week at 60 °C) and an averaged sediment density of 2.65 g cm−3. The sediment porosity was then corrected by the bottom water salinity.
A second sediment core was processed within 1–2 h after sampling for the extraction of pore waters by its introduction in a glove bag flushed with N2 atmosphere in order to avoid oxidation of reduced compounds. The extraction of pore waters was conducted by rhizons with a mean pore size of 0.1 μm (Seeberg-Elverfeldt et al. 2005). After collection, the pore water Fe2+ and the total dissolved Fe were measured using the ferrozine method (Stookey 1970). Pore waters were poisoned using HgCl2 for total alkalinity (TA) and dissolved inorganic carbon (DIC), acidified with HCl for SO42− measurements and stored at 4 °C until laboratory analysis. Subsamples for NH4+, NO3− and DIP measurements were frozen on board. The pore water NH4+ concentration was diluted and quantified by the indophenol blue method (Grasshof et al. 1983), while the pore water NO3− concentration was measured using the vanadium chloride reductant after dilution, followed by the addition of NEDD/sulphanilamide and the colorimetric method (Hansen and Koroleff 1999; Schnetger and Lehners 2014). The uncertainties of the methods for NH4+ and NO3− were 5% and 2%, respectively.
The pore water DIC concentrations were quantified with a DIC analyser (Apollo SciTech®), by the use of 1 ml of the sample volume with 4–7 replicates as described previously by Rassmann et al. (2016). The DIC method was calibrated using certified reference materials (oceanic CO2 measurements, Scripps Institution of Oceanography-batch no. 136) which provided an uncertainty of 0.3%. The pore water SO42− concentrations were diluted by a factor of 100 and measured using ion chromatography on an ICS 1000 chromatograph with an AS14 column and an AG14 guard column, both in 4 mm diameter (Dionex) (Rassmann et al. 2020). The uncertainty of this method was of ± 1.6%.
For the pore water DIP, the measurements were performed spectrophotometrically (Murphy and Riley 1962) at 880 nm on 3 ml of each sample, using a mixed reagent prepared daily from sulfuric acid (H2SO4), potassium antimony(III) oxytartrate hemihydrate (K(SbO)C4H4O6·1/2H2O), ammonium molybdate ((NH4)6Mo7O24·4H2O) and ascorbic acid (C6H8O6). The precision of the method was 1%.
Sequential Extraction of P Forms
Another sediment core was sampled and freeze-dried for 48 h and ground for further analysis of the P solid forms. The sedimentary P pools were separated using the sequential extraction method as detailed in Andrieux-Loyer et al. (2008) and adapted from widespread extraction methods (Psenner et al. 1988; Ruttenberg 1992).
Fe-bound P (including the Fe oxide–bound P and the vivianite) was extracted by 0.1 mol l−1 of dithionite–bicarbonate (8 h, 20 °C). In a second step, authigenic Ca-bound P (including carbonate fluorapatite Ca5(PO4, CO3)3F, biogenic hydroxyapatite Ca10(PO4)6(OH)2 and CaCO3 bound P) was quantified on the remaining fraction by 1 mol l−1 of Na acetate buffer (pH = 4, 6 h, 20 °C) then washed by 1 mol l−1 of MgCl2 (pH = 8, 0.5 h, 20 °C). Then, the detrital P was extracted using 1 mol l−1 of HCl (overnight). Total P was determined non-sequentially by 1 mol l−1 HCl treatment overnight after sediment ignition at 550 °C (4 h), while the inorganic P referring to the sum of P forms (Fe-bound P, Ca-bound P and detrital P), was extracted with 1 mol l−1 HCl before sediment ignition, in order to ensure the measurements’ efficiency. Organic P was then quantified by the difference between total P and inorganic P (Andrieux-Loyer et al. 2008).
Model Description
The modelling was performed by a steady-state version of the diagenetic model OMEXDIA (Soetaert et al. 1996; Khalil et al. 2018), implemented in the R software, using the R package ReacTran (Soetaert and Meysman 2012) and extended to include the benthic P cycle (Slomp et al. 1996; Ait Ballagh et al. 2020). The steady-state model assumption in our study allowed to understand the relative contribution of biogeochemical processes involved in the benthic P cycle in dynamic system outside flood events. This assumption is based on a stationary state observed over several years (2007–2015) during the spring–summer season, which allows typical diagenetic situation to be investigated using a steady-state model (Rassmann et al. 2020). This condition was achieved by setting the rates of changes to 0 (i.e. ∂Ci/∂t = 0). The model outputs were fitted to the measurements of organic carbon, terminal electron acceptors (O2, NO3−, SO42−), products of mineralization (NH4+, DIP, DIC and oxygen demand units (ODU; reduced substances during anoxic mineralization)) and solid P fractions (organic P, Fe-bound P and Ca-bound P). The model solves a set of general diagenetic equations (Berner 1980; Boudreau 1997) for dissolved Cd and solid Cs concentrations (Eqs. (1) and (2)), as described in the following:
where Cd and Cs denote the concentrations of the dissolved and solid species, respectively (in mol per volume of pore water or dry sediment−1). ∅ is the sediment porosity (cm3 cm−3), x is the sediment depth (cm), Ds is the diffusion coefficient (cm2 day−1), DB is the bioturbation coefficient (cm2 day−1), ω represents the sedimentation rate (cm day−1) and ∑Rd and ∑Rs denote the sum of the biogeochemical reactions occurring to dissolved and solid species, respectively. α is the depth-profile bioirrigation rate, while \( {\mathrm{C}}_{{\mathrm{d}}_0} \)is the bottom water concentration of dissolved phases (μmol l−1).
The dissolved species are transported by molecular diffusion and bioirrigation, whereas bioturbation is the main transport process for solid phases. In addition, advection is a common process for all species (Berner 1980; Boudreau 1997; Rabouille and Gaillard 1991). The sediment compaction is represented in the model by the exponential decrease of porosity (Berner 1980; Rabouille and Gaillard 1991). Below the sediment–water interface, all the mineralization pathways produce NH4+ and DIP (Table 2). The labile (FDet) and semi-labile (SDet) fractions of organic carbon are mineralized in the oxic zone by oxygen (OxicMin; Table 2, R1). Once the oxygen is totally consumed, the denitrification takes place in suboxic zone and is limited by nitrate concentration (Denitrif; Table 2, R2; Emerson et al. 2004). In the anoxic zone, metal oxides and sulfate are used as next terminal electron acceptors in the mineralization (AnoxicMin; Table 2, R3–R4). The ammonium and the other reduced compounds expressed as ODU (Berner 1989; Soetaert et al. 1996) are reoxidized by oxygen in the oxic zone (Nitrif and ODUoxid, respectively; Table 2, R5–R6). The stoichiometry of organic matter is represented by coefficients x and y denoting molar C/P and N/P ratios, respectively.
The diagenetic equations in Table 2 take into account all transport and reactive processes for the compounds; the labile and semi-labile fractions of organic carbon (Eqs. (3) and (4)), oxygen (Eq. (5)), nitrate (Eq. (6)), sulfate (Eq. (7)), ammonium (Eq. (8)), the dissolved inorganic carbon (Eq. (9)) and the ODU that are produced by anoxic mineralization and may be lost from the system to form solid substances as a pyrite (Eq. (10); Berner 1970; Soetaert et al. 1996; Rassmann et al. 2020). These equations are depending on the conversion between solid and liquid units (ϑ).
where ρs denotes the average density of the sediment particles (g cm−3).
Benthic Phosphorus Cycle
The benthic P cycle is included in the model to obtain more insight into the P transformation in the Rhône prodelta sediments. The present model describes the concentration change with depth of pore water DIP (μmol l−1) and three forms of particulate P (organic P, Fe-bound P and Ca-bound P (μmol g−1)). All the equations are detailed in Table 3 (see also Ait Ballagh et al. 2020). Figure 2 illustrates the biogeochemical processes of the benthic P cycle.

Schematic representation of the benthic P cycle
In the model, the P settling at the sediment–water interface is either associated to the organic matter by C/P ratios for the labile and semi-labile OC fractions or bound to Fe oxides (Emsley 1980; Sundby et al. 1992; Ruttenberg and Berner 1993; Slomp et al. 1996; Delaney 1998; Andrieux-Loyer and Aminot 2001; Slomp et al. 2004; Ruttenberg 2014). A refractory part Prefrac (μmol g−1) which is not degraded in the sediments is added to the labile fraction (Table 3, Eq. (11)).
Below the sediment–water interface, organic matter mineralization releases DIP (cm−3 liquid day−1) in the pore waters (Table 3, Eq. (12)). In the oxic zone, the DIP may diffuse to the overlying waters or be adsorbed onto sediment Fe oxide surfaces, which leads to Fe-bound P formation (μmol cm−3 liquid day−1) (Table 3, Eq. (13)) (Syers et al. 1973; Froelich 1988; Fox 1990; Balls 1994; Filippelli and Delaney 1996; Anschutz et al. 1998; Slomp et al. 1998; Cade-Menun et al. 2019). The Fe oxides are used as next terminal acceptors once oxygen, nitrate and manganese oxides are totally consumed (Canavan and Slomp 2006; Krom and Berner 1981). Therefore, the adsorbed DIP is released by the dissolution of Fe-bound P (μmol cm−3 solid day−1) (Table 3, Eq. (14)).
In the anoxic zone, the concentrations of DIP increase through organic matter mineralization and Fe-bound P dissolution. The pore waters are supersaturated with respect to carbo-fluorapatite (DIPbw > DIPeq) and form an authigenic mineral with Ca2+ (Ruttenberg and Berner 1993). The authigenic Ca-bound P (Ca-bound Pauth) does not dissolve once it is formed, and represents a permanent sink for reactive P in the sediments (μmol cm−3 liquid day−1) (Table 3, Eq. (15); Slomp et al. 1996). The coupling of the biogeochemical processes takes into account the conversion from solid units to the liquid ones (ϑ) when it is necessary, and the stoichiometric conversion factors (Table 3, Eqs. (16)–(18); Soetaert et al. 1996).
