
Abstract
Three sediment stations in Himmerfjärden estuary (Baltic Sea, Sweden) were sampled in May 2009 and June 2010 to test how low salinity (5–7 ‰), high primary productivity partially induced by nutrient input from an upstream waste water treatment plant, and high overall sedimentation rates impact the sedimentary cycling of methane and sulfur. Rates of sediment accumulation determined using 210Pbexcess and 137Cs were very high (0.65–0.95 cm year−1), as were the corresponding rates of organic matter accumulation (8.9–9.5 mol C m−2 year−1) at all three sites. Dissolved sulfate penetrated <20 cm below the sediment surface. Although measured rates of bicarbonate methanogenesis integrated over 1 m depth were low (0.96–1.09 mol m−2 year−1), methane concentrations increased to >2 mmol L−1 below the sulfate–methane transition. A steep gradient of methane through the entire sulfate zone led to upward (diffusive and bio-irrigative) fluxes of 0.32 to 0.78 mol m−2 year−1 methane to the sediment–water interface. Areal rates of sulfate reduction (1.46–1.92 mol m−2 year−1) integrated over the upper 0–14 cm of sediment appeared to be limited by the restricted diffusive supply of sulfate, low bio-irrigation (α = 2.8–3.1 year−1), and limited residence time of the sedimentary organic carbon in the sulfate zone. A large fraction of reduced sulfur as pyrite and organic-bound sulfur was buried and thus escaped reoxidation in the surface sediment. The presence of ferrous iron in the pore water (with concentrations up to 110 μM) suggests that iron reduction plays an important role in surface sediments, as well as in sediment layers deep below the sulfate–methane transition. We conclude that high rates of sediment accumulation and shallow sulfate penetration are the master variables for biogeochemistry of methane and sulfur cycling; in particular, they may significantly allow for release of methane into the water column in the Himmerfjärden estuary.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Biogenic methane produced in marine sediments is one of the largest reservoirs of methane on Earth (Claypool and Kvenvolden 1983). In most marine systems, very little of this methane is released into the seawater and atmosphere because it is efficiently oxidized by sulfate reduction coupled to the anaerobic oxidation of methane (AOM) within the sulfate–methane transition (SMT) (Reeburgh 1975; Boetius et al. 2000; Orphan et al. 2001; Treude et al. 2003). The amount of methane that escapes from continental margin sediments through the water column and into the atmosphere accounts for only 2 % of global methane emission (Judd et al. 2002).
The largest part of the marine methane emission, about 75 %, is probably released from near-shore coastal environments (Bange et al. 1994). This is because coastal regions (estuaries, bays, and other shallow areas) are often characterized by high rates of organic matter deposition due to large amounts of terrestrial and riverine runoff, high primary production in the water column, and the discharge of anthropogenic waste, e.g., from sewage treatment plants (Meybeck et al. 1989; Smith et al. 2010; Borges and Abril 2011). These factors support high rates of carbon mineralization in the sediment by oxygen respiration, denitrification, metal oxide reduction, sulfate reduction, and, ultimately, methanogenesis (Borges and Abril 2011). The depletion of sulfate allows methanogenesis to occur at shallow depths. In addition to a high organic carbon load that drives high rates of organic carbon mineralization, rates of sulfate reduction and methanogenesis depend on the season, the sediment temperature, the salinity, and the sulfate concentrations in the water column (Martens and Klump 1980a; Kipphut and Martens 1982; Heyer and Berger 2000; Valentine 2002). In coastal systems with low salinity, sulfate depletion often occurs in the topmost tens of centimeters; methane is less efficiently oxidized, and a significant amount of methane can escape as bubbles (Chanton et al. 1989; Heyer and Berger 2000).
The purpose of this study is to understand how low salinity, high organic matter input, and high overall sedimentation rates impact carbon mineralization rates and the turnover of methane and sulfur in eutrophic, littoral Baltic Sea sediments. Himmerfjärden, a large estuarine system on the Swedish Baltic coast connecting Lake Mälaren with the central Baltic Sea is ideal for this study due to its point–source anthropogenic loading, high sediment accumulation rates, and low salinity.
Study Site
The Baltic Sea is the largest brackish water body in the world. Salinity decreases from 25 ‰ in the Danish Straits to 2 ‰ in the Gulf of Bothnia. Over the past 50 years, the Baltic Sea has experienced eutrophication with increased primary production supported by increased discharge of inorganic and organic nutrients (Bartnicki and Valiyaveetil 2008; Larsson et al. 1985; Rosenberg et al. 1990; Stigebrandt 1991), which has led to an expansion of coastal and open-water anoxia (Conley et al. 2009, 2011).
Himmerfjärden covers an area of 174 km2 (Engqvist and Omstedt 1992). The estuary has a mean water depth of 18 m and a maximum depth of 52 m (Fig. 1). It consists of multiple silled sub-basins with limited water exchange (Engqvist and Omstedt 1992). The system receives freshwater discharge from surface runoff, Lake Mälaren, and a local sewage treatment plant, which treats the sewage of approximately 250,000 people within the greater Stockholm metropolitan region. This has led to eutrophication of the estuary for the past 60 years (Savage et al. 2010). The total nitrogen discharge from the sewage treatment plant has been shown to have a direct effect on plankton productivity in the estuary (Bianchi et al. 2002; Larsson et al. 1985).

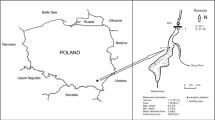
Location of stations H5, H3, and H2 in Himmerfjärden estuary, Sweden
Three stations, H2, H3, and H5, were selected for this study (Fig. 1). They belong to a suite of long-term monitoring stations (http://www2.ecology.su.se/dbHFJ/index.htm) and are located in water depths of 25–50 m. Salinity in the bottom water at the stations varies between 5.5 and 6.7 ‰, bottom water oxygen concentrations are in the range of 0.06–0.44 mM, and surface chlorophyll α concentrations vary between 1 and 12.5 mg m−3 (Table 1). Station H5 is closest to the waste water treatment plant and is characterized by the lowest salinity (6–6.7 ‰) and highest chlorophyll α content (1–12.5 mg m−3) (Table 1). Oxygen concentrations at 25 m depth vary between 0.12 mM in the summer to 0.41 mM in the winter when the stratified bottom waters mix (Table 1). Station H3 is in 50 m depth and generally has oxygenated bottom water year round. Station H2 is furthest downstream and is the least affected by the waste water treatment plant as indicated by the lowest chlorophyll α content (1–6.5 mg m−3). Bottom water oxygen concentrations at station H2 range from 0.06 to 0.44 mM year round (Table 1).
The sediment at the three stations consists of organic-rich clays with a one- to several-centimeter-thick brown iron oxyhydroxide-rich layer at the top. The Holocene organic-rich mud overlays a compact layer of clay below 120 cm below sea floor (cmbsf ) at station H2 that probably represents the upper boundary of the brackish glacial lake deposits (8,500–7,500 BP) (Heinsalu et al. 2000). Additionally, at station H5, laminated intervals occur in the topmost 20 cm, suggesting historical anoxic bottom water conditions.
Materials and Methods
Sediment Sampling
Sediments from the three stations were sampled during two campaigns with the research vessel R/V Limanda in May 2009 and June 2010. The sediments were collected with a multicorer and a small gravity corer (Rumohrlot corer) with lengths of approximately 40 and 140 cm, respectively. Methane samples were immediately collected and fixed on board as described below. Otherwise, the cores were capped with rubber stoppers, transported to the Marine Research Center of Stockholm University on Askö Island and kept cold (+4 °C) until utilized for experiments and sampling.
Subsamples for methane concentration measurements were taken using 3-cm3 cutoff syringes that were immediately inserted into predrilled holes in gravity cores on board the R/V Limanda. The sediments were transferred to serum 20-ml vials containing 5 mL of 5 M NaCl. Pore water was directly extracted using Rhizons connected to 10-mL syringes (Elverfeldt et al. 2005) at a resolution of 1 cm for the Multicorer and 5 cm for the Rumohrlot cores. The total volume of pore water extracted was 8 to 10 mL, of which the first 1 mL was discarded to clean the syringe and remove air trapped in the syringe, and the Rhizon. Sediment samples were collected for total carbon (TC), total inorganic carbon (TIC), total sulfur (TS), total nitrogen, reactive iron, density, and porosity. Samples for 210Pb and 137Cs analyses were retrieved from multicores by slicing the core at a resolution of 1 to 2 cm.
