
Abstract
Introduction
Cell stretch is a method which can rapidly apply mechanical force through cell-matrix and cell-cell adhesions and can be utilized to better understand underlying biophysical questions related to intracellular force transmission and mechanotransduction.
Methods
3D printable stretching devices suitable for live-cell fluorescent imaging were designed using finite element modeling and validated experimentally. These devices were then used along with FRET based nesprin-2G force sensitive biosensors as well as live cell fluorescent staining to understand how the nucleus responds to externally applied mechanical force in cells with both intact LINC (linker of nucleoskeleton and cytoskeleton) complex and cells with the LINC complex disrupted using expression of dominant negative KASH protein.
Results
The devices were shown to provide a larger strain ranges (300% uniaxial and 60% biaxial) than currently available commercial or academic designs we are aware of. Under uniaxial deformation, the deformation of the nucleus of NIH 3T3 cells per unit of imposed cell strain was shown to be approximately 50% higher in control cells compared to cells with a disrupted LINC complex. Under biaxial deformation, MDCK II cells showed permanent changes in the nuclear morphology as well as actin organization upon unloading, indicating that failure, plastic deformation, or remodeling of the cytoskeleton is occurring in response to the applied stretch.
Conclusion
Development and open distribution of low-cost, 3D-printable uniaxial and biaxial cell stretching devices compatible with live-cell fluorescent imaging allows a wider range of researchers to investigate mechanical influences on biological questions with only a minimal investment of resources.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The model of cellular tensegrity predicts that cell-matrix and cell-cell forces are readily transferred across the cytoskeleton and applied to intracellular structures such as the nucleus.10 Relatively recent work in the field of cell biology has identified a group of nuclear membrane-associated proteins, known as the LINC (linker of nucleoskeleton and cytoskeleton) complex, that mediate the connection of the cytoskeleton to the nuclear membrane.2 The LINC complex is formed by nuclear envelope spectrin repeat protein (nesprin) and Sad1/UNC-84 (SUN) proteins that cross both the inner and outer nuclear membranes to mechanically tether the cytoskeleton to the nuclear lamina.14 The LINC complex is evolutionarily conserved across all eukaryotes, suggesting that the mechanical linkage of the cytoskeleton to the nucleus may be essential to cell function and homeostasis.2 One potential function of the LINC complex is the transfer of forces from the cytoskeleton onto the nucleus as a means of cellular signaling. The nucleus has long been proposed to be a mechanosensor,22 and recent work has demonstrated that forces applied to the nucleus regulate cellular functions, including nuclear transport,8 DNA structure,19 and gene expression.19 Disruption of the LINC complex has been shown to perturb cell migration, nuclear shape, and position.12,13 In turn, dysregulation of cell organization, proliferation, and migration is a hallmark of cancer development and progression. Therefore, a better understanding of the role of nuclear mechanical signaling would provide significant insight into clinically relevant processes such as wound healing and cancer.
Epithelial and fibroblast cells types are a prominent component of tissues which are subject to mechanical forces under normal physiological conditions, such as the lining of the lungs, bladder, and stomach for epithelial cells as well as skin and tendons for fibroblasts. Therefore their behavior in response to forces is an important biological question. NIH 3T3 and MDCK II cells were chosen in particular because of their use in previous mechanobiology studies (allowing our results to be more directly compared), their ability to be easily transfected, and their cell line immortality (allowing stable expressing cell lines to be generated when necessary rather than relying on transient transfections).