Benthic Budget Calculation
The steady-state benthic budgets of all simulated dissolved and solid compounds were calculated by the model with a focus on P budget: the deposition flux, the integrated rates of the biogeochemical processes and burial for each state variable. Total oxic mineralization, total denitrification, total anoxic mineralization, total organic P mineralization, total Fe-bound P formation, total Fe-bound P release and total Ca-bound P precipitation, expressed in μmol m−2 day−1, are the depth-integrated values of oxic mineralization (OxicMin), denitrification (Denitrif), anoxic mineralization (AnoxicMin), organic P mineralization (\( {\mathrm{P}}_{{\mathrm{org}}_{\mathrm{min}}} \)), Fe-bound Pformation, Fe-bound Prelease and Ca-bound Pprecipitation depth profiles expressed in the model in nmol cm−3 day−1, respectively.
Results
Model-Data Comparison of Dissolved and Solid Phases
The measured and simulated concentration-depth profiles of oxygen, nitrate, sulfate, DIC, ammonium, ODU and total organic carbon (TOC) are presented in Fig. 3 for all sampling stations. The O2 concentrations depleted rapidly from 185 to 235 μmol l−1 in bottom waters (at stations C and L, respectively; Table 5) to 0 μmol l−1 (at all stations). The O2 penetration depth into the sediment ranged from 2 mm in the proximal domain at station Z to 12.5 mm in the distal domain at station E, a pattern which was well represented by the model (Fig. 3). Nitrate concentrations were low at all stations and showed an increase with the distance from the river mouth to the adjacent continental shelf, without exceeding 15 μmol l−1 (Fig. 3). The measured nitrate concentrations in the bottom waters never exceeded 3 μmol l−1 (Table 5) and showed a peak below the sediment–water interface, indicating nitrification, followed by denitrification, which was reproduced by the model (Fig. 3). The depth of the nitrate peak increased in line with the increase of oxygen penetration from the river mouth to continental shelf, an evolution that was well reproduced by the model. The measured SO42− profiles highlighted a sharp decrease with sediment depth at stations Z and A in the proximal zone; the SO42− concentrations ranged from about 30 mmol l−1 in the bottom waters to concentrations below 5 mmol l−1 in deeper layers. The SO42− profiles showed a slighter decrease with sediment depth in the prodelta and distal domains (Fig. 3b, c; stations AK, B, K, L, C and E). The model was in good agreement with the SO42− measurements.

a Simulated (grey lines) and measured (symbols (grey circles)) O2, NO3−, SO42−, DIC, NH4+, ODU and TOC in the proximal domain (stations Z, A and AZ). b Simulated (grey lines) and measured (symbols (grey circles)) O2, NO3−, SO42−, DIC, NH4+, ODU and TOC in the prodelta domain (stations AK, B, K and L). c Simulated (grey lines) and measured (symbols (grey circles)) O2, NO3−, SO42−, DIC, NH4+, ODU and TOC in the distal domain (stations C and E)
The measured and simulated DIC profiles indicated an increase with depth at all stations: concentrations were extremely high in the proximal domain (around 50 mmol l−1 at stations Z and A below 15 cm, Fig. 3). DIC concentrations in pore waters at depth decreased according to distance from the river mouth to about 5 mmol l−1 in the distal domain (Fig. 3c, station E). This pattern linked to the diagenetic intensity was also well reproduced by the model. The same biogeochemical gradient was observed for the pore water NH4+ profiles, with concentrations increasing with sediment depth at all stations. A gradient of NH4+ concentrations at sediment depth (15 cm) from up to 3000 μmol l−1 (station Z) until about 100 μmol l−1 (station E) was visible, similar to the DIC profiles. Therefore, a larger production of DIC and NH4+ was clearly observed in the proximal domain compared to the prodelta and distal domains. Overall, the model was in good agreement with pore water DIC and NH4+ profiles at all stations (Fig. 3). The ODU concentrations (mainly Fe2+) displayed an increase with sediment depth, with about 150 μmol l−1 close to the river mouth, but this maximum decreased in continental shelf stations to less than 5 μmol l−1 (station E; Fig. 3c). The simulated TOC profiles never exceeded 2% at the sediment–water interface and showed a decrease with sediment depth at all stations (Fig. 3).
Figure 4 shows the measured and simulated profiles of DIP, organic P, Fe-bound P and Ca-bound P. DIP concentrations were low near the sediment–water interface then showed an increase with sediment depth. In deeper sediment layers of the proximal domain, DIP concentrations were extremely high (about 250 μmol l−1 at stations Z, A and AZ). These deep DIP maximum concentrations decreased with water depths and distance from the river mouth until about 25 μmol l−1 at station E (Fig. 4). Overall, the model reproduced the increasing shape of DIP with sediment depth, except at stations Z, B and K below 10 cm depth (Fig. 3). The DIP evolution followed a similar pattern as DIC and NH4+ for all stations. The measured organic P profiles never exceeded 10 μmol g−1 at the sediment–water interface at all stations. Organic P showed a slight decrease with sediment depth and showed a generally constant concentration offshore. Stations Z and A displayed a larger spread of organic P values with sediment depth. The model showed a good agreement with average measurements for this phase but showed a large gradient near the sediment–water interface (Fig. 4). Similarly, the observed Fe-bound P concentrations at stations A, Z and AZ showed dispersed values, while the model reproduced the average Fe-bound P concentrations and showed a slight decrease with sediment depth (Fig. 4). Concerning the other stations, the average Fe-bound P concentrations were well represented by the model (Fig. 4). A similar pattern was found for Ca-bound Pauth concentrations and showed also dispersed values in stations A, Z and AZ. This phase showed very consistent values at the profile scale and between profiles at all stations from the river mouth to the continental shelf, which was overall reproduced by the model (Fig. 4).


Simulated (grey lines) and measured (symbols (grey circles)) DIP, organic P, Fe-bound P and Ca-bound P in the sampled stations
Boundary Conditions and Model Parameters
The fixed parameters of the model listed in Table 4 were ammonium adsorption coefficient (NH3Ads), maximum oxidation of ODU (rODUox), nitrification rate constant (rnit), half-saturation constants for O2 limitation for oxic mineralization (\( {\mathrm{Ks}}_{{\mathrm{O}}_2\mathrm{oxic}} \)) and nitrification (\( {\mathrm{Ks}}_{{\mathrm{O}}_2\mathrm{nitri}} \)), nitrate limitation for nitrate reduction (\( {\mathrm{Ks}}_{{\mathrm{NO}}_3\mathrm{denit}} \)), O2 limitation for the oxidation of reduced substances (\( {\mathrm{Ks}}_{{\mathrm{O}}_2\mathrm{ODUox}} \)), O2 inhibition for nitrate reduction (\( {\mathrm{Kin}}_{{\mathrm{O}}_2\mathrm{denit}} \)) and anoxic mineralization (\( {\mathrm{Kin}}_{{\mathrm{O}}_2\mathrm{anox}} \)), nitrate inhibition for anoxic mineralization (\( {\mathrm{Kin}}_{{\mathrm{NO}}_3\mathrm{anox}} \)), sulfate limitation for anoxic mineralization (\( {\mathrm{Ks}}_{{\mathrm{SO}}_4} \)) and finally the C/N and C/P fixed for labile and semi-labile fractions of organic carbon for all the sampling stations (Table 4). The C/Nfast and C/Nslow were fixed to 7.1 (Pastor et al. 2011) and 10 (Khalil et al. 2018), respectively. The C/P ratio was fixed by assuming that N/P was equal to 16. Therefore, the estimated C/P for labile fraction (C/Pfast) was averaged to 114, while the C/P for semi-labile fraction (C/Pslow) was 160. It is noteworthy that because the proportion of the labile fraction increases offshore (from 50% to 75%), the overall C/P ratio decreases as well from 137 in the proximal zone to 126 in the distal zone which is consistent with the aquatic nature of the organic matter degraded in these sediments.
Table 5 includes the parameters that were measured at each station. The bottom water temperature ranged between 14.7 °C (at station C) and 20 °C (at station AZ), which is typical of the area at this season. Salinity varied between 37.8 at stations A and AZ and 38.5 at station K (Table 5). The bottom water concentrations of all dissolved phases did not follow a spatial pattern (Table 5). The bottom water concentrations of NO3−, NH4+ and ODU were low for all stations. The SO42− concentrations were nearly constant in bottom waters and close to 30 mmol l−1 at all stations (Table 5). The porosity at surface sediment and at depth was lowest in station K (0.76 and 0.58, respectively) and highest in station C (0.86 and 0.64, respectively). The sedimentation rates adopted for this modelling study were obtained from the literature (Charmasson et al. 1998; Radakovitch et al. 1999; Miralles et al. 2005; Lansard et al. 2007), ranging from 10 cm year−1 in proximal domain to 0.1 cm year−1 in distal domain (Pastor et al. 2011).
The parameters estimated by fitting the model to the data are listed in Table 6. The reactive OC flux (OCflux) followed a spatial pattern and decreased from 160 mmol m2 day−1 at station Z (the river mouth) to 10 mmol m−2 day−1 at station E on the continental shelf. The first-order decay rate constants for labile fraction of OC (Rfast) showed a clear increase with distance to the continental shelf (0.06 day−1 at station Z to 0.2 day−1 at stations B, K, L, C and E). Contrarily, the first-order decay rate constants of the semi-labile fraction (Rslow) decreased with distance to the river from 0.0035 day−1 at station Z to 0.0002 day−1 at stations B and C (Table 6). The labile OC represented an important proportion of OC (Pfast) that increased from 50% near the river mouth (stations Z and A) to 75% on the continental shelf. The refractory carbon varied between 0.9 and 1.4% (Table 6). The proportion of ODU deposition (pdepo) ranged between 90 and 99%. The bioturbation layer thickness (xb) increased from 5 cm for stations A, Z and AZ to 10 cm for other stations, while the bioturbation coefficient (Db0) was relatively invariant (from 0.01 to 0.015 cm2 day−1). The bioirrigation at the sediment surface (αirr0) was needed for all stations but did not indicate a clear spatial trend according to the distance from the Rhône mouth (0.05–0.2 day−1), with bioirrigation depths (xirr) ranging between 6 and 14.5 cm of depth (Table 6).