35S-sulfate reduction rates (SRR) were measured in intact subcores (28 mm in diameter) at a resolution of 1 to 2 cm. Samples for rate measurements of 14C-AOM and 14C-bicarbonate methanogenesis incubations were taken with 5-mL cutoff syringes, plugged with butyl rubber stoppers, and stored in N2-filled plastic bags before injection of the radiotracer.
Analytical Procedures
Methane Measurement
Methane concentration was measured in the headspace by gas chromatography with a flame ionization detector using a Shimadzu GC-8a gas chromatograph. Nitrogen was used as carrier gas at a flow rate of 15 mL min−1 at 40 °C. The methane concentration was calculated per volume sediment corrected for the sediment porosity according to the following equation:
where V head is the volume of the headspace in the sample vial (cubic centimeter), φ is the sediment porosity, A is the peak area of methane eluted at 0.8 min, α is the slope of the standard curve (parts per million volume basis), and V sed is the volume of the sediment sample (cubic centimeter). The molar volume of methane at 20 °C (24.1 L mol−1) was used to convert from partial volume CH4 to mole CH4.
Pore Water Analyses
The extracted pore water was subsampled by preserving 1 mL with 100 μL ZnCl2 (5 %) for hydrogen sulfide and sulfate analyses, and collecting 2 mL headspace-free pore water for dissolved inorganic carbon (DIC) and chloride measurements.
Sulfate (10- to 20-fold dilution) and chloride (200-fold dilution) samples were measured by ion chromatography (761 Compact IC, Ω Metrohm using 838 Advanced Sample Processor Ω Metrohm) with 3.2 mM Na2CO3 and 1 mM NaHCO3 as eluent. The detection limit of sulfate in pore waters was 50 μM. Sulfide was determined using the photometric methylene blue method after Cline (1969) (Shimadzu UV120 spectrophotometer, 2 μM limited detection).
Dissolved inorganic carbon concentrations were determined by flow injection analysis (Hall and Aller 1992). Due to the high sulfide concentrations in the samples, 100 μL of 0.5 M NaMoO4·2H2O solution was added to trap H2S (Lustwerk and Burdige 1995). The detection limit is 0.1 mM for these measurements.
Dissolved iron (Fe2+) in the pore water was measured using the Ferrozine method (Viollier et al. 2000) and determined on a Pharmacia LKB Ultraspec III spectrophotometer, 0.3 μM detection limit.
Solid-Phase Analyses
Total nitrogen, total sulfur, and organic carbon (Corg) concentrations in the sediment were determined with an elemental analyzer (GC–FISONS 1500) at 1,052 °C. Total inorganic carbon was measured on a CM 5012 CO2 Coulometer (UIC) after acidification with H3PO4. Organic carbon was calculated as the difference between TC and TIC. The C:N molar ratio was also calculated. Based on organic carbon profiles and sedimentation rates, organic carbon accumulation (J Corg) rates were calculated as:
where J Corg is the organic carbon accumulation rate, and Corg content, ω, and ρ are the organic carbon content (percent dry weight), sedimentation rate (centimeter per year), and density of dry bulk sediment (grams per cubic centimeter).
Sediments for porosity measurements were taken using 5-cm3 cutoff syringes. A 3-cm3 subsample of wet sediment was weighed, the density was determined from the wet mass per cubic centimeter, and the sediments were dried at 60 °C until they reached a constant mass. The difference between wet and dried mass was used to calculate the porosity.
For chlorin measurement, 10–20 mg of freeze-dried sediment was extracted three times in the dark with 5 mL 100 % acetone in an ice-bath according to Schubert et al. (2005). The extracted solution was immediately analyzed with a Hitachi F-2000 fluorometer at 428 nm. Chlorophyll α was used as calibration standard. The extracted solutions then were acidified and remeasured. The chlorin index (CI) was calculated based on the ratio of the fluorescence intensity of non-acidified to acidified extracts (Schubert et al. 2005).
Total reactive iron in the sediment was extracted by a two-step ascorbate–dithionite extraction under anoxic conditions according to März et al. (2008). The reactive ascorbate extractable iron (Feasc) and reactive dithionite extractable iron (Fedithio) extractions were measured for total dissolved iron (Fe2+ and Fe3+) by the atomic absorption spectroscopy, Thermo Scientific iCE 3000 series using the ASX-520 Autosampler. The extractions of Feasc and Fedithio represent of reactive amorphous iron and crystalline iron (oxyhydro)oxides, respectively (März et al. 2008).
Process Rate Measurements
Sulfate reduction rates were determined by injecting 35S-sulfate tracer (50 kBq) into the retrieved subcores followed by 6 to 8 h of incubation. The incubation was stopped by transferring the sediment into 50-mL plastic centrifuge tubes containing 20 mL zinc acetate (20 %, v/v). The total amount of 35S-labeled reduced inorganic sulfur was determined using the single-step cold distillation method of Kallmeyer et al. (2004) by counting on a Tricarb 2500 liquid scintillation counter. Sulfate reduction rates (nanomoles per cubic centimeter per day) were calculated using the following equation (Jørgensen 1978):
where {SO 2−4 } is the pore water sulfate concentration corrected for porosity (φ) (nanomoles per cubic centimeter of wet sediment), TRI35S and 35SO 2−4 are the radioactivities (becquerel) of sulfate and total reduced sulfur species, respectively, and t is the incubation time in days. The factor 1.06 is the estimated fractionation factor between 35S and the natural isotope 32S (Jørgensen and Fenchel 1974). SRR were determined in three parallel cores for all depth intervals, and the values reported are the median values of the triplicates.
Bicarbonate methanogenesis rates were measured by injecting 14C-HCO −3 tracer (20 kBq) into the sediment in the 5-cm3 cutoff syringes taken from the Rumohrlot cores and incubating for 16 h at in situ temperatures (the same temperature at the coring time, 4 °C). The incubations were stopped by transferring the sediment into 25 mL of 2.5 % NaOH in glass jars (50 mL). In the laboratory, the headspace gas was flushed by a carrier gas of 95 % N2:5 % O2 at 30 mL/min for 30 min through 850 °C copper oxide columns as catalyst to oxidize 14CH4 to 14CO2. The CO2 was trapped in a series of two scintillation vials containing 10 mL of Carbosorb solution (OptiPhase HiSafe-3 plus β-phenylethylamine in v/v ratio of 4:1). The radioactivity was measured on a liquid scintillation counter (Tricarb 2500). The methanogenesis rate (ME) was calculated using the following equation:
where {DIC} is the concentration of dissolved inorganic carbon per cubic centimeter sediment corrected for porosity ({DIC} = DIC × φ) in the pore water, 14CH4 and H14CO −3 are the activities (kilobecquerel) of labeled methane and bicarbonate, respectively, and t is the incubation time in days. The bicarbonate methanogenesis rates were measured on three parallel samples and the values presented here are the median rates of the triplicates.
Rates of anaerobic oxidation of methane were determined by injecting 14C-methane tracer (10 kBq) directly into 3 cm−3 sediment samples in cutoff syringes and incubating the sediment for 14 h at in situ temperature. After the incubation, the microbial activity was stopped by transferring the sediments to 50-mL glass tubes containing 20 mL NaOH (2.5 %). AOM rates (nanomoles per cubic centimeter per day) were calculated based on the ratio of 14C-bicarbonate and 14C-methane using the methane concentration in each sample (Treude et al. 2003) according to the following equation:
where {CH4} is the porosity-corrected concentration of methane at the beginning of the incubation. 14CO2 is the activity (becquerel) of carbon dioxide, 14CH4 is the activity of methane also trapped as 14CO2, and t is the incubation time (day). The rate of anaerobic oxidation of methane is calculated as nanomoles per cubic centimeter per day. Similar to the SRR, AOM rates were measured in three parallel cores and the values presented here are the median values.