As actin filaments are integral features of the cytoskeleton and connect integrins to the LINC complex, this work aims to quantify the magnitude of force directly propagated from external mechanical stimulation, through actin filaments, and to the nuclear envelope. In previous work by Arsenovic et al.,4 Förster resonance energy transfer (FRET) based force sensitive biosensors were integrated into mini-nesprin-2G, measuring actin-mediated force at the LINC complex. These sensors are constructed by genetically encoding two fluorescent proteins linked with an elastic peptide chain (together called a tension sensor module), into a protein of interest. When the two fluorophores are in close proximity (less than 10 nm) energy is transferred non-radiatively from the shorter wavelength fluorophore (donor) to the longer wavelength fluorophore (acceptor). The efficiency of this energy transfer is strongly dependent on the fluorophore spacing (\(r^6\)), allowing an applied force causing elastic deformation of the peptide linker to be related to a relative change in brightness of the two fluorophores. There are multiple approaches to quantifying the amount of energy transfer, the two used in this study are a simple ratio of acceptor to donor signal magnitude (denoted FRET Index), which provides a relative FRET measurement,4 and sensorFRET (denoted FRET efficiency), which accounts for the spectral bleed-through to provide an absolute measure of the FRET efficiency.5 In both cases a lower FRET efficiency or index indicates a higher force, however a FRET efficiency measurement is independent of instrumentation settings such as the laser power and gain. Many tension sensor module variations have been used, with the fluorophore pairing and linker length being able to be changed to optimize imaging conditions and force sensitivity.3,7 Two variants of the force sensitive biosensor were used in this study, one consisting of mTFP and Venus fluorophores linked with 40 spectrin repeats used previously in Arsenovic et al.,4 and an updated version consisting of Cerulean and Venus fluorophores linked with 25 spectrin repeats, which is optimized for the dual wavelength excitation needed for sensorFRET,5 which will be referred to throughout this article as nesprin-2G T40V and nesprin-2G C25V, respectively. Although the previous work by Arsenovic et al. demonstrated the mini-nesprin-2G biosensor can quantify changes in nuclear force due to myosin activation and inhibition, it was not shown whether the sensitivity of the measurements are sufficient to characterize changes in force due to external mechanical stimuli, such a substrate deformation.
Many in vitro models have been developed to apply mechanical stimuli to cells by stretching an elastic cell culture substrate.11,17,18,21 These types of experiments are in many cases preferable to alternative means of mechanical stimulation such as AFM or micro pipette aspiration, as it can be applied evenly to a large number of cells simultaneously. However, commercially available stretching apparatuses are typically limited to relatively small strains in addition to being either expensive, not conducive to in situ imaging, or inflexible with respect to the applied strain ratios. To remedy these shortcomings, we developed two cell stretching apparatuses which can apply uniaxial strains of up to 300% in one dimension or biaxial strains of up to 60% in both dimensions. We previously used these devices to apply mechanical forces to adherens junctions16 and desmosomes,6 two principal structures of cell-cell contacts. Both devices are suitable for in situ imaging, and the components of which can be made using a 3D printer and assembled using hand tools. We have made the design, stereolithography files (.STL) for 3D printing, and assembly instructions for each apparatus available in the supplemental material of this manuscript, to enable use by other researchers. Using these devices, we show 1) that the nuclear LINC complex contributes significantly to nuclear strain induced by extracellular stretch and 2) that large mechanical forces applied to the cell result in permanent nuclear shape changes suggesting permanent deformation or failure.
Results and Discussion
Stretcher Design
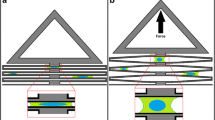
Stretching apparatuses for both uniaxial and biaxial deformation were developed with the design constraints that they should be capable of imposing large substrate deformations, allow imaging with an inverted fluorescent microscope, and are able to be reproduced relatively cheaply in order to allow testing of multiple experimental groups in parallel. Although the biaxial stretcher design can be used for both uni-directional and bi-directional deformation, the uniaxial stretcher is a smaller and simpler design that may be useful for researchers who do not need the flexibility of the biaxial stretcher (Fig. 1).

Real and computer aided drafting (CAD) model images of the uniaxial (a, b) and biaxial (c, d) stretcher designs. Stereolithography (STL) files for printing are provided in the supplemental information. Labels included on CAD images highlight specific components referred to in the article.