The Fe-bound P fluxes at the sediment–water interface (Fe-bound Pflux) decreased offshore with values as high as 730 μmol m−2 day−1 at station Z and decreased to 10 μmol m−2 day−1 at stations C and E. The Fe-bound P formation rate constants (\( {\mathrm{R}}_{\mathrm{Fe}\hbox{-} \mathrm{bound}\ {\mathrm{P}}_{\mathrm{form}}} \)) were 0.8 day−1 at all stations, except for stations C and E where they were only 0.1 day−1. Concerning the Fe-bound P release, the rate constants (\( {\mathrm{R}}_{\mathrm{Fe}\hbox{-} \mathrm{bound}\ {\mathrm{P}}_{\mathrm{rel}}} \)) were similar at all stations between 0.0005 day−1 (at stations B and K) and 0.0008 day−1 (at the other stations; Table 6). The Fe-bound P asymptotic concentrations (Fe-bound Pasymp) ranged between 2 μmol g−1 (at station A) and 4 μmol g−1 (at station C). The Ca-bound P precipitation rate constants (\( {\mathrm{R}}_{\mathrm{Ca}\hbox{-} \mathrm{bound}\ {\mathrm{P}}_{\mathrm{precip}}} \)) ranged between 0.002 day−1 (stations K and C) and 0.006 day−1 (stations Z and L) (Table 6). Inherited concentrations of Ca-bound P (Ca-bound Pher) were between 3.6 and 5.3 μmol g−1.
Discussion
The application of the model to the dataset from sediments on all stations (from Z to E) along the Rhône River prodelta allowed quantifying total organic matter mineralization rates and calculating the contribution of different diagenetic pathways to the mineralization processes. Moreover, the addition of the benthic P cycle allowed identifying the sources and sinks of internally produced DIP and calculating sedimentary P budgets in proximal, prodelta and distal domains.
Spatial Variability of Organic Matter Mineralization
The Organic Matter Properties
The OC fluxes (OCflux) ranged between 160 and 10 mmol m−2 day−1 (Fig. 5) and were characteristic of organic-rich ecosystems (Rabouille et al. 2008). They were similar to the obtained OC fluxes for the same stations at the Rhône River prodelta by Pastor et al. (2011). Moreover, the OC fluxes decreased exponentially as a function of distance from the Rhône River mouth (Fig. 5). This decrease was already observed by Cathalot et al. (2010) for the oxygen diffusive fluxes. During flood events, occurring mostly in fall–winter, the majority of organic-rich particulate inputs from the Rhône River are accumulated in the proximal and prodelta domains. Therefore, these areas are characterized by large OC mineralization rates during spring (Lansard et al. 2009; Cathalot et al. 2010; Pastor et al. 2011; Rassmann et al. 2016). This is also evidenced in the results by complete removal of sulfate during sulfate reduction and the intense production of DIC especially in the proximal domain.

Metabolized OC fluxes at the sediment–water interface along the Rhône River prodelta
In front of the Rhône River mouth, Rassmann et al. (2016, 2020) showed that pore waters were supersaturated with Ca2+, preventing carbonate dissolution in these deltaic sediments. As a consequence, the DIC was mainly produced by OC mineralization processes (i.e. bacterial respiration). The obtained fluxes (10–160 mmol m−2 day−1) were in the same range as those from the Louisiana shelf (35 mmol m−2 day−1; Morse and Eldridge 2007), although higher in the proximal domain. They were higher also compared to the Guadalquivir River mouth (Ferrón et al. 2009) and the Po River (Hammond 1999).
In addition to refractory carbon, the model takes into account two fractions of organic matter with variable reactivities: the labile and semi-labile fractions. Each labile and semi-labile fraction has a first-order decay rate constant (Rfast and Rslow, respectively; Soetaert et al. 1996), while the proportion of labile OC (Pfast) determines the relative abundance of both fractions in the OC inputs. The reactive compound was the dominant fraction in the organic carbon input (Corg) as it increased from 50% in the proximal zone to 75% in the continental shelf (Table 6), being negatively correlated to the OC fluxes as has been already mentioned in Pastor et al. (2011). The first-order decay rate constant of the labile OM fraction (Rfast) was higher in the prodelta and distal domains compared to the proximal domain and increased with distance offshore from 0.06 day−1 at the river mouth to 0.2 day−1 on the continental shelf (Fig. 6a) which indicated a progressive increase in lability and proportion of labile organic matter with the distance offshore. The rate constants in the proximal zone (0.06 to 0.15 day−1) were slightly higher than those calculated for the Mississippi Delta and the Louisiana shelf (0.02–0.05 day−1; Morse and Eldridge 2007). However, the rate constants calculated for the continental shelf were in the same range as the eutrophic Scheldt estuary (0.15 day−1; Hofmann et al. 2008) and the Thau lagoon (0.1 day−1), which are characterized by labile organic matter from phytoplankton and macrophyte debris (Dedieu et al. 2007).

First-order decay rate constants of labile (a) and semi-labile (b) OM fractions as a function of distance from the river mouth
The first-order decay rate constant of the semi-labile OM fraction (Rslow) decreased with distance from the Rhône River mouth to the continental shelf by a factor of 10 (0.0035 to 0.0002 day−1, respectively; Fig. 6b). This could be related to the supply of older and less reactive material as indicated by 14C dating (Cathalot et al. 2013). The Rslow values were in the same range as the values from the eutrophic environments, such as Scheldt estuary (0.002 day−1; Hofmann et al. 2008) and the Thau lagoon (0.001 day−1; Dedieu et al. 2007). This trend matched with the results of Pastor et al. (2011) and highlighted that the mix of labile and semi-labile organic matter in the proximal and prodelta domains contained essentially terrestrial fine and coarse organic matter including a small proportion of riverine organic matter (Dumoulin et al. 2018). At the same time, the sediments in the distal domains probably receive labile marine organic matter (Pozzato et al. 2018) which is entirely mineralized together with aged terrestrial organic particles (Lansard et al. 2009).
The Rfast, Rslow and Pfast values highlighted that the terrestrial organic matter in the proximal domain was composed of fine and coarse continental material rich in organics (Cathalot et al. 2013). It was less reactive than those at other stations offshore probably due to prior degradation during transport or storage on land. Stations offshore receive a larger proportion of marine organic material such as phytoplankton (Pastor et al. 2011). This evolution is confirmed by the results obtained using δ13COC by Lansard et al. (2009). δ13COC varied from − 26.8‰ in the proximal domain sediments to − 23.9‰ on the shelf, showing a significant increase in the distal domain, linked to the limited dispersal of terrestrial organic matter on the continental shelf. The limited transport of terrestrial organic matter led to the highest values of Rfast with distance from the river mouth linked to the increase of marine material proportion (Fig. 6a).
Organic Matter Mineralization Processes
The decreasing mineralization rates of organic matter from the proximal to distal domain were reflected in the penetration depths of oxygen that were less than 2 mm at station A or Z while they reached 12.5 mm in the distal domain (Cathalot et al. 2010; Rassmann et al. 2016). At these stations, the large increase of DIC production and the high consumption of sulfate indicated the large mineralization of reactive organic matter (Fig. 3).
The pore water NH4+, DIC and DIP were indicators of organic matter mineralization, and their concentrations increased as a function of sediment depth (Fig. 3; Berg et al. 2003). Overall, the prodelta stations showed lower concentrations of these compounds in deep sediments compared to the proximal domain (e.g. the NH4+ with 300–1000 μmol l−1 and 2000–3500 μmol l−1, respectively) but were higher than the distal domain (200–500 μmol l−1). The same pattern was shown by the other components (Fig. 3). The observed decrease of asymptotic concentrations of DIC, NH4+ and DIP with distance to the shore was related to the decrease of OC mineralization rates in these areas, causing the decrease of anoxic mineralization. This is well reflected in Table 6 where OC flux represents the metabolizable organic matter deposition which, in the model calculation, is completely mineralized in surficial sediments. The values of this integrated mineralization varies from over 100 mmol C m−2 day−1 in the proximal zone to 10 mmol C m−2 day−1 on the distal shelf, with a gradual decrease over the prodelta.
In the proximal domain, and after the total consumption of O2 and NO3−, the ODU mainly consisted of dissolved Fe2+, as H2S is very low (Rassmann et al. 2020). The ODU showed an increase of concentrations with sediment depth, corresponding to the Fe-bound P decrease (Fig. 4). This indicated a production of dissolved Fe and a potential release of DIP through the reductive dissolution of Fe oxides, used as a terminal electron acceptor in the anoxic zone of sediments (Krom and Berner 1981; Canavan and Slomp 2006). This Fe-bound P decrease was less important in prodelta and distal domains, as shown by rather constant Fe-bound P profiles with depth, which indicated that the released DIP in the sediments mainly originated from the organic matter release without or with a minor contribution of net Fe-bound P release (Fig. 4).
Overall, the model calculations showed that oxic mineralization followed a spatial pattern along the biogeochemical gradient, increasing its contribution to the total mineralization from 13.7% (station A, proximal domain) to 80% (station E, the distal domain; Fig. 7). The oxic mineralization contribution showed the same pattern as Pastor et al. (2011) and was in the same range (8–67%). The oxygen below the sediment–water interface was used also for the reoxidation of NH4+ and the reduced substances that were produced in the anoxic zone. However, the nitrification process was not very important in the Rhône prodelta sediments, and only a small fraction of ODU was reoxidized, since more than 90% of ODU was buried at depth (pdepo, Table 6). Therefore, the main process responsible for oxygen consumption was oxic mineralization. The denitrification represented the smallest contribution compared to the other mineralization pathways (0.7 to 3.7%) and showed an increase from proximal to distal domains (Fig. 7). This low contribution was due to the fact that denitrified NO3− originated only from nitrification below the sediment–water interface, due to the low concentration of NO3− in bottom waters (Table 5). Anoxic mineralization was related to the Mn oxides, Fe oxides and sulfate reduction in the anoxic zone. It exhibited a clear spatial gradient, accounting for more than 85% of total mineralization near the river mouth, decreasing to 16% near the continental shelf (Fig. 7). Sensitivity analysis of the model showed that the variations of the sedimentation rates in the Rhône proximal and prodelta domains do not impact significantly the contributions of mineralization pathways. Indeed, an increase by a factor of 10 of the sedimentation rates (from 1 to 10 cm year−1 in the proximal zone and from 0.1 to 1 cm year−1 for the prodelta domain) induced slightly lower contribution of oxic mineralization (from 18 to 14% of the total mineralization) in the proximal domain and was negligible in the prodelta domain.