210Pb and 137Cs Analyses and Calculation of Sedimentation Rates
Dry and ground sediment samples for radiochemical measurement (2l0Pbexcess and l37Cs activities) were sealed in polysulfone vials and equilibrated for at least 3 weeks. Activities of the radionuclides were determined using ultra-low level gamma spectroscopy on a closed-end coaxial well detector (Ge Coaxial Type N gamma detector) for 1 to 3 days. The total 2l0Pb radioactivity was determined directly by measuring the 210Pb at 46.5 KeV gamma peak and 224Ra that was indirectly determined by measuring the gamma activity of 2l4Pb (at 295 and 352 KeV) and 2l4Bi (609 KeV). The 210Pbexcess was determined by the total 210Pb minus the supported 210Pb that derives from 226Ra. Self-absorption corrections were made on each sample following the technique of Cutshall et al. (1983).
137Cs activities were determined by measurement of the 662 KeV gamma peak intensity. Elevated 137Cs is an artificial tracer (produced from nuclear bomb testing) introduced to the Baltic Sea environment in the l950s and in 1963. Additionally, the study area received a large amount of 137Cs as a result of the Chernobyl catastrophe in 1986.
Sediment accumulation rates are assumed to be constant over time. A geochronology was established using the down-core distribution of 2l0Pbexcess activities (λ = 22.3 a half-life) using a constant initial concentration model (Appleby and Oldfield 1983):
where C (0) is the unsupported 210Pb activity at the sediment surface, C is the activity at the depth of age determination, and λ is the 210Pb decay constant. In addition, a date of 1986 was assigned to the peak 137Cs activity as an independent chronostratigraphic marker.
Results
Pore Water Chemistry
Pore water chloride concentrations were between 95 and 105 mM and remained constant with depth at all stations (data not shown).
At stations H2 and H5, methane concentrations increased from 0.1 mmol L−1 at 1 cm sediment depth to the saturation concentration of 1.95 mmol L−1 (1 atm, 10 °C) at a depth of 20 cm and continued to increase linearly with depth to the bottom of the core (Fig. 2a). Similarly, methane concentrations increased in almost linear fashion at station H2, but concentrations >2 mmol L−1 at station H2 were reached below 30 cm depth, whereas these concentrations at station H5 were found below 10 cm depth (Fig. 2a). By contrast, at station H3, methane concentrations remained stable below 57 cm depth and slightly varied between 4.3 and 4.8 mmol L−1 (Fig. 2a). Calculation of the saturation concentration of methane at the respective water depths for the three stations indicated that all methane concentrations remained below the solubility limit at the in situ pressures. At all three stations, the sulfate–methane transition was very broad and not marked by a distinct decrease in methane concentrations at the depth of sulfate depletion (Fig. 2a).

a Concentrations of methane and sulfate, b concentrations of sulfide and iron (II), and c concentrations of dissolved inorganic carbon in pore water
Sulfate concentrations in the surface sediments increased from 4.3 mM at H5 to 4.5 mM at H3 and 4.8 at H2. At all three stations, sulfate concentrations decreased in a nearly linear fashion to concentrations of 0.1 mM at 17–20 cm depth. Although the penetration depth (defined as the depth at which sulfate concentrations reached 0.1 mM) at station H5 was shallowest, 17 cm, sulfate gradients were steepest at station H2. The pore water gradients were very sensitive to surface sulfate concentrations. This is also reflected in the calculated sulfate fluxes (see “Methane and Sulfate Fluxes Based on Reaction-Transport Modeling”). Below 20 cm, sulfate concentrations remained <0.1 mM down to the bottom of the cores (ca. 120 cm depth) (Fig. 2a).
The highest dissolved sulfide concentrations were measured at station H5 (871 μM) and the lowest at station H3 (387 μM). At all three stations, dissolved sulfide concentrations showed a maximum between 9 and 19 cm before decreasing with depth. Sulfide concentrations decreased to very low values near the detection limit of 1 μM at stations H3 and H5, whereas they remained between 65 and 180 μM at below the SMT at station H2 (Fig. 2b).
Dissolved iron concentrations followed the general pattern of elevated or very high concentration in the topmost centimeter decreasing to near detection in the sulfidic zone. In the sulfide-free zone at depths below 47 cm, dissolved iron increased again at station H3, whereas no increase was observed at station H5 and H2. At station H5, the dissolved iron concentrations were the highest of all three stations in the topmost centimeter (Fig. 2b).
Dissolved inorganic carbon concentrations increased with depth reaching values between 21.3 and 22.7 mM. The steepest increase occurred in the upper 20 cm in the sulfate reduction zone, whereas in the methanogenesis zone below, DIC concentration remained more or less constant (station H3) or increased only very gradually (stations H5 and H2) (Fig. 2c).
Solid-Phase Geochemistry
The TIC content at all stations was very low (0.01–0.02 dry wt.%) and close to detection. We therefore assume that TC is almost entirely comprised of Corg and only present the Corg data (Fig. 3a). The Corg content at station H5 steadily decreased from 2.7 dry wt.%. at the top of the sediment core to 1.9 dry wt.% at the bottom of the core. At station H3, the Corg content decreased down core from 3.2 to 2.5 dry wt.% (7.5 to 107.5 cm depth). At station H2, the Corg content decreased with depth from 3.7 to 2.6 dry wt.% (Fig. 3a). Sedimentary C:N ratios showed a low variation between 9.6 and 10.6 throughout the core (Fig. 3a).

a Solid-phase Corg, TS, and C: N ratios, b chlorin content and index (CI), and c reactive iron in Himmerfjärden sediments
Total sulfur (TS) contents at station H5 ranged between 0.5 and 1.2 dry wt.% with a pronounced minimum between 30 and 50 cm depth. The TS content at H3 was almost constant throughout the core (0.5–0.6 dry wt.%), whereas at station H2, it gradually increased from 0.4 dry wt.% at the surface to 1.5 dry wt.% at 45 cm depth. Further below, the contents slightly decreased to 1.1 dry wt.% at 75 cm depth (Fig. 3a).
In general, the chlorin content gradually decreased with depth at all stations (Fig. 3b). At station H5, the chlorin content decreased from 9.5 to 3.0 μg g−1 (dry weight) between 37.5 and 88.5 cm depth. At station H3, the concentration decreased from 15.2 to 5.0 μg g−1 between 7.5 and 97.5 cm depth, with the exception at the depth of 37.5 cm, where the chlorin concentration was 40.5 μg g−1. At station H2, concentrations decreased from 9.8 to 3 μg g−1 between12.5 and 72.5 cm depth (Fig. 3b). The calculated chlorin index (CI) varied between 0.65 and 0.99 with values close to 1 indicating refractory material at all stations, except at 37.5 cm depth at station H3, where the CI value was 0.54.
High concentrations of reactive Fe were observed throughout the cores at all three sites (Fig. 3c). Total reactive iron, as defined by Feasc + Fedithio, was much greater at stations H3 and H5 compared to station H2, where total Fe reactive was <10 μmol cm−3 (Fig. 3c). Concentrations of total reactive iron showed a distinct peak of >18 μmol cm−3 at 45 cmbsf at station H5. In general, concentrations of reactive Fe were >10 μmol cm−3 at both stations H3 and H5. Stations H3 and H5 were also similar in that the reactive Fe was almost equally divided between the dithionite and ascorbate reducible fractions. At station H2, easily reducible Feasc was only significant in the upper 0–5 cm of the sediment. The low concentrations of Feasc correlated with the presence of high dissolved H2S concentrations.
Rates of Sulfate Reduction, Anaerobic Oxidation of Methane, and Methanogenesis
We could not calculate accurate specific activities necessary for precisely determining sulfate reduction rates below 14 cm, due to the very low sulfate concentrations (expectedly <50 μM). At station H5, peak SRR (46 nmol cm−3 day−1) in the upper part were found within the surface 0–1 cm (Fig. 4a), whereas peak rates in the upper layer of sediment varied from 25 nmol cm−3 day−1 (station H3, 3.5 cm) to 29 nmol cm−3 day−1 (station H2, 5.5 cm). At all three sites, a second distinct SRR peak rate was observed near the bottom of the sulfate penetration. These rates varied from 33 nmol cm−3 day−1 at station H2 to 76 nmol cm−3 day−1 at stations H3 and H5. This deep peak of SRR is consistent with the peak of AOM measured at H3 (16 nmol cm−3 day−1) at 14 cm depth. Integrated SRR over 14 cm were nearly similar at all three sites and ranged from 1.46 mol m−2 year−1 (station H3) to 1.92 mol m−2 year−1 (station H2) (see Table 2).