The stress-strain behavior of the PDMS substrate (Product: 0.005” NRV G/G 40D 12”x12”, Specialty Manufacturing Inc., MI, USA) was characterized under uniaxial loading until failure at 318% strain and 5.15 MPa (Fig. 2a). Residual strain after failure was only 1% of the total, indicating that deformation is almost entirely elastic, albeit non-linear. This experimentally determined behavior was used as an input to finite element modelling of the cruciform geometry, shown in Figs. 2b, 2c, and 2d, used for the biaxial stretch device. The FEA model results show that as the cruciform geometry inherently causes a stress concentration between the cruciform legs, the center of the cruciform is only able to be strained to approximately 60% in both dimensions before the stress exceeds the 5 MPa failure stress at the corners. However, 60% biaxial strain is still sufficient to emulate or exceed the deformation requirements of most physiologically relevant mechanical loading. One disadvantage of the cruciform geometry is that the applied strain deviates from biaxiality at positions further away from the center, as shown in Fig. 2d, which plots the two principal strains as a function of position along the red arrow in Fig. 2b. The shaded region denotes where the deviation from biaxiality is less than 2%, indicating there is a 6mm diameter region where the strain state is nearly uniformly biaxial. Tracking of fiduciary markings in 3 independent experiments were used to determine the membrane strain as a function of applied rotation to the worm gear (Fig. 2e). The variability of the strain applied increases as the applied strain increases and is due to a number of experimental factors including the dimensional accuracy when cutting out the membrane cruciform, centering and angular alignment of the membrane when installing it, pre-strain applied to remove slack from the membrane, and the constraint imposed by the adhesion of the media well. No loss of structural integrity has been observed due to regular use over more than 1 year, although autoclave sterilization caused significant warping (ethanol sterilization is recommended).

(a) Experimentally determined uniaxial stress strain behavior of the PDMS membrane material showing linear behavior up to 100% strain (obtained by attaching one end of the membrane to a lab scale and applying. (b) Shape of 1/4 cruciform geometry used for finite element modeling. (c) Max principal strain distribution in the cruciform geometry after deformation. (d) Divergence of principal strain components as a function of distance from the cruciform center (along red arrow in b and c) showing deviations from biaxiality are less than 2% within a 6 mm diameter. (e) Experimental measurement of the membrane strain behavior as a function of worm gear rotation, with the overlaid boxplots showing the variability in applied strain.
The use of 3D printing allows these devices to be manufactured quickly, cheaply, and without specialized manufacturing equipment (excluding the 3D printer itself). The uniaxial stretcher requires no post processing and can be assembled immediately following printing. The biaxial stretcher design utilizes tapped threads and additional aluminum axles and clamps, therefore some additional post processing with basic hand held tools is required. As the precision of 3D printers varies, the STL file available for the biaxial stretcher in the supplemental information utilizes slightly undersized holes which need to be drilled and tapped to obtain a tighter tolerance fit on the components. Detailed assembly instructions are provided in the supplemental information.
The base plate of both apparatuses is nominally 110 mm by 160 mm and designed to fit in place of the Zeiss Universal Mounting Frame K (Product: 451352-0000-000, Carl Zeiss Microscopy LLC, Oberkochen, Germany). This secure fixturing of the base helps minimize sample movement which is necessary for in situ imaging. In situ imaging of live cells also requires that the cell culture remain covered in media, which necessitates the use of a water sealed chamber adhered to the PDMS substrate. For both stretchers we utilized an annular shaped culture well cut from cast PDMS adhered to the substrate with a generous coating of petroleum jelly, which allows the membrane to deform independently of the culture well while maintaining a water-tight seal.
The rollers to which the PDMS substrate is attached are controlled using commercially available 14:1 ratio worm gears (Rotomatic ®102C, Grover Musical Products Inc., OH, USA). While both worm gears on the uniaxial stretcher are operated independently, a timing belt and pulley can be used on the biaxial design in order to maintain a fixed strain ratio. The equal size pulleys shown in Fig. 1c ensure a 1:1 strain ratio, although varying the relative pulley size on two of the worm gears allows other fixed strain ratios to be applied.