Contribution of mineralization pathways (%) in the sampled stations
The model showed a decreasing loss of ODU by burial from 99% at station Z to 90% at station C (Table 6). The ODU deposition highlighted a loss of reduced products of mineralization at deeper depths as solid substances, such as pyrite (Berner 1970; Soetaert et al. 1996). Similarly, high removal rates were found previously by Pastor et al. (2011) and Rassmann et al. (2020) in the Rhône River prodelta.
Benthic P Transformation Along the Rhône River Prodelta
At the proximal stations, DIP concentrations showed an increase with sediment depth until a maximum of 300 μmol l−1. The DIP concentrations were larger compared with concentrations obtained by Rassmann et al. (2020) for the same stations (50–160 μmol l−1). This was probably linked to the intensity of OC inputs from 2015 (Rassmann et al. 2020) to 2018 (this study). As pointed out in the literature, the secondary conversion of Fe oxides to FeS certainly contributed to the net release of adsorbed DIP on Fe oxides with sediment depth (Anschutz et al. 1998). Indeed, Rassmann et al. (2020) described a clear precipitation of FeS at the proximal stations as shown by the spatial pattern of solid FeS, decreasing from the proximal domain to the adjacent continental shelf sediments. Both the increase of DIP concentrations and the decrease of Fe-bound P with sediment depth in the proximal and prodelta zones accompanied a slight increase of Ca-bound P with depth, characterized by dispersed values of measurements (Fig. 4). The high DIP concentrations in pore waters likely contributed to the precipitation of Ca-bound Pauth (Fig. 4). Despite the decrease of OC fluxes to the seafloor and thus of organic P, the Fe-bound P and the Ca-bound P concentrations were almost similar at the sediment–water interface in the proximal, prodelta and distal domains (Figs. 3 and 4). This can probably be related to a compensation of flux versus sedimentation, as both decrease at a similar rate from the River mouth to offshore.
Sources of Pore Water DIP
The organic P input to the sediment was calculated using the organic C fluxes and the C/Pfast and C/Pslow assumed in this study (Table 4). The obtained integrated rates of organic P mineralization, extremely high at the River mouth (1196 μmol m−2 day−1; Fig. 8), were characteristic of organic-rich ecosystems. Their decrease from the proximal to the distal domain (1196 to 81 μmol m−2 day−1; Fig. 8), was linked to the decrease of organic matter inputs (Pastor et al. 2011). The values obtained from the Rhône prodelta sediments were higher than the integrated rates of organic P mineralization from the eutrophic Elorn and Aulne estuaries (262–757 μmol m−2 day−1 and 172–414 μmol m−2 day−1, respectively; Ait Ballagh et al. 2020). Figure 8 shows that the organic P mineralization represented the main source by up to 90% of the total DIP production, while the net Fe-bound P release had a minor contribution in benthic DIP production at all stations (Z to E).

Integrated rates of DIP production (μmol m−2 day−1), for organic P mineralization and net Fe-bound P release
Concerning the Fe-bound P fluxes deposited at the sediment–water interface (Fe-bound Pflux), Table 6 and Fig. 8 highlighted their importance in the proximal domain (730 μmol m−2 day−1 at station AZ), which were more than 10 times higher in the prodelta and distal domains (82 and 10 μmol m−2 day−1 at stations B and E, respectively). The Fe-bound P fluxes in the distal domain were similar to the fluxes from Vilaine Bay (4–25 μmol m−2 day−1; Ratmaya 2018), due to the similarity of sedimentation regimes at the sampling stations of the distal zone (0.1 cm year−1) and the Vilaine Bay (0.2 cm year−1; Ratmaya 2018).
Except at station A (Fig. 9), the model indicated that the formation of Fe-bound P in the oxic zone and its release below were very similar along the Rhône River prodelta. This indicated that, at all stations, Fe-bound P acted as a temporary sink for DIP in the oxic zone, which was thereafter released in the anoxic zone. Therefore, Fe-bound P acted as a shuttle between organic P mineralization and Ca-bound Pauth precipitation (sink-switching) along the Rhône River prodelta (Ruttenberg and Berner 1993; Slomp et al. 1998; Kraal et al. 2015).

Integrated Fe-bound P (μmol m−2 day−1) formation and release rates
Consumption/Export of Internally Produced DIP
The model calculations showed that DIP produced by mineralization and net Fe-bound P release was essentially lost by recycling to the overlying waters rather than burial through Ca-bound Pauth formation. Figure 10 indicates that the sum of both processes, in terms of DIP consumption/export fluxes, decreased from the river mouth to the continental shelf similar to the organic matter mineralization. A difference by a factor of 3 was observed between the proximal and the prodelta domains while a factor of 10 was calculated between the proximal and the distal domains. In the prodelta and distal domains, the recycled DIP accounted for up to 90% of total DIP losses, while the burial of reactive P as Ca-bound Pauth form was higher in the proximal domain but never exceeded 28% of the total DIP loss (Fig. 10).

DIP losses (μmol m−2 day−1) along the Rhône River prodelta
The recycling of DIP to the overlying waters (Fig. 10) decreased from the river mouth to the continental shelf (931 to 74 μmol m−2 day−1) and occurred mainly through molecular diffusion (> 70%) at all stations, while bioirrigation process accounted for a minor part. These recycled DIP fluxes to the overlying waters were in the same range as those from the Elorn and Aulne estuaries (85–738 μmol m−2 day−1; Ait Ballagh et al. 2020), the Haringvliet Lake (370 μmol m−2 day−1; Canavan and Slomp 2006) and those from the Gulf of Finland 0–1032 μmol m−2 day−1 (Lehtoranta et al. 2017). The eutrophic Haringvliet Lake, the Vilaine bay and the Elorn and Aulne estuaries were similar to the Rhône River prodelta in terms of the role of organic P mineralization as the main benthic DIP source, unlike the Gulf of Finland, where the DIP release mostly originated from the Fe-bound P release (Canavan and Slomp 2006; Lehtoranta et al. 2017).
Sedimentary Biogeochemical P Budgets
Figure 11 represents vertically integrated P budgets for 3 selected stations, 1 from each domain: the proximal (station A), prodelta (station K) and distal (station E) domains. For these budgets, the benthic P processes were integrated over the sedimentary column of each sampling station. Overall, it is clear that DIP was mainly released by organic P mineralization, as it was the main source of DIP in the Rhône prodelta sediments. However, this intensity was enhanced by the Fe-bound P phase, with a minor contribution of net Fe-bound P release. As the organic P mineralization fluxes decreased from the proximal (1004–1196 μmol m−2 day−1; Fig. 11, station A) to the distal (113 and 81 μmol m−2 day−1, station E) domains, the Fe-bound P fluxes also decreased by a factor of 50 between the river mouth and offshore in relation with the decrease of the river material deposition offshore. Therefore, the iron-related P cycle decreased in intensity towards the shelf with an overall minor contribution in the proximal zone (8–81 μmol m−2 day−1) and no net Fe-bound P release contribution in the distal domain (Fig. 11).

Sedimentary P budgets (μmol m−2 day−1) for typical stations of the different zones of the Rhône River prodelta: proximal (station A), prodelta (station K) and distal (station E) domains
The largest part of the intense DIP production in the sediment was recycled to the overlying waters by molecular diffusion at all domains. A significant sink was calculated as Ca-bound Pauth burial in the proximal zone (Fig. 11, station A) with rapid levelling off offshore as the production of DIP in the sediment decreased. Sensitivity tests showed that uncertainty on the sedimentation rates may impact the calculated rate of DIP recycling to the water column. Indeed, increasing sedimentation rates by a factor of 10 (from 1 to 10 cm year−1 in the proximal zone) decreased DIP production in the sediment essentially by lowering the contribution of Fe-bound P dissolution to DIP and contributed to lower overall DIP flux to the water column (− 40%). This increase of sedimentation rate also induced an increase of burial as Ca-bound P (+34%) in the proximal domain. Furthermore, these sensitivity tests showed that the C/P ratios which control the input of organic P to the sediment have an influence on the mass balance of the P cycle. An increase of the C/P of semi-labile organic matter from 160 (used in this study) to 200 would decrease the recycling flux by 20% in the proximal domain, essentially because of the lower flux of organic P deposited in the sediment. Ca-bound P burial would also be lowered by 15% because of the decreased DIP availability. Lower variations were calculated for the prodelta domain with less than 10% variation of DIP recycling and Ca-bound P burial for similar variations of C/P in the semi-labile fraction. The change of C/P for the labile fraction (from 114, adopted in this study, to 100) would have a lower impact on the P mass balance with decreases of 9% for DIP recycling and 3% for Ca-bound P burial.
The P Budgets at a Deltaic Scale
In order to assess the contribution of the Rhône River prodelta as a source/sink of DIP to or from the Mediterranean Sea, the P budgets (recycling/burial) along the Rhône River prodelta were calculated. For that purpose, we averaged the benthic DIP fluxes and authigenic Ca-bound P burial fluxes obtained using the model-data approach for each zone of interest (proximal, prodelta and continental shelf-distal zones). As these zones were rather homogeneous regarding the benthic DIP fluxes and Ca-bound Pauth burial (for ± 20% standard variation, see Fig. 10) compared to the overall variation between the zones (1 order of magnitude between proximal and distal zones, Fig. 10), averaging did not bring significant bias in the final calculations. Surface areas of the proximal, prodelta and distal domains (Table 7) were adopted from Lansard et al. (2009) in agreement with the Thiessen polygon method, used in Cathalot (2008).