a Sulfate reduction rate and rate of anaerobic oxidation of methane (St. H3 only), and b bicarbonate–methanogenesis rate
Rates of bicarbonate methanogenesis varied between 0.2 and 1.2 nmol cm−3 day−1 and 0.1 to 3.2 nmol cm−3 day−1 at stations H2 and H5, respectively. Methanogenesis rates increased below the sulfate zone. At station H5, methanogenesis rates peaked immediately right below the SMT and decreased again further below (Fig. 4b). Integrated bicarbonate methanogenesis rates over 100 cm depths were variable between 1.09 mol m−2 year−1 at station H5 and 0.96 mol m−2 year−1 at station H2 (Table 2).
Sedimentary 210Pbexcess and 137Cs Distribution, and Sedimentation Rates
At all stations, 210Pbexcess decreased with depth but did not reach zero levels in the top 27 cm. Based on the distribution of 210Pbexcess, and assuming steady-state input of 210Pb and minimal sediment mixing, we calculated sedimentation rates of 0.98 cm year−1 at station H5, 0.82 cm year−1 at station H3, and 0.77 cm year−1 at station H2 (Table 3).
137Cs activities were high in the top 20 cm at stations H5 and H3 and decreased abruptly below this depth. In addition, there were distinct peaks of 137Cs activity at 23 cm depth at station H5 and 21 cm depth at station H3 (Fig. 5b). The 137Cs peak at station H2 is shallower, at 13 cm depth, and less distinct. The calculated sedimentation rates of 0.91, 0.82, and 0.65 cm year−1 for station H5, H3, and H2, respectively, were slightly lower than 210Pbexcess -based sedimentation rates (Table 3).

a 210Pb excess and b 137Cs profiles in Himmerfjärden estuary sediments
Methane and Sulfate Fluxes Based on Reaction-Transport Modeling
Utilizing the methane, sulfate, and DIC profiles, the reaction-transport model of Wang et al. (2008) was applied to calculate the fluxes between the water column and underlying sediments, using diffusion coefficients for 5 °C obtained from Schulz and Zabel (2006). Due to the relatively high rates of sediment accumulation, advective pore water transport was also considered. Average sedimentation rates obtained from the 210Pb and 137Cs approaches for each station were used. A value of 0.05 for significance level was employed for the fitting program. The results indicate that methane fluxes (in moles per square meter per year) from the sediment to the sediment–water interface were 0.37 mol m−2 year−1 for H5, 0.25 mol m−2 year−1 for H3, and 0.11 mol m−2 year−1 for H2. The sulfate fluxes from water column into the sediment were 0.34, 0.30, and 0.45 mol m−2 year−1 at stations H5, H3, and H2, respectively (Table 4, Fig. 6). The DIC fluxes in the sulfate reduction zone were 1.18, 1.15, and 0.89 mol m−2 year−1 for stations H5, H3, and H2, respectively (Table 4).

Upward flux of methane to the sediment–water interface, downward flux of sulfate based on the reaction-transport model, and deposition of Corg in Himmerfjärden estuary sediments
Discussion
Sediment Accumulation in Himmerfjärden
To understand methane and sulfur biogeochemistry of Himmerfjärden, it is important to appreciate the high rates of sediment accumulation and, ultimately, the delivery of organic carbon from the water column to the underlying sediment. 210Pbexcess and 137Cs distributions indicate that Himmerfjärden sediments accumulate at very high rates from 0.98 cm year−1 at station H5 in the upper estuary to 0.65 cm year−1 at the lower end (Table 2). These sediment accumulation rates agree well with those calculated by Reuss et al. (2005) (0.89 cm year−1 based on 210Pbexcess and 137Cs profiles). Our rates are slightly lower than rates determined by Bianchi et al. (2002) (1.32 cm year−1 at station H5 based on 210Pbexcess and 137Cs profiles) and Meili et al. (1998) who estimated a rate of 1 cm year−1 in Himmerfjärden archipelago based on 137Cs profiles. The uppermost sediment layers may now be accumulating at a lower rate, based on the 210Pbexcess distributions, within 0.15–0.17 cm year−1 in the top 5 cm at all stations. Nevertheless, sediment accumulation rates in Himmerfjärden estuary are still 1.5–3.5-fold higher than in the open Baltic Sea basins, for example the Bothnian Sea, Bothnian Bay, Finland Bay, and Baltic Proper (0.26–0.62 cm year−1, Mattila et al. 2006).
The 137Cs profiles represent a pulsed input from the Chernobyl catastrophe in 1986 and earlier inputs from above-ground atomic bomb tests, with a fallout peak in 1963. We assigned the peak in the 137Cs distributions to the Chernobyl event (see Fig. 5b). The overall distribution of 137Cs can also be affected by sediment mixing, which would broaden the peak. Sediment mixing does occur at station H2 as indicated by the broadening of the 137Cs peak as compared to the sharp peaks at stations H3 and H5 (Fig. 5b). Input of allochthoanous post-Chernobyl 137Cs-bearing particles also affects the distribution of 137Cs. The elevated 137Cs activities above the putative 1986 Chernobyl peak in Fig. 5b are likely derived from 137Cs containing particles from the Himmerfjärden watershed. Satellite imagery shows that agricultural activities dominate the watershed land-use. 137Cs activities measured in Himmerfjärden (1–2.8 Bq g−1 dry) fall into the range given for Chernobyl impacted soils and sediments in Finland and Scandinavia (0.3 to 46 Bq g−1 dry) (Table 5). The enhanced 137Cs activities above the Chernobyl (1986) peak indicate that the sedimentation patterns in Himmerfjärden are dominated by resuspended sediments and sediment delivered from upstream and soils in the Himmerfjärden watershed.
Sediment accumulation rates are greatest in the inner Himmerfjärden at station H5. Bianchi et al. (2002) attributed high sediment accumulation rates at station H5 to the presence of the nearby sewage treatment plant. By implication, one would expect the highest rates of Corg accumulation at station H5, which our data confirm (Table 2, Fig. 6). In contrast, the highest Corg contents in the core top were measured at station H2 (3.8 %), compared to 3.2 % at stations H3, and 2.8 % at station H5. Although the station H2 sediments have the highest Corg contents, this organic carbon exhibits the greatest degree of chlorin degradation, as indicated by the chlorin index value near unity (Fig. 3b). This is consistent with lower sedimentation rates and possibly sediment mixing due to bioturbation. We observe that the sediment accumulation rates along Himmerfjärden decrease by 30 % from station H5 to station H2. Thus, the combination of decreasing sedimentation rates from the head (station H5) to the mouth (station H2) and greater Corg contents towards lower end of the estuary result in estimated Corg burial rates greatest at station H5 and correspondingly lowest station H2 (Table 2, Fig. 6). The relatively high sediment accumulation rates and organic carbon burial fluxes have, as will be discussed below, important implications for the biogeochemistry of sulfur and methane throughout the Himmerfjärden estuary.
Sulfate Reduction and Sulfur Fluxes in Himmerfjärden
Sulfate reduction is usually the dominant anaerobic pathway of organic carbon decomposition in organic-rich, marine sediments (Jørgensen 2006). The downward flux of sulfate from the sediment surface to bottom sulfate zone drives the organoclastic sulfate reduction (Eq. 7) and sulfate-dependent AOM (Eq. 8)
and
Organoclastic sulfate reduction (Eq. 7) usually exceeds methanotrophic sulfate reduction (Eq. 8) driven by upward diffusing methane (Martens and Klump 1980b). In spite of the high sedimentation rates and organic carbon burial rates, sulfate reduction rates only varied between 1.46 and 1.92 mol m−2 year−1 in the upper 14 cm, with the greatest areal rates at station H2 and the least at station H3 (Table 2). Integrated sulfate reduction rates in Himmerfjärden estuary sediments are similar to those of Bornholm Basin and Gotland Deep sediments in the Baltic Sea (Lapham, Brüchert unpublished data). The rates of sulfate reduction, however, are lower than those measured in shallow, high-deposition sedimentary environments (13.0 mol m−2 year−1) and in estuaries and embayments (2.6 mol m−2 year−1) (Canfield 2005) (Table 6). We attribute the relatively low rates of integrated sulfate reduction to the shallow sulfate penetration depth. Salinity in Himmerfjärden is only 6 to 6.5 ‰, leading to surface sulfate concentrations that are also low (<4.7 mM). Additionally, and perhaps more importantly, the high sediment burial rates result in a limited residence time of the Corg in the sulfate reduction zone (20 to 30 years).