Uniaxial Stretch
MDCK II cell lines which continually express nesprin-2G T40V were developed so that variability in the expression levels would not skew the measurements and the FRET could be quantified on many cells in a single experiement. The shape of the cell-substrate interface affects both the magnitude and orientation of the forces exerted by and on the cell.20 Therefore, cells were seeded onto 20 μm wide fibronectin lines micropatterned onto the PDMS substrate (Fig. 3a). This patterning served two purposes: (1) to systematically orient the long-axis of the cell nucleus in the direction of applied membrane strain, as any anisotropic properties of the nucleus would effect the measured nuclear deformation in a direction-dependent manner, and (2) to provide visually distinct features, helping to aid tracking of cells subject to focal drift and/or deformation induced translations. Individual cell nuclei were tracked before (red outline) and after (green outline) 30% applied uniaxial membrane strain (Fig. 3b) in order to quantify the nuclear strain imparted to each cell. Raw images of the MDCK cells before and after deformation are included in the supplementary material (Fig. S1). Nuclei were masked such that only pixels at the perimeter of the nucleus contribute to the FRET quantification, helping to exclude free and improperly localized nesprin-2G T40V (which are not expected to be load bearing) from the analysis. Nuclei with increasing strain area reported statistically insignificant negative changes in the FRET ratio. Similarly, increasing aspect ratios (long-axis/short-axis) of strained nuclei tended to have larger decreases in the FRET ratio, however this relationship was statistically insignificant. A notable exception to the generally negative relationship between the aspect ratio and nesprin-2G-TS FRET ratio was nuclei #15 shown in Figs. 3b and 3d. For this nucleus, the aspect ratio increased nearly 60% (more than double the average) while the FRET ratio unexpectedly increased and the area strain decreased. The average FRET index (a ratio of the acceptor magnitude to the donor magnitude) of the strained nuclei was significantly lower than the initial unstrained nuclei (Fig. 3e), indicative of higher average force on nesprin-2G T40V when strained. This is to be expected if load is being transfered from focal adhesions to the LINC complex through the actin cytoskeleton, as reported in the literature.2,15

(a) Schematic representation of the micropatterned fibronectin lines applied to the membrane. (b) Change in nuclear shape of 16 nuclei (MDCK cells) before and after straining the substrate. (c) Change in FRET index as a function of nuclear area strain. (d) Change in FRET index as a function of nuclear aspect ratio. (e) Boxplot of FRET index distributions at both strain conditions (p = 0.015 for T-Test).
There are a number of biological factors which contribute to the variability in the strain vs. FRET behavior on a cell to cell basis (Figs. 3c and 3d). The first of these is the sensor expression level. Since the change in FRET efficiency is related to the force applied to each molecule, if the FRET sensors are being produced at too high of a rate and cannot all be integrated into the LINC complex, any signal will be diluted by unloaded sensors, lowering the measured change in FRET. Masking the images to only include the nucleus perimeter in the analysis reduces but cannot eliminate this effect. Second, it is expected that the organization and mechanical properties of the cytoskeleton connecting the substrate and nucleus can be strongly affected by the point the cell is in within its cell division cycle and thereby alters the force propagated to the LINC complex. Finally, since the deformation is uniaxial it is expected that the ends of the nuclei (aligned with the applied strain) could be subject to significantly higher forces than the sides of the nuclei, leading to increased variability.
In addition to measuring the change in FRET due to applied strain, differences in the nuclear deformation behavior were also characterized in NIH 3T3 fibroblast cells with disrupted and intact LINC complexes. As the initial cell condition (eg.: orientation, shape and confluency) can significantly alter the forces applied to the cell, these were controlled for using micropatterned fibronectin rectangles (100 × 30 μm, shown in Fig. 4a) designed such that single cells could be deformed in isolation. NIH 3T3 cells were used because they are able to be isolated as single cells and confined on the pattern much more easily than MDCK cells. Disruption of the LINC complex was carried out using DN-KASH,12 a protein which binds to the KASH domain of the SUN component of the LINC complex, effectively displacing nesprin proteins and removing the physical connection between the cytoskeleton and the nucleus.

Effect of LINC complex disruption on strain propagation to the nucleus. (a) Example images of control cells (NIH 3T3) before and after stretch used for strain quantification, ellipse fitting was used on the DAPI channel to quantify nuclear deformation, CellTracker\(^{\mathrm{TM}}\) Green CMFDA Dye was used to quantify the overall cell strain, and Alexa Fluor 647 labeled fibronectin pattern used to constrain the cell shape and orientation. Equivalent representative images for the DN-KASH group are available in Supplemental Fig. S2. (b) Quantification