By using these surface areas, we obtained the total benthic DIP fluxes (a source to the water) and the sink due to precipitated Ca-bound P burial in the sediments of the Rhône River prodelta and its adjacent continental shelf. These fluxes were then compared to the riverine DIP discharge. According to Malagó et al. (2019), the Rhône River delivered 1465 tons DIP year−1 to the Mediterranean Sea from 2003 to 2007, whereas Ludwig et al. (2009) estimated a discharge of 2750 tons DIP year−1, in agreement with direct measurements by Moutin et al. (1998). These separate estimations varied by a factor of 2 which could be related to the decrease of P inputs to the rivers from the 1990s to the 2000s to the end of the 2010s. It may also be due to differences in assumptions for the calculations. It is out of the scope of this paper to further discuss these issues, and we used the two values for bracketing our estimates of the prodelta contribution in the DIP source and sink.
As can be seen from Table 7, the DIP fluxes recycled from the sediment accounts for 19–35% of the riverine discharge input of DIP which indicates that this source could represent an additional source corresponding to 25% of the Rhône input. We propose here that it can be considered as an additional source of DIP to the Mediterranean Sea since the largest part of this benthic flux originates from the recycling of organic P of terrestrial origin. These calculated proportions surely contain uncertainties on some assumptions such as the DIP inputs and the extrapolation of summer values of P budgets over the year. However, the calculation at the deltaic scale over the entire year was necessary in order to compare the sediment source to the river input. These calculations are important because the contribution of sediments was substantial compared to riverine inputs, especially near the river mouth, and no estimations were available before this study for the Rhône River prodelta. Finally, we estimate that the obtained DIP flux from the sediment may be overestimated by less than one-third due to the extrapolation over the year.
In the proximal and prodelta zones, it corresponds to the recycling of terrestrial organic matter (Cathalot et al. 2013) and is not related to internal recycling of marine (or deltaic) production based on riverine DIP input. It thus constitutes a real addition to the DIP input from the Rhône River. This is probably less true for the continental shelf sediments (33% of benthic DIP recycling) where marine organic matter produced by DIP originating from the Rhône River is partly recycled in surface sediments, therefore not totally contributing as an additional source. Considering that, the 25% addition to the riverine flux by benthic recycling of organic P can be seen as an upper boundary.
Burial of Ca-bound Pauth phase accounts for around 3–6% of the riverine DIP flux, which is minor sink. Furthermore, as most of this precipitation and burial of Ca-bound Pauth is linked to DIP production by mineralization in the sediment (see Fig. 11), it does not constitute a true sink of DIP delivered from the Rhône River. Indeed, as stated above, most of the mineralized organic P in the proximal and prodelta zones originates from the continent and is independent of the DIP source from the Rhône River. Only the share of Ca-bound Pauth burial in continental shelf sediments can be considered a sink of DIP originating from the Rhône River as organic matter mineralized in these sediments is of marine origin based on nutrients from the Rhône River. As it represents only 20% of the total Ca-bound Pauth burial, it can be considered that a very limited fraction of the DIP input by the river (< 1%) is precipitated as Ca-bound P and buried in sediments.
Conclusion
In order to gain new insights about organic matter and phosphorus transformation in the sediments along the Mediterranean Rhône River prodelta, we used a modelling approach based on an early diagenetic model (OMEXDIA) coupled to the dataset from proximal, prodelta and distal domains. The model application highlighted the intensity of mineralization processes from the Rhône River mouth to the adjacent continental shelf. This intensity was mainly driven by high deposition of OC in the sediments, as a characteristic of organic-rich ecosystems, with a large proportion of terrestrial organic matter, hence providing a biogeochemical gradient. The combined model parameters indicated that the OC fluxes to the seafloor led to the increase of oxic mineralization rates and reduction of anoxic mineralization rate contribution, as a function of distance from the Rhône River mouth. According to the model, the anoxic mineralization contributed to the large concentrations of mineralization products at the sediment depth such as DIC, DIP and NH4+, especially in the proximal domain. Our modelling approach was extended to include the benthic P cycle along the Rhône River prodelta. It showed that there was an intense production of pore water DIP, mainly by organic P mineralization. The Fe-bound P was a small net source of DIP in the proximal zone and was negligible in the prodelta and distal domains, while this phase had a key role in trapping and transferring DIP in sediment, promoting the Ca-bound P precipitation in anoxic sediments in the proximal domain. However, the sediments along the Rhône River prodelta acted mainly as a secondary source of DIP to the overlying waters, corresponding to a maximum of 25% of the riverine input, as only a minor fraction was precipitated to Ca-bound P.
Data Availability
The entire dataset is provided on the SeaNoe database (https://doi.org/10.17882/73204).
References
Ait Ballagh, F. E., C. Rabouille, F. Andrieux-loyer, K. Soetaert, K. Elkalay, and K. Khalil. 2020. Spatio-temporal dynamics of sedimentary phosphorus along two temperate eutrophic estuaries: A data-modelling approach. Continental Shelf Research 193. Elsevier Ltd: 104037. https://doi.org/10.1016/j.csr.2019.104037.
Andrieux-Loyer, F., and A. Aminot. 2001. Phosphorus forms related to sediment grain size and geochemical characteristics in french coastal areas. Estuarine, Coastal and Shelf Science 52 (5): 617–629. https://doi.org/10.1006/ecss.2001.0766.
Andrieux-Loyer, F., X. Philippon, G. Bally, R. Kérouel, A. Youenou, and J. Le Grand. 2008. Phosphorus dynamics and bioavailability in sediments of the Penzé estuary (NW France): In relation to annual P-fluxes and occurrences of Alexandrium Minutum. Biogeochemistry 88 (3): 213–231. https://doi.org/10.1007/sl0533-008-9199-2.
Anschutz, P., S. Zhong, B. Sundby, A. Mucci, and C. Gobeil. 1998. Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments. Limnology and Oceanography 43 (1): 53–64. https://doi.org/10.4319/lo.1998.43.1.0053.
Antoine, D., and A. Morel. 1995. Algal pigment distribution and primary production in the eastern Mediterranean as derived from coastal zone color scanner observations. Journal of Physical Oceanography Research 100: 16,193–16,209.
Antonelli, C., M. Provansal, and C. Vella. 2004. Recent morphological channel changes in a deltaïc environment. The case of the Rhône River, France. Geomorphology 57 (3-4): 385–402. https://doi.org/10.1016/S0169-555X(03)00167-3.
Antonelli, C., F. Eyrolle, B. Rolland, M. Provansal, and F. Sabatier. 2008. Suspended sediment and 137Cs fluxes during the exceptional December 2003 flood in the Rhone River, Southeast France. Geomorphology 95 (3-4): 350–360. https://doi.org/10.1016/j.geomorph.2007.06.007.
Arnau, P., C. Liquete, and M. Canals. 2004. River mouth plume events and their dispersal in the northwestern Mediterranean Sea. Oceanography 17: 23–31.
Balls, P.W. 1994. Nutrient inputs to estuaries from nine Scottish east coast rivers; influence of estuarine processes on inputs to the North Sea. Estuarine, Coastal and Shelf Science. 39 (4): 329–352. https://doi.org/10.1006/ecss.1994.1068.
Batker, D., I. De la Torre, R. Costanza, J.W. Day, P. Swedeen, R. Boumans, and K.J. Bagstad. 2014. The threats to the value of ecosystem goods and services of the Mississippi Delta. In Perspectives on the restoration of the Mississippi Delta: The once and future delta, 155–173.
Benitez-Nelson, C.R. 2000. The biogeochemical cycling of phosphorus in marine systems. Earth Science Reviews 51 (1-4): 109–135. https://doi.org/10.1016/S0012-8252(00)00018-0.
Berg, P., S. Rysgaard, and B. Thamdrup. 2003. Dynamic modeling of early diagenesis and nutrient cycling. A case study in an arctic sediment. American Journal of Science 303: 905–955.
Berner, R.A. 1970. Sedimentary pyrite formation. American Journal of Science 268 (1): 1–23.
Berner, R. A. 1980. Early diagenesis: a theoretical approach. Princeton: Princeton University Press. https://doi.org/10.1016/j.giq.2006.06.002, 23, 2, 330, 331 241 pp.
Berner, R.A. 1989. Biogeochemical cycles of carbon and sulfur and their effect on atmospheric oxygen over phanerozoic time. Global and Planetary Change 1 (1-2): 97–122. https://doi.org/10.1016/0921-8181(89)90018-0.
Bianchi, T.S., and M.A. Allison. 2009. Large-river delta-front estuaries as natural “recorders” of global environmental change. Proceedings of the National Academy of Sciences 106 (20): 8085–8092. https://doi.org/10.1073/pnas.0812878106.
Boudreau, Bernard P. 1996. A method-of-line code for carbon and nutrient daigenesis in aquatic sediments. Computer & Geosciences 22 (5): 479–496.
Boudreau, B.P. 1997. Diagenetic models and their implementation. Modelling transport and reactions in aquatic sediments. Springer. https://doi.org/10.1016/S0264-8172(98)80005-6.
Burdige, D.J. 2007. Preservation of organic matter in marine sediments: Controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chemical Reviews 107 (2): 467–485. https://doi.org/10.1021/cr050347q.
Cade-Menun, B.J., S. Duhamel, R.J. Dodd, C. Lønborg, C.T. Parsons, and W.D. Taylor. 2019. Editorial: Phosphorus along the soil-freshwater-ocean continuum. Frontiers in Marine Science 10 (6): 1–3. https://doi.org/10.1071/en13151.
Cai, W.J., and F.L. Sayles. 1996. Oxygen penetration depths and fluxes in marine sediments. Marine Chemistry 52 (2): 123–131. https://doi.org/10.1016/0304-4203(95)00081-X.