In Himmerfjärden, organoclastic sulfate reduction and AOM appear to overlap within the upper 20 cm of the sediment (Fig. 4a). In the low-sulfate environment (<5 mM), the thermodynamics of both the organoclastic and the methanotrophic sulfate reduction are favorable (Jørgensen 2006; Knab et al. 2008). At station H3, we measured AOM rates throughout the sulfate zone and the SMT at 20 cm depth (Fig. 4a). Based on the integrated rates of AOM over the upper 14 cm (0.3 mol m−2 year−1), 20 % of the overall sulfate reduction can be attributed to AOM (Eq. 8). Overall, in the sulfate reduction zone, 25 % of the organic carbon was degraded via organoclastic sulfate reduction, which is consistent with the decrease of Corg (16 %) in solid phase (3.7 % in the 0–5 cm sediment interval to 3.1 % below the SMT and the chlorin concentration decline (25 %), (17.6 μg g−1 at 5 cm depth to 13.5 μg g−1 at 22.5 cm depth). Therefore, at station H3, 75 % of Corg that reached the surface sediment was buried into the methanogenic zone. At stations H2 and H5, organic matter burial below the sulfate reduction zone was similar to station H3, which is a very high proportion for marine sediments (Hartnett et al. 1998).
Sulfate fluxes into the sediment based on diffusion and pore fluid advection due to burial are inversely proportional to the organic carbon burial and methane fluxes as illustrated in Fig. 6. They are also significantly lower than the total sulfate reduction rates estimated for the upper 14 cm of sediment; the modeled diffusion–advection flux is only about 16–23 % of the total gross sulfate reduction. This suggests that another transport mechanism of sulfate into the sediments must exist. We have not accounted for sulfate transported into the upper 10–15 cm by bio-irrigating organisms. The sediments of Himmerfjärden are populated by Marenzelleria, a widely distributed invasive polychaete (Kautsky 2008; Blank et al. 2008). Hedman et al. (2011) have demonstrated that Marenzelleria enables solute transport down to more than 15 cm depth.
The impact of bio-irrigation on pore fluid exchange can be estimated by the use of a simple, one-dimensional, non-local exchange model. In this case, the sediment interval of 0 to 14 cm is considered as a discrete layer. The irrigation coefficient, α, is used in Eq. 9 (Fossing et al. 2000), which describes the fraction of pore fluid exchanged with the surface per unit time:
where SRRmeas = the GSR (the integrated gross sulfate reduction rate; Table 6) times the sulfate reduction layer (14 cm) and SRRdiff = calculated flux from the fitting model (Table 4). C 0 is the sulfate concentration of the overlying surface sediment, C 14 cm is the concentration at 14 cm depth (moles per cubic meter), and φ (porosity) was set to 0.9 (the average porosity between surface sediment (0.93) and 14-cm interval (0.86)). We calculate values of α between 2.85 and 3.12 year−1. These values are at the lower range of values estimated for coastal regions (12–180 year−1) in the top 20 cm depth (Boudreau 1997) and suggest that, on average, the pore fluids are exchanged with the overlying surface water three times per year. Albeit not vigorous, bio-irrigation may serve to maintain the 10- to 15-cm-deep sulfate penetration depth observed throughout Himmerfjärden.
While Marenzelleria probably do not mix the sediment much (Hedman et al. 2011), they will significantly enhance the overall flux of not only sulfate but also other dissolved components such as DIC and methane (Table 4). We use the value for α to estimate the flux of other dissolved constituents using the relationship (Boudreau 1997, p. 143):
where F I is the flux to the sediment–water interface, assuming constant pore water irrigation (constant α) over the zone L I (in our case, 14 cm). This is a very simple approach to a complex phenomenon, but the resulting fluxes are significantly greater than the diffusive/burial advection fluxes calculated for methane (about twofold) and DIC (about fourfold). Moreover, these DIC fluxes out of the sediment are more consistent with measured carbon turnover rates (sulfate reduction and methanogenesis) than those determined without bio-irrigation. Although bio-irrigation enhances dissolved fluxes, the presence of laminated sediment in the top 20 cm at station H5 does suggest that local and seasonal hypoxia in Himmerfjärden limit sediment mixing and to a certain degree bio-irrigation (i.e., the α values are not very large). As a consequence, regeneration of sulfate by reoxidation of sulfide and the advection supply of sulfate are limited in these sediments. This is also reflected in the relatively large fraction of sulfur that is buried as reduced solid-phase S (Table 2).
The accumulated reduced sulfur in pyrite and organic sulfur below the SMT corresponds to 41–81 % of the gross sulfate reduction rate (Table 2). The large sediment accumulation rates at stations H5 and H3 play a decisive role. Although free dissolved sulfide is present in the sulfate reduction zone, the reduced sulfide is completely scavenged from the pore waters (Fig. 2b). Essentially, the rapid burial rate removes a significant fraction of sulfur out of the surface layers.
At both stations, H5 and H3, high amounts of reactive iron are available below the sulfate reduction zone (Fig. 3c) and ferrous iron is released into the pore water. In contrast, at station H2, where the burial flux of reactive iron is significantly lower, free sulfide is not only present as a peak in the sulfate reduction zone but is never completely titrated by reactive iron in the lower 100 cm of the core (70–240 μM) (Fig. 2b). The Feasc may exist in part as FeS, which is very likely to be dissolved under the strong Fe complexing conditions used in the ascorbate–citrate treatment. At stations H3 and H5, sulfide below the SMT is very efficiently scavenged and a large inventory of reactive iron, both Feasc and Fedithio, remains below the sulfate zone. This may be related to the lower sedimentation rates at station H2, and ultimately the rate of reactive iron delivery relative to the sulfate reduction rate. In contrast, high rates of reactive iron delivery below the sulfate zone result in an effective scavenging of sulfide from the deeper pore waters and potentially allow for continued organic carbon degradation via iron reduction.
The presence of Fe2+ in pore water below the sulfate zone suggests that iron reduction occurs and may be a result of the interaction between dissolved sulfides (H2S, FeS) and FeOOH species leading to ferrous iron production and the formation of elemental S (Riedinger et al. 2010; Holmkvist et al. 2011; Tarpgaard et al. 2011). Dissolved reduced iron may result from the direct coupling of anaerobic oxidation of methane (AOM) to iron reduction (Beal et al. 2009). The flux ratios of DIC and SO 2−4 range from 2.3 (station H2) to 3.3 (stations H3 and H5), thus exceeding the maximum ratio of 2 expected for organoclastic sulfate reduction (Jørgensen and Parkes 2010; Burdige and Komada 2011). This also suggests an additional terminal electron acceptor process, e.g., iron reduction, in the Himmerfjärden sediments.
Controls on Methane Fluxes
In the Himmerfjärden sediments, the methane-bearing, sulfate-free (methanogenic) zone is within the upper 20 cm of the sediment column due to both high organic matter accumulation rates and low sulfate concentrations (4.3–4.8 mM). In spite of the presence of sulfate at all three sites, methane concentrations exhibit linear gradients extending to the sediment–water interface. As shown in Fig. 6, the calculated flux of methane by diffusive transport to the sediment–water interface is greatest at station H5 (0.37 mol m−2 year−1) and lowest at station H2 (0.11 mol m−2 year−1), consistent with the organic carbon burial rates. These upward methane fluxes to the sediment–water interface are in the range of previously calculated methane release rates from other brackish sediments (0–2.5 mol m−2 year−1) (Hariss et al. 1988; Lyimo et al. 2002; Middelburg et al. 2002), from the northern Baltic Sea (0–0.2 mol m−2 year−1, Gotland Deep, Bothnian Sea and Bothnian Bay, Brüchert et al. unpublished data), as well as from the coastal southern Baltic Sea (0.02–133 mol m−2 year−1) (Heyer and Berger 2000). Due to bio-irrigation, the actual methane fluxes (0.32–0.78 mol m−2 year−1) may be greater than the estimated advective–diffusive methane fluxes to the sediment–water interface (Table 4). These values are much lower than values for methane fluxes from sediments from freshwater or wetland ecosystems (1.25–3.75 mol m−2 year−1) (Crill and Martens 1986; Miller and Oremland 1988; Purvaja and Ramesh 2001; Nahlik and Mitsch 2011). Nevertheless, the brackish Himmerfjärden estuary sediments have the potential for significant release of methane to the sediment–water interface, as in other low-salinity coastal regions.
Interestingly, the rates of bicarbonate methanogenesis in the sulfate-depleted sediments are less than 3.2 nmol cm−3 day−1 and generally less than 1 nmol cm−3 day−1 in the methanic zone. Although only measured at two stations (H5 and H2), rates of bicarbonate reduction to methane are also slightly higher at the upstream station H5. In comparison to methanogenesis rates previously determined for coastal brackish sediments in the southern Baltic Sea (55–200 nmol cm−2 day−1, Heyer et al. 1990), Gotland Deep (0.3–2.8 nmol cm−2 day−1 down to 20 cm depth; Piker et al. 1998), and Eckernförde Bay (up to 37 nmol cm−3 day−1; Treude et al. 2005), the methanogenesis rates in Himmerfjärden sediments are low. If data are integrated over the cored depth interval, however, the resulting flux of bicarbonate methanogenesis (0.96 and 1.09 mol m−2 year−1) fits well to the calculated upward fluxes of methane to the sediment–water interface (0.32 and 0.78 mol m−2 year−1) (Tables 2 and 4).
Methanogenesis can also occur in the sulfate zone if sufficient non-competitive substrates are available for both sulfate reduction and methane production processes (Lovley and Klug 1983; Oremland and Polcin 1982). This has been observed in marine sediments of the Skagerrak (Parkes et al. 2007; Knab et al. 2009) and Limfjorden where sulfate was <5 mM (Jørgensen and Parkes 2010). Indeed, we measured low rates of bicarbonate methanogenesis (0.2–0.8 and 0.1–1 nmol cm−3 day−1) in the active sulfate reduction zones at stations H5 and H2, respectively. This suggests that, in addition to the methane flux from below, methane is being produced in the surface sediment. This methane is either transported out of the sediment or may be, albeit slowly, oxidized. However, measured rates of bicarbonate methanogenesis may in part represent a back reaction of tracer of up to 5 % during anaerobic oxidation of methane (Holler et al. 2011). Methanogenesis within the surface sulfate-bearing zone needs further detailed investigation.
At all three stations, methane concentrations linearly decrease from the methanic sediments through the sulfate-bearing zone to the sediment–water interface. Although AOM was measured, this overlap of methane and sulfate suggests that the oxidation of methane is rather “sluggish,” as also observed in Black Sea sediments (Knab et al. 2009), and that a substantial methane flux to the water column may occur. An intriguing possibility is that the methane-oxidizing community may not be able to keep pace with the high rates of sediment accumulation. Methane oxidizing consortia are notoriously slow growing (Nauhaus et al. 2007). Regnier et al. (2011) have demonstrated that the microbial response to rapid changes in the pore water methane and sulfate gradients, e.g., due to rapid burial or ebullition events, can be delayed over years (typically 80 years for a sudden gas ebullition event). Due to the high sedimentation rates in Himmerfjärden estuary, the residence time of a microbial community in the sulfate zone is only 20 to 30 years. Thus, the broad overlapping zones of organoclastic and methanotrophic sulfate reduction in the sulfate zone in Himmerfjärden may occur as a consequence of a small, inefficient AOM community at the bottom of the sulfate zone.
Conclusions
Himmerfjärden is a littoral benthic ecosystem typical for the brackish coastal waters of the central Baltic Sea. Eutrophication and low concentrations of sulfate in the overlying water impact methane production, consumption, and release from the sediment to the water column. The unusual controlling variable for sulfur and methane biogeochemistry in Himmerfjärden, however, is the high rate of sediment accumulation. We propose that the depth of the sulfate zone (ca. 15 cm) is controlled by the irrigating activity of the invasive polychaete Marenzelleria. High rates of sediment accumulation as they occur in the Himmerfjärden estuary shorten the residence time of the sediment in the sulfate reduction zone. Such a short residence time (ca. 20 to 30 years) has important consequences for sulfur, iron, and carbon biogeochemistry. Thus, the Himmerfjärden sediments have certain characteristics that distinguish them from central basin sediments of the Baltic and continental shelf margins. Reduced sulfur produced within the sulfate reduction zone escapes to a large extent by burial, which reduces the reoxidation of sulfide in the surface sediments. In addition, unusually large amounts of reactive iron become buried into the methanic zone. The role of this reactive iron in organic carbon and methane at depth is unclear.
Although methane production rates are not unusually high when compared to other marine sediments, the narrow sulfate zone allows for a significant upward flux of methane to the sediment–water interface. This methane flux from the methanogenic zone to the sediment–water interface correlates with the burial flux of organic carbon and may be enhanced by bio-irrigation. The oxidation of methane in the sulfate zone appears to be sluggish. The slow-growing methane-oxidizing communities that are responsible for methane oxidation may not be able to keep up with the fast sediment accumulation. Therefore, the Himmerfjärden estuary (Sweden) is regarded as a model area for study, and the early-stage diagenesis of biogeochemical processes is certainly a prime candidate to achieve this goal.
References
Appleby, P.G., and F. Oldfield. 1983. The assessment of 210Pb data from sites with varying sediment accumulation rates. Hydrobiologia 103: 29–35.
Bange, H.W., U.H. Bartel, S. Rapsomanikis, and M.O. Andreae. 1994. Methane in the Baltic and North Seas and a reassessment of the marine emissions of methane. Global Biogeochemistry Cycle 8: 465–480.
Bartnicki, J., and S. Valiyaveetil. 2008. Estimation of atmosphere Nitrogen deposition to the Baltic Sea in the periods 1997-2003 and 2003-2006. The report for HELCOM (http://www.helcom.fi/stc/files/Publications/OtherPublications/EMEP_Estimation_of_atmospheric_N_deposition_%20to_the_BS.pdf).
Beal, J.H., H.C. House, and J.V. Orphan. 2009. Manganese- and iron-dependent marine methane oxidation. Science 325: 184–187.
Blank, M., A.O. Laine, K. Jürss, and R. Bastrop. 2008. Molecular identification key based on PCR/RFLP for three polychaete sibling species of the genus Marenzelleria, and the species’ current distribution in the Baltic Sea. Helgoland Marine Resource 62: 129–141.
Bianchi, T.S., E. Engelhaupt, B.A. Mckee, S. Miles, R. Elmgren, A. Hajdu, C. Savage, and M. Baskaran. 2002. Do sediments from coastal site accurately reflect time trends in water column phytoplankton? A test from Himmerfjärd Bay (Baltic Sea proper). Limmology and Oceanography 47: 1537–1544.
Boetius, A., K. Ravenschlag, C.J. Schubert, D. Rickert, F. Widdel, A. Gleseke, R. Amann, B.B. Jørgensen, U. Witte, and O. Pfannkuche. 2000. A marine microbial consurtium aooarently mediating anaerobic oxidation of methane. Nature 407: 623–626.
Borges, A.V., and G. Abril. 2011. Carbon dioxide and methane dynamics in estuaries. In Treatise on estuarine and coastal science 5, ed. E. Wolanski and D.S. McLusky, 119–161. Waltham: Academic.
Burdige, D.J., and T. Komada. 2011. Anaerobic oxidation of methane and the stoichiometry of remineralization processes in continental margin sediments. Limmology and Oceanography 56(5): 1781–1796.
Boudreau, P. Bernard. 1997. Diagenetic models and their implementation. Heidelberg: Springer-Verlag.
Callaway, J.C., R.D. DeLaune, and W.H. Patrick Jr. 1996. Chernobyl 137Cs used to determine sediment accretion rates at selected northern European coastal wetlands. Limmology and Oceanography 41(3): 444–450.
Canfield, E.Donald. 2005. The sulfur cycle. In Aquatic geomicrobiology: 48 (Advances in marine biology), ed. D.E. Canfield, B. Thamdrup, and E. Kristensen, 314–374. California: Elsevier Academic.