Canavan, R.W., and C.P. Slomp. 2006. Phosphorus cycling in the sediment of a coastal freshwater lake and response to salinization. In Biogeochemical cycling of nutrients and trace metals in the sediment of Haringvliet Lake: Response to salinization, 95–113.
Canavan, Richard W., Caroline P. Slomp, Parisa Jourabchi, Philippe Van Cappellen, Anniet M. Laverman, and Gerard A. van den Berg. 2006. Organic matter mineralization in sediment of a coastal freshwater lake and response to salinization. Geochimica et Cosmochimica Acta 70 (11): 2836–2855. https://doi.org/10.1016/j.gca.2006.03.012.
Canfield, D.E., E. Kristensen, and B. Thamdrup. 2005. The iron and manganese cycles. Advances in Marine Biology 48. https://doi.org/10.1016/S0065-2881(05)48008-6.
Cathalot, C. 2008. Devenir et impact des apports fluviaux sur les marges continentales: importance biogéochimique et environnementale du recyclage dans les sédiments du prodelta du Rhône. Ph.D. thesis, Université Pierre et Marie Curie, Paris, France.
Cathalot, C., C. Rabouille, L. Pastor, B. Deflandre, E. Viollier, R. Buscail, A. Grémare, C. Treignier, and A. Pruski. 2010. Temporal variability of carbon recycling in coastal sediments influenced by rivers: Assessing the impact of flood inputs in the Rhone River prodelta. Biogeosciences 7 (3): 1187–1205.
Cathalot, C., C. Rabouille, N. Tisnérat-Laborde, F. Toussaint, P. Kerhervé, R. Buscail, K. Loftis, M.Y. Sun, J. Tronczynski, S. Azoury, B. Lansard, C. Treignier, L. Pastor, and T. Tesi. 2013. The fate of river organic carbon in coastal areas: A study in the Rhône River delta using multiple isotopic (δ13C, δ14C) and organic tracers. Geochimica et Cosmochimica Acta 118: 33–55. https://doi.org/10.1016/j.gca.2013.05.001.
Charmasson, S., O. Radakovitch, M. Arnaud, P. Bouisset, and A.S. Pruchon. 1998. Long-core profiles of 137Cs, 134Cs, 60Co and 210Pb in sediment near the Rhône River (northwestern Mediterranean Sea). Estuaries 21 (3): 367–378. https://doi.org/10.1007/BF02690450.
Clayton, T.D., and R.H. Byrne. 1993. Spectrophotometric seawater pH measurements: Total hydrogen results. Deep-Sea Research 40 (10): 2115–2129. https://doi.org/10.1016/0967-0637(93)90048-8.
Cloern, J.E. 2001. Our evolving conceptual model of the coastal eutrophication problem. Marine Ecology Progress Series 210: 223–253.
Dedieu, K., C. Rabouille, F. Gilbert, K. Soetaert, E. Metzger, C. Simonucci, D. Jézéquel, F. Prévot, P. Anschutz, S. Hulth, S. Ogier, and V. Mesnage. 2007. Coupling of carbon, nitrogen and oxygen cycles in sediments from a Mediterranean lagoon: A seasonal perspective. Marine Ecology Progress Series 346: 45–59. https://doi.org/10.3354/meps07031.
Delaney, M.L. 1998. Phosphorus accumulation in marine sediments and the oceanic phosphorus cycle. Global Biogeochemical Cycles 12 (4): 563–572.
Diaz, R.J., and R. Rosenberg. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321 (5891): 926–929. https://doi.org/10.1126/science.1156401.
Diaz, F., P. Raimbault, B. Boudjellal, N. Garcia, and T. Moutin. 2001. Early spring phosphorus limitation of primary productivity in a NW Mediterranean coastal zone (Gulf of Lions) 211: 51–62.
Dickson, A G, C. L. Sabine, and J. R. Christian. 2007. Guide to best practices for ocean CO2 measurements. In PICES Special Publication, 3:191 pp.
Dijkstra, N., C. P. Slomp, and T. Behrends. 2016. Vivianite is a key sink for phosphorus in sediments of the Landsort Deep, an intermittently anoxic deep basin in the Baltic Sea. Chemical Geology 438. Elsevier B.V.: 58–72. https://doi.org/10.1016/j.chemgeo.2016.05.025.
Downing, J.A., M. McClain, R. Twilley, J.M. Melack, J. Elser, N.N. Rabalais, W.M. Lewis, et al. 1999. The impact of accelerating land-use change on the N-cycle of tropical aquatic ecosystems: Current conditions and projected changes. Biogeochemistry 46 (1-3): 109–148. https://doi.org/10.1007/BF01007576.
Dugdale, R.C., and F.P. Wilkerson. 1988. Nutrient sources and primary production in the eastern Mediterranean. Oceanologica Acta: 179–184.
Dumoulin, J.P., L. Pozzato, J. Rassman, F. Toussaint, M. Fontugne, N. Tisnérat-Laborde, L. Beck, I. Caffy, E. Delqué-Količ, C. Moreau, and C. Rabouille. 2018. Isotopic signature (δ13C, δ14C) of DIC in sediment pore waters: An example from the Rhone River delta. Radiocarbon 60 (5): 1465–1481. https://doi.org/10.1017/RDC.2018.111.
Egger, M., T. Jilbert, T. Behrends, C. Rivard, and C. P. Slomp. 2015. Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments. Geochimica et Cosmochimica Acta 169. Elsevier Ltd: 217–235. https://doi.org/10.1016/j.gca.2015.09.012.
Emerson, S., and G. Widmer. 1978. Early diagenesis in anaerobic lake sediments—II. Thermodynamic and kinetic factors controlling the formation of iron phosphate. Geochimica et Cosmochimica Acta 42 (9): 1307–1316. https://doi.org/10.1016/0016-7037(78)90035-2.
Emerson, S., J. Hedges, H.D. Holland, and K.K. Turekian. 2004. Sediment diagenesis and benthic flux. The Oceans and Marine Geochemistry 6: 293–319.
Emsley, J. 1980. The phosphorus cycle. In The handbook of environmental chemistry, 20:147–167. https://doi.org/10.1016/0143-1471(82)90111-8, 2.
Estournel, C., V. Kondrachoff, P. Marsaleix, and R. Vehil. 1997. The plume of the Rhone: Numerical simulation and remote sensing. Continental Shelf Research 17 (8): 899–924. https://doi.org/10.1016/S0278-4343(96)00064-7.
Eyrolle, F., O. Radakovitch, P. Raimbault, S. Charmasson, C. Antonelli, E. Ferrand, D. Aubert, G. Raccasi, S. Jacquet, and R. Gurriaran. 2012. Consequences of hydrological events on the delivery of suspended sediment and associated radionuclides from the Rhône River to the Mediterranean Sea. Journal of Soils and Sediments 12 (9): 1479–1495. https://doi.org/10.1007/s11368-012-0575-0.
Ferrón, S., T. Ortega, and J.M. Forja. 2009. Benthic respiration on the northeastern shelf of the Gulf of Cádiz (SW Iberian Peninsula). Marine Ecology Progress Series 392: 69–80. https://doi.org/10.3354/meps08240.
Filippelli, G.M., and M.L. Delaney. 1996. Phosphorus geochemistry of equatorial Pacific sediments. Geochimica et Cosmochimica Acta 60 (9): 1479–1495. https://doi.org/10.1016/0016-7037(96)00042-7.
Fox, L.E. 1990. Geochemistry of dissolved phosphate in the Sepik River and Estuary, Papua, New Guinea. Geochimica et Cosmochimica Acta 54 (4): 1019–1024. https://doi.org/10.1016/0016-7037(90)90435-N.
Froelich, P.N. 1988. Kinetic control of dissolved phosphate in natural rivers and estuaries: A primer on the phosphate buffer mechanism. Limnology and Oceanography 33: 649–668.
Gatti, J., A. Petrenko, J.L. Devenon, Y. Leredde, and C. Ulses. 2006. The Rhone river dilution zone present in the northeastern shelf of the Gulf of Lion in December 2003. Continental Shelf Research 26 (15): 1794–1805. https://doi.org/10.1016/j.csr.2006.05.012.
Got, H., and J.C. Aloisi. 1990. The Holocene sedimentation on the Gulf of Lions margin: A quantitative approach. Continental Shelf Research 10 (9-11): 841–855. https://doi.org/10.1016/0278-4343(90)90062-Q.
Grasshof, K., M. Ehrhardt, and K. Kremling. 1983. Methods of seawater analysis, Third, Completely Revised and Extended Edition. Verlag Chemie GmbH. https://doi.org/10.1016/0304-4203(78)90045-2, 7, 1, 86, 87.
Gypens, Nathalie, Christiane Lancelot, and Karline Soetaert. 2008. Simple parameterisations for describing N and P diagenetic processes: Application in the North Sea. Progress in Oceanography 76 (1): 89–110. https://doi.org/10.1016/j.pocean.2007.10.003.
Hammond, D.E. 1999. Diagenesis of carbon and nutrients and benthic exchange in sediments of the northern Adriatic Sea. Marine Chemistry 66 (1-2): 53–79.
Hansen, H. P., and F. Koroleff. 1999. Determination of nutrients. In Methods of seawater analysis, Third Edition, Completely Revised and Extended Edition, 159–228. https://doi.org/10.1002/9783527613984.
Hofmann, A.F., K. Soetaert, and J.J. Middelburg. 2008. Present nitrogen and carbon dynamics in the Scheldt estuary using a novel 1-D model. Biogeosciences 5 (4): 981–1006. https://doi.org/10.5194/bg-5-981-2008.
Howarth, R.W., G. Billen, D. Swaney, A. Townsend, N. Jaworski, K. Lajtha, J.A. Downing, R. Elmgren, N. Caraco, T. Jordan, F. Berendse, J. Freney, V. Kudeyarov, P. Murdoch, and Z. Zhao-Liang. 1996. Regional nitrogen budgets and riverine N & P fluxes for the drainages to the North Atlantic Ocean: Natural and human influences. Biogeochemistry 35 (1): 75–139. https://doi.org/10.1590/2318-0331.011616030.