Chanton, P.J., C.S. Martens, and C.A. Kelley. 1989. Gas transport from methane-saturated, tidal freshwater and wetland sediments. Limnology and Oceanography 34(5): 807–819.
Claypool, G.E., and I.R. Kvenvolden. 1983. Methane and other hydrocarbon gases in marine sediment. Annual Review of Earth and Planetary Sciences 11: 299–327.
Cline, D.Joel. 1969. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnology and Oceanography: Methods 14: 454–458.
Conley, D.J., S. Björck, E. Bonsdorff, et al. 2009. Hypoxia–related processes in the Baltic Sea. Environmental Science and Technology 43(10): 3412–3420.
Conley, D.J., J. Carstensen, J. Aigars. et al. 2011 Hypoxia is increasing in the coastal zone of the Baltic Sea. Environmental Science & Technology 45: 6777–6783.
Crill, P.M., and C.S. Martens. 1986. Methane production from bicarbonate and acetate in an anoxic marine sediment. Geochimica et Cosmochimica Acta 50: 2089–2097.
Cutshall, N.H., I.L. Larsen, and C.R. Olsen. 1983. Direct analysis of 210Pb in sediment samples: self-absorption corrections. Nuclear Instruments and Methods A306: 309–312.
Elverfeldt, J.S., M. Schlüter, T. Feseker, and M. Kölling. 2005. Rhizon sampling of porewaters near the sediment–water interface of aquatic systems. Limnology and Oceanography: Methods 3: 361–371.
Engqvist, A., and A. Omstedt. 1992. Water exchange and density structure in a multi-basin estuary. Continental Shelf Research 12(9): 1003–1026.
Fossing, H., T.G. Ferdelman, and P. Berg. 2000. Sulfate reduction and methane oxidation in continental margin sediments influenced by irrigation (South–East Atlantic off Namibia. Geochimica et Cosmochimica Acta 64(5): 897–910.
Hall, P.O.J., and R.C. Aller. 1992. Rapid, small-volume, flow injection analysis for ∑ CO2 and NH +4 in marine and freshwaters. Limnology and Oceanography 37: 1113–1119.
Hariss, R.C., D.I. Sebacher, K.B. Bartlett, D.S. Bartlett, and P.M. Crill. 1988. Sources of atmospheric methane in the south Florida environment. Global Biogeochemial Cycles 2: 231–243.
Hartnett, H.E., R.G. Keil, J.I. Hedges, and A.H. Devol. 1998. Influence of oxygen exposure time on organic carbon preservation in continental margin sediments. Nature 391: 572–574.
Hedman, J.E., J.S. Gunnarsson, G. Samuelsson, and F. Gilbert. 2011. Particle reworking and solute transport by the sediment-living polycheates Marenzelleria neglecta and Hediste diversicolor. Journal of Experimental Marine Biology and Ecology 407: 294–301.
Heinsalu, A., S. Veski, and J. Vassiljer. 2000. Paleoenvironment and shoreline displacement on Suursaari island, the Gulf of Finland. Bulletin of he Geological Society of Finland 71(part 1–2): 24–26.
Heyer, J., U. Berger, and R. Suckow. 1990. Methanogenesis in different parts of a brackish water ecosystem. Limmologica 20: 135–139.
Heyer, J., and U. Berger. 2000. Methane emission from the coastal area in the southern Baltic Sea. Estuarine, Coastal and Shelf Science 51: 13–30.
Holby, O., and S. Evans. 1996. The vertical distribution of Chernobyl-derived radionuclides in a Baltic Sea sediment. Journal of Environmental Radioactivity 33(2): 129–145.
Holler, T., G. Wegener, H. Niemann, C. Deusner, T.G. Ferdelman, A. Boetius, B. Brunner, and F. Widdel. 2011. Carbon and sulfur back flux during anaerobic oxidation of methane and coupled sulfate reduction. Proceedings of the National Academy of Sciences of the USA 108(52): E1484–E1490.
Holmkvist, L., T.G. Ferdelman, and B.B. Jørgensen. 2011. A cruptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochimica et Cosmochimica Acta 75: 3581–3599.
Ilus, E., and R. Saxén. 2005. Accumulation of Chernobyl-derived 137Cs in bottom sediments of some Finnish lakes. Journal of Environmental Radioactivity 82: 199–211.
Ilus, E., J. Mattila, S.P. Nielsen, E. Jakobson, J. Herrmann, V. Graveris, V. Vilimaite-Silobritiene, M. Suplinska, A. Stepanow, and M. Lüning. 2007. Long-lived radionuclides in the seabed of the Baltic Sea. Report of the sediment Baseline study of HELCOM MORS-PRO in 2000–2005. Baltic Sea Environment Proceeding 10.
Iversen, N., and B.B. Jørgensen. 1985. Anaerobic methane oxidation rates at the sulfate–methane transition in marine sediments from Kattegat and Skagerrak (Denmark). Limnology and Oceanography 30: 944–955.
Jørgensen, B.B., and T. Fenchel. 1974. The sulfur cycle of a marine sediment model system. Marine Biology 24: 189–201.
Jørgensen, B.Bo. 1978. Comparison of methods for quantification of bacterial sulfate reduction in coastal marine sediments 1. Measurement with radiotracer techniques. Geomicrobiology Journal 1(1): 11–27.
Jørgensen, B.Bo. 2006. Bacteria and marine geochemistry. In Marine geochemistry, 2nd ed, ed. H.D. Schulz and M. Zabel, 169–201. Berlin: Springer.
Jørgensen, B.B., and R.J. Parkes. 2010. Role of sulfate reduction and methane production by organic carbon degradation in eutrophic fjord sediments (Limfjorden, Denmark). Limnology and Oceanography 55(3): 1338–1352.
Judd, A.G., M. Hovland, L.I. Dimitrov, S. Garci, A. Gil, and V. Jukes. 2002. Geological methane budget at continental margins, and its influence on climate change. Geofluids 2: 109–126.
Kallmeyer, J., T.G. Ferdelman, A. Weber, H. Fossing, and B.B. Jørgensen. 2004. A cold chromium distillation procedure for radiolabeled sulphide applied to sulphate reduction measurements. Limnology and Oceanography: Methods 2: 171–180.
Kautsky, Hans. 2008. Askö and Himmerfjärden. In Ecology of Baltic coastal waters, ed. U. Schiewer, 335–357. Berlin: Spinger.
Kipphut, G.W., and C.S. Martens. 1982. Biogeochemical cycling in an organic-rich coastal marine basin—3. Dissolved gas transport in methane-saturated sediments. Geochimica et Cosmochimica Acta 46: 2049–2060.
Knab, N. J., A. D. Dale, K. Lettmann, H. Fossing, and B.B. Jørgensen. 2008. Thermodynamic and kinetic control on anaerobic oxidation of methane in marine sediments. Geochimica et Cosmochimica Acta 72: 3746–3757.
Knab, N.J., B.A. Cragg, R.R.C. Hornibrook, L. Holmvist, R.D. Pancost, C. Borowski, R.K. Parkes, and B.B. Jørgensen. 2009. Regulation of anaerobic methane oxidation in sediments of the Black Sea. Biogeosciences 6: 1505–1518.
Larsson, U., R. Elmgren, and F. Wulff. 1985. Eutrophication and the Baltic Sea—causes and consequences. Ambio 14: 9–14.
Lovley, D.R., and M.J. Klug. 1983. Sulfate reducers can outcompete methanogens at freshwater sulfate concentrations. Applied and Environmental Microbiology 45: 187–192.
Lustwerk, R.L., and D.J. Burdige. 1995. Elimination of dissolved sulphide interface in the flow injection determination of ∑CO2 by addintion of molybdate. Limnology and Oceanography 40: 1011–1012.
Lyimo, T.J., A. Pol, and H.J.M. Op den Camp. 2002. Methane emission, sulfide concentration and redox potential profiles in Mtoni mangrove sediment, Tanzania. Western Indian Ocean Journal of Marine Science 1(1): 71–80.
Mattila, J., H. Kankaanpää, and E. Ilus. 2006. Estimation of recent sedimentary accumulation rates in the Baltic Sea using artificial radionuclides 137Cs and 239, 240 Pu as time markers. Boreal Environment Research 11: 95–107.