Ibañez, C., D. Pont, and N. Prat. 1997. Characterization of the Ebre and Rhone estuaries: A basis for defining and classifying salt-wedge estuaries. Limnology and Oceanography 42 (1): 89–101. https://doi.org/10.4319/lo.1997.42.1.0089.
Jackson, J.B.C., M.X. Kirby, W.H. Berger, K.A. Bjorndal, L.W. Botsford, B.J. Bourque, R.H. Bradbury, et al. 2001. Historical overfishing and the recent collapse of coastal ecosystems. Science 293 (5530): 629–637. https://doi.org/10.1126/science.1059199.
Joshi, S.R., R.K. Kukkadapu, D.J. Burdige, M.E. Bowden, D.L. Sparks, and D.P. Jaisi. 2015. Organic matter remineralization predominates phosphorus cycling in the mid-bay sediments in the Chesapeake Bay. Environmental Science and Technology 49 (10): 5887–5896. https://doi.org/10.1021/es5059617.
Khalil, K, A M Laverman, M Raimonet, and C Rabouille. 2018. Importance of nitrate reduction in benthic carbon mineralization in two eutrophic estuaries: Modeling, observations and laboratory experiments. Marine Chemistry 199. Elsevier: 24–36. https://doi.org/10.1016/j.marchem.2018.01.004.
Kraal, P., E. D. Burton, A. L. Rose, B. D. Kocar, R. S. Lockhart, K. Grice, R. T. Bush, E. Tan, and S. M. Webb. 2015. Sedimentary iron-phosphorus cycling under contrasting redox conditions in a eutrophic estuary. Chemical Geology 392. Elsevier B.V.: 19–31. https://doi.org/10.1016/j.chemgeo.2014.11.006.
Krom, M.D., and R.A. Berner. 1981. The diagenesis of phosphorus in a nearshore marine sediment. Geochimica et Cosmochimica Acta 45 (2): 207–216. https://doi.org/10.1016/0016-7037(81)90164-2.
Krom, M.D., N. Kress, S. Brenner, and L.I. Gordon. 1991. Phosphorus limitation of primary productivity in the eastern Mediterranean Sea. Limnology and Oceanography 36 (3): 424–432. https://doi.org/10.4319/lo.1991.36.3.0424.
Lansard, B., S. Charmasson, C. Gascó, M.P. Antón, C. Grenz, and M. Arnaud. 2007. Spatial and temporal variations of plutonium isotopes (238Pu and 239,240Pu) in sediments off the Rhone River mouth (NW Mediterranean). Science of the Total Environment 376 (1-3): 215–227. https://doi.org/10.1016/j.scitotenv.2007.01.069.
Lansard, B., C. Rabouille, L. Denis, and C. Grenz. 2008. In situ oxygen uptake rates by coastal sediments under the influence of the Rhône River (NW Mediterranean Sea). Continental Shelf Research 28 (12): 1501–1510. https://doi.org/10.1016/j.csr.2007.10.010.
Lansard, B., C. Rabouille, L. Denis, and C. Grenz. 2009. Benthic remineralization at the land-ocean interface: A case study of the Rhône River (NW Mediterranean Sea). Estuarine, Coastal and Shelf Science 81. Elsevier Ltd: 544–554. https://doi.org/10.1016/j.ecss.2008.11.025.
Lebo, M.E. 1991. Particle-bound phosphorus along an urbanized coastal plain estuary. Marine Chemistry 34 (3-4): 225–246. https://doi.org/10.1016/0304-4203(91)90005-H.
Lehtoranta, J., O. P. Savchuk, J. Elken, K. Dahlbo, H. Kuosa, M. Raateoja, P. Kauppila, A. Räike, and H. Pitkänen. 2017. Atmospheric forcing controlling inter-annual nutrient dynamics in the open Gulf of Finland. Journal of Marine Systems 171. Elsevier B.V: 4–20. https://doi.org/10.1016/j.jmarsys.2017.02.001.
Lenstra, W.K., M. Egger, N.A.G.M. van Helmond, E. Kritzberg, D.J. Conley, and C.P. Slomp. 2018. Variations in river input of iron impact sedimentary phosphorus burial in an oligotrophic Baltic Sea estuary. Biogeosciences Discussions 15 (10): 6979–6996. https://doi.org/10.1021/pr100554m.
Lochet, F., and M. Leveau. 1990. Transfers between a eutrophic ecosystem, the river Rh8ne, and an oligotrophic ecosystem, the North-Western Mediterranean Sea. Hydrobiologia 207 (1): 95–103.
Ludwig, W., and J.L. Probst. 1998. River sediment discharge to the oceans: Present-day controls and global budgets. American Journal of Science 298 (4): 265–295. https://doi.org/10.2475/ajs.298.4.265.
Ludwig, W., E. Dumont, M. Meybeck, and S. Heussner. 2009. River discharges of water and nutrients to the Mediterranean and Black Sea: Major drivers for ecosystem changes during past and future decades? Progress in Oceanography 80. Elsevier Ltd: 199–217. https://doi.org/10.1016/j.pocean.2009.02.001, 80, 3-4.
Malagó, A., F. Bouraoui, B. Grizzetti, and A. De Roo. 2019. Modelling nutrient fluxes into the Mediterranean Sea. Journal of Hydrology: Regional Studies 22. Elsevier: 100592. https://doi.org/10.1016/j.ejrh.2019.01.004.
McKee, B.A., R.C. Aller, M.A. Allison, T.S. Bianchi, and G.C. Kineke. 2004. Transport and transformation of dissolved and particulate materials on continental margins influenced by major rivers: Benthic boundary layer and seabed processes. Continental Shelf Research 24 (7-8): 899–926. https://doi.org/10.1016/j.csr.2004.02.009.
Meybeck, M. 1982. Carbon, nitrogen, and phosphorus transport by world rivers. American Journal of Science 282 (4): 401–450.
Milliman, J.D., and R.H. Meade. 1983. World-wide delivery of river sediment to the oceans. The Journal of Geology 91 (1): 1–21.
Miralles, J., O. Radakovitch, and J.C. Aloisi. 2005. 210Pb sedimentation rates from the Northwestern Mediterranean margin. Marine Geology 216 (3): 155–167. https://doi.org/10.1016/j.margeo.2005.02.020.
Morse, J.W., and P.M. Eldridge. 2007. A non-steady state diagenetic model for changes in sediment biogeochemistry in response to seasonally hypoxic/anoxic conditions in the “dead zone” of the Louisiana shelf. Marine Chemistry 106 (1-2): 239–255. https://doi.org/10.1016/j.marchem.2006.02.003.
Moutin, T., and P. Raimbault. 2002. Primary production, carbon export and nutrients availability in western and eastern Mediterranean Sea in early summer 1996 (MINOS cruise). Journal of Marine Systems 33–34: 273–288.
Moutin, T., P. Raimbault, H.L. Golterman, and B. Coste. 1998. The input of nutrients by the Rhône river into the Mediterranean Sea: Recent observations and comparison with earlier data. Hydrobiologia 373 (374): 237–246 10.1023/A.
Moutin, T., F. Van Wambeke, and L. Prieur. 2012. Introduction to the biogeochemistry from the oligotrophic to the ultraoligotrophic Mediterranean (BOUM) experiment. Biogeosciences 9 (10): 3817–3825. https://doi.org/10.5194/bg-9-3817-2012.
Muller-Karger, F.E., R. Varela, R. Thunell, R. Luerssen, C. Hu, and J.J. Walsh. 2005. The importance of continental margins in the global carbon cycle. Geophysical Research Letters 32 (1): 1–4. https://doi.org/10.1029/2004GL021346.
Murphy, J., and J.P. Riley. 1962. A modified single solution method for determination of phosphate in natural waters. Analytica Chimica Acta 27: 31–36. https://doi.org/10.1057/9781137461131.
Naudin, J.J., G. Cauwet, M.J. Chrétiennot-Dinet, B. Deniaux, J.L. Devenon, and H. Pauc. 1997. River discharge and wind influence upon particulate transfer at the land-ocean interaction: Case study of the Rhone River plume. Estuarine, Coastal and Shelf Science 45 (3): 303–316. https://doi.org/10.1006/ecss.1996.0190.
Nixon, S.W. 1995. Coastal marine eutrophication: A definition, social causes, and future concerns. Ophelia 41 (1): 199–219. https://doi.org/10.1080/00785236.1995.10422044.
Ollivier, P., B. Hamelin, and O. Radakovitch. 2010. Seasonal variations of physical and chemical erosion: A three-year survey of the Rhone River (France). Geochimica et Cosmochimica Acta 74. Elsevier Ltd: 907–927. https://doi.org/10.1016/j.gca.2009.10.037.
Pastor, L., C. Cathalot, B. Deflandre, E. Viollier, K. Soetaert, F.J.R. Meysman, C. Ulses, E. Metzger, and C. Rabouille. 2011. Modeling biogeochemical processes in sediments from the Rhǒne River prodelta area (NW Mediterranean Sea). Biogeosciences 8 (5): 1351–1366. https://doi.org/10.5194/bg-8-1351-2011.
Pont, D., J.P. Simonnet, and A.V. Walter. 2002. Medium-term changes in suspended sediment delivery to the ocean: Consequences of catchment heterogeneity and river management (Rhône River, France). Estuarine, Coastal and Shelf Science 54 (1): 1–18. https://doi.org/10.1006/ecss.2001.0829.
Powley, H.R., M.D. Krom, and P. Van Cappellen. 2017. Understanding the unique biogeochemistry of the Mediterranean Sea: Insights from a coupled phosphorus and nitrogen model. Global Biogeochemical Cycles 31 (6): 1010–1031. https://doi.org/10.1002/2017GB005648.
Pozzato, L., J. Rassmann, B. Lansard, J-P. Dumoulin, P. van Breugel, C. Rabouille. 2018. Origin of remineralized organic matter in sediments from the Rhone River prodelta (NW Mediterranean) traced by Δ14C and δ13C signatures of pore water DIC. Progr. Oceanogr., 163: 112–122.
Provansal, M., O. Radakovitch, F. Sabatier, and A. Clémens. 2012. Le Rhône aval en 21 questions. Villeurbanne: Rapport ZABR, Graie Editors.
Psenner, R., B. Bostrom, M. Dinka, K. Pettersson, R. Pucsko, and M. Sager. 1988. Fractionation of phosphorus in suspended matter and sediment. Ergebnisse der Limnologie 22: 219–228.
Rabouille, C., and J.F. Gaillard. 1991. Towards the EDGE: Early diagenetic global explanation. A model depicting the early diagenesis of organic matter, O2, NO3, Mn, and PO4. Geochimica et Cosmochimica Acta 55 (9): 2511–2525. https://doi.org/10.1016/0016-7037(91)90369-G.
Rabouille, C., L. Denis, K. Dedieu, G. Stora, B. Lansard, and C. Grenz. 2003. Oxygen demand in coastal marine sediments: Comparing in situ microelectrodes and laboratory core incubations. Journal of Experimental Marine Biology and Ecology 285–286: 49–69. https://doi.org/10.1016/S0022-0981(02)00519-1.
Rabouille, C., D.J. Conley, M.H. Dai, W.J. Cai, C.T.A. Chen, B. Lansard, R. Green, K. Yin, P.J. Harrison, M. Dagg, and B. McKee. 2008. Comparison of hypoxia among four river-dominated ocean margins: The Changjiang (Yangtze), Mississippi, Pearl, and Rhône rivers. Continental Shelf Research 28 (12): 1527–1537. https://doi.org/10.1016/j.csr.2008.01.020.
Radakovitch, O., S. Charmasson, M. Arnaud, and P. Bouisset. 1999. 210Pb and caesium accumulation in the Rhône Delta. Estuarine, Coastal and Shelf Science 48 (1): 77–92.
Rassmann, J., B. Lansard, L. Pozzato, and C. Rabouille. 2016. Carbonate chemistry in sediment porewaters of the Rhône River delta driven by early diagenesis (northwestern Mediterranean). Biogeosciences 13 (18): 5379–5394. https://doi.org/10.5194/bg-13-5379-2016.
Rassmann, J., E.M. Eitel, C. Cathalot, C. Brandily, B. Lansard, M. Taillefert, and C. Rabouille. 2020. Benthic alkalinity and DIC fluxes in the Rhône River prodelta generated by decoupled aerobic and anaerobic processes. Biogeosciences Discussions 17 (1): 13–33.
Ratmaya, W. 2018. Rôle des sédiments dans le cycle des nutriments et impacts sur l’eutrophisation des écosystèmes côtiers. Ph.D. thesis, Univ. Bretagne Occidentale, Brest, France.
Revsbech, N.P. 1989. An oxygen microsensor with a guard cathode. Limnology and Oceanography 34 (2): 474–478. https://doi.org/10.4319/lo.1989.34.2.0474.
Ruttenberg, K.C. 1992. Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnology and Oceanography 37 (7): 1460–1482. https://doi.org/10.4319/lo.1992.37.7.1460.
Ruttenberg, K.C. 2014. 10.13 – the global phosphorus cycle. Treatise on Geochemistry: 499–558. https://doi.org/10.1016/B978-0-08-095975-7.00813-5.
Ruttenberg, K.C., and R.A. Berner. 1993. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochimica et Cosmochimica Acta 57 (5): 991–1007. https://doi.org/10.1016/0016-7037(93)90035-U.
Sabatier, F., G. Maillet, M. Provansal, T.J. Fleury, S. Suanez, and C. Vella. 2006. Sediment budget of the Rhône delta shoreface since the middle of the 19th century. Marine Geology 234 (1-4): 143–157. https://doi.org/10.1016/j.margeo.2006.09.022.
Schnetger, B., and C. Lehners. 2014. Determination of nitrate plus nitrite in small volume marine water samples using vanadium(III) chloride as a reduction agent. Marine Chemistry 160. Elsevier B.V.: 91–98. https://doi.org/10.1016/j.marchem.2014.01.010.
Seeberg-Elverfeldt, J., M. Schlüter, T. Feseker, and M. Kölling. 2005. Rhizon sampling of porewaters near the sediment-water interface of aquatic systems. Limnology and Oceanography: Methods 3 (8): 361–371. https://doi.org/10.4319/lom.2005.3.361.
Slomp, C.P., E.H.G. Epping, W. Helder, and W.V. Raaphorst. 1996. A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments. Journal of Marine Research 54 (6): 1179–1205. https://doi.org/10.1357/0022240963213745.
Slomp, C.P., J.F.P. Malschaert, and W.V. Raaphorst. 1998. The role of adsorption in sediment-water exchange of phosphate in North Sea continental margin sediments. Limnology and Oceanography 43 (5): 832–846.
Slomp, C.P., J. Thomson, and G.J. De Lange. 2004. Controls on phosphorus regeneration and burial during formation of eastern Mediterranean sapropels. Marine Geology 203 (1-2): 141–159. https://doi.org/10.1016/S0025-3227(03)00335-9.
Smith, V.H., G.D. Tilman, and J.C. Nekola. 1999. Eutrophication: Impacts of excess nutrient inputs on freshwater, marine, and terrestrial ecosystems. Environmental Pollution 100 (1-3): 179–196.
Soetaert, K., and F. Meysman. 2012. Reactive transport in aquatic ecosystems: Rapid model prototyping in the open source software R. Environmental Modelling and Software 32. Elsevier Ltd: 49–60. https://doi.org/10.1016/j.envsoft.2011.08.011.
Soetaert, K., P.M.J. Herman, and J.J. Middelburg. 1996. A model of early diagenetic processes from the shelf to abyssal depths. Geochimica et Cosmochimica Acta 60 (6): 1019–1040. https://doi.org/10.1016/0016-7037(96)00013-0.
Stookey, L.L. 1970. Ferrozine-A new spectrophotometric reagent for iron. Analytical Chemistry 42 (7): 779–781.
Sundby, B., C. Gobeil, N. Silverberg, and A. Mucci. 1992. The phosphorus cycle in coastal marine sediments. Limnology and Oceanography 37 (6): 1129–1145. https://doi.org/10.4319/lo.1992.37.6.1129.
Syers, J.K., R.F. Harris, and D.E. Armstrong. 1973. Phosphate chemistry in lake sediments. Journal of Environment Quality 2 (1): 1–14. https://doi.org/10.2134/jeq1973.00472425000200010001x.
Syvitski, J.P.M., A.J. Kettner, I. Overeem, E.W.H. Hutton, M.T. Hannon, G.R. Brakenridge, J.W. Day, et al. 2009. Sinking deltas. Nature Geoscience 2 (10): 681–686.
Thamdrup, B., H. Fossing, and B.B. Jorgensen. 1994. Manganese, iron, and sulfur cycling in a coastal marine sediment, Aarhus Bay, Denmark. Geochimica et Cosmochimica Acta 58 (23): 5115–5129.
Tromp, T.K., P. Van Cappellen, and R.M. Key. 1995. A global model for the early diagenesis of organic carbon and organic phosphorus in marine sediments. Geochimica et Cosmochimica Acta 59 (7): 1259–1284. https://doi.org/10.1016/0016-7037(95)00042-X.
Tyrrell, T. 1999. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400 (6744): 525–531.
Van Den Broeck, N., and T. Moutin. 2002. Phosphate in the sediments of the Gulf of Lions (NW Mediterranean Sea), relationship with input by the river Rhone. Hydrobiologia 472 (1/3): 85–94. https://doi.org/10.1023/A:1016308931115.
Vitousek, P.M., H.A. Mooney, J. Lubchenco, and J.M. Melillo. 1997. Human domination of Earth’s ecosystems. Science 277 (5325): 494–499. https://doi.org/10.1126/science.277.5325.494.
Wang, Y.F., and P. Van Cappellen. 1996. A multicomponent reactive transport model of early diagenesis: Application to redox cycling in coastal marine sediments. Geochimica et Cosmochimica Acta 60 (16): 2993–3014. https://doi.org/10.1016/0016-7037(96)00140-8.
Wijsman, J.W.M., P.M.J. Herman, J.J. Middelburg, and K. Soetaert. 2002. A model for early diagenetic processes in sediments of the continental shelf of the Black Sea. Estuarine, Coastal and Shelf Science 54 (3): 403–421. https://doi.org/10.1006/ecss.2000.0655.
Wilkinson, G.M. 2017. Eutrophication of freshwater and coastal ecosystems. Encyclopedia of sustainable technologies. Vol. 4. Elsevier. https://doi.org/10.1016/B978-0-12-409548-9.10160-5.
Acknowledgements
We would like to thank the captain and the crew members of N.O. Tethys II (INSU/CNRS) for their assistance during sampling at sea. We are grateful to Laurie Brethous and Anouk Villedieu who provided help at sea during sampling and in the laboratory for measurements. Florian Caradec and Emilie Rabiller are acknowledged for their help during P speciation measurements at IFREMER’s laboratory. We thank the two anonymous reviewers whose comments greatly improved the quality of this manuscript. This is LSCE publication number 7409.
Funding
This work was supported by the INSU/EC2CO-MissRhoDia project, the French State program “Investissement d’avenir” run by the National Research Agency (AMORAD project ANR-11-RSNR-0002) and the CEDoc cooperation research project between Morocco and France.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Lijun Hou
Rights and permissions
About this article

Cite this article
Ait Ballagh, F.E., Rabouille, C., Andrieux-Loyer, F. et al. Spatial Variability of Organic Matter and Phosphorus Cycling in Rhône River Prodelta Sediments (NW Mediterranean Sea, France): a Model-Data Approach. Estuaries and Coasts 44, 1765–1789 (2021). https://doi.org/10.1007/s12237-020-00889-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-020-00889-9