Martens, C.S., and J.V. Klump. 1980a. Biogeochemical cycling in an organic-rich coastal marine basin—I. Methane sediment–water exchange processes. Geochimica et Cosmochimica Acta 44: 471–490.
Martens, C.S., and J.V. Klump. 1980b. Biogeochemical cycling in an organic-rich coastal marine basin—4. An organic carbon budget for sediments dominated by sulfate reduction and methanogenesis. Geochimica et Cosmochimica Acta 48: 1987–2004.
März, C., J. Hoffmann, U. Bleil, G.J. de Lange, and S. Kasten. 2008. Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambesi deep-sea fan (SW Indian Ocean). Marine Geology 255: 118–130.
Meili, M., P. Jonsson, and R. Carman. 1998. 137Cs dating of laminated sediments in Swedish archipelago areas of the Baltic Sea. In Dating of sediments and determination of sedimentation rate, ed. Erkki Ilus, 127–130. Helsinki: STUK–Radiation and Nuclear Safety Authority (Finland).
Meybeck, M., D. Chapman, and R. Helmer (eds.). 1989. Global freshwater quality. A first assessment, 306. Oxford: Blackwell.
Middelburg, I.I., I. Nieuwenhuize, N. Iversen, N. Høgh, H. De Wilde, W. Heider, R. Seifert, and O. Chirstof. 2002. Methane distribution in European tidal estuaries. Biogeochemsitry 59: 95–119.
Miller, L.G., and R.S. Oremland. 1988. Methane efflux from the pelagic regions of four lakes. Global Biogeochemical Cycles 2: 269–277.
Nahlik, A.M., and W.J. Mitsch. 2011. Methane emission from tropical freshwater wetlands located in different climatic zones of Costa Rica. Global Change Biology 17: 1321–1334.
Nauhaus, K., M. Albrecht, M. Elvert, A. Boetius, and F. Widdel. 2007. In vitro cell growth of marine archaeal-bacterial consortia during anaerobic oxidation of methane. Environmental Microbiology 9(1): 187–196.
Oremland, R.M., and S. Polcin. 1982. Methanogenesis and sulphate reduction: competitive and noncompetitive sustrates in estuarine sediments. Applied and Environmental Microbiology 44: 1270–1276.
Orphan, V.T., C.H. House, K.U. Hinrichs, K.D. McKeegan, and E.F. Delong. 2001. Methane-consuming Archaea releaved by directly coupled isotope and phylogentic analysis. Science 293: 484–486.
Parkes, R.J., B.A. Cragg, N. Banning, et al. 2007. Biogeochemistry and biodiversity of methane cycling in subsurface marine sediments (Skagerrak, Denmark). Environmental Microbiology 9: 1146–1161.
Piker, L., R. Schmaljohann, and J.F. Imhoff. 1998. Dissimilatory sulfate reduction and methane production in Gotland Deep sediments (Baltic Sea) during a transition period from oxic to anoxic bottom water (1993–1996). Aquatic Microbial Ecology 14: 183–193.
Purvaja, R., and R. Ramesh. 2001. Natural and anthropogenic methane emission from coastal wetlands of south India. Environmental Management 27(4): 547–557.
Reeburgh, S.William. 1975. Methane consumption in Cariaco trench waters and sediments. Earth and Planetary Science Letters 28: 337–344.
Regnier, P., A.W. Dale, S. Arndt, D.E. LaRowe, J. Mogollón, and P. Van Cappellen. 2011. Quantitative analysis of anaerobic oxidation of methane (AOM) in marine sediments: a modeling perspective. Earth-Science Reviews 106: 105–130.
Reuss, N., D.J. Conley, and T.S. Bianchi. 2005. Preservation conditions and the use of sediment pigments as a tool for recent ecological reconstruction in four Northern European estuaries. Marine Chemistry 95: 283–302.
Riedinger, N., B. Brunner, M.J. Formolo, E. Solomon, S. Kasten, M. Strasser, and T.G. Ferdelman. 2010. Oxidative Sulfur cycling in the deep biosphere of the Naikai Trough, Japan. Geology Society of America 38: 851–854.
Rosenberg, R., R. Elmgren, S. Fleisher, P. Jonsson, G. Persson, and H. Dahlin. 1990. Marine eutrophication case studies in Sweden. Ambio 19: 102–108.
Savage, C., P.R. Reavitt, and R. Elmgren. 2010. Distribution and retention of effluent nitrogen in surface sediment of a coastal bay. Limnology and Oceanography 49: 1503–1511.
Schubert, C.J., J. Niggemann, G. Klockgether, and T.G. Ferdelman. 2005. Chlorin index: a new parameter for organic matter freshness in sediments. Geochemistry, Geophysics, Geosystems 6(3): 1–12.
Schulz, H.D. 2006. Quantification of early diagenesis: dissolved consituents in marine porewater. In Marine Geochemistry 2, ed. H.D. Schulz and M. Zabel, 73–124. Berlin: Springer.
Smith, F.S., S.M. Elliott, and S.K. Lyons. 2010. Methane emissions from extinct megafauna. Nature Geoscience 3: 374–375.
Stigebrandt, Anders. 1991. Computations of oxygen fluxes through the sea surface and the net production of organic matter with application to the Baltic and adjacent seas. Limmology and Oceanography 36(6): 444–454.
Tarpgaard, I.H., H. Røy, and B.B. Jørgensen. 2011. Concurrent low and high affinity sulfate reduction kinetics in marine sediment. Geochimica et Cosmochimica Acta 75(11): 2997–3010.
Thode-Andersen, S., and B.B. Jørgensen. 1989. Sulfate reduction and the formation of 35S labeled FeS, FeS2 and SO in coastal marine sediments. Limmology and Oceanography 34: 793–806.
Treude, T., A. Boetius, K. Knittel, K. Wallmann, and B.B. Jørgensen. 2003. Anaerobic oxidation of methane above gas hydrates at Hydrate Ridge, NE Pacific Ocean. Marine Ecological Progess 264: 1–14.
Treude, T., M. Krüger, A. Boetius, and B.B. Jørgensen. 2005. Environmental control on anaerobic oxidation of methane in the gassy sediment of Eckernförde Bay (German Baltic). Limmology and Oceanography 50(6): 1771–1786.
Valentine, L.David. 2002. Biogeochemistry and microbial ecology of methane oxidation in anoxic environments: a review. Antonie Van Leeuwenhoek 81: 271–282.
Viollier, E., P.W. Inglett, K. Hunter, A.N. Roychoudhury, and P. Van Cappellen. 2000. The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Applied Geochemistry 15: 785–790.
Walling, Q.H., and P.N. Owens. 1996. Interpreting the 137Cs profiles observed in several small lakes and reservois in southern England. Chemical Geology 129: 115–131.
Wang, G., J. Spivack, S. Rutherford, U. Monor, and S. D’Hondt. 2008. Quantification of co-occurring reaction rates in deep subseafloor sediments. Geochimica et Cosmochimica Acta 72: 3479–3488.
Acknowledgments
This work has received funding from the European Community’s Seventh Framework Programme (FP/2007-2013) under grant agreement 217246 of the ERANET BONUS project BALTIC GAS (BMBF 03F0488D to TF), the Max Planck Society, and the Stockholm University, and a Ph.D. scholarship from the Vietnam Ministry of Education and Training (MOET) and the German Academic Exchange Service (DAAD) to N. M. Thang. We would like to thank these institutions for their financial support. We thank the crew and staff at the Äsko Laboratory and Kirsten Imhoff, Andrea Schipper, and Gabriele Schüßler for assistance in the laboratory. The comments of Dr. M. Holmer, Dr. S. Henkel, and one anonymous reviewer were very constructive and highly appreciated. N.M. Thang especially thanks Laura M. Wehrmann for many interesting discussions and for English corrections.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Thang, N.M., Brüchert, V., Formolo, M. et al. The Impact of Sediment and Carbon Fluxes on the Biogeochemistry of Methane and Sulfur in Littoral Baltic Sea Sediments (Himmerfjärden, Sweden). Estuaries and Coasts 36, 98–115 (2013). https://doi.org/10.1007/s12237-012-9557-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-012-9557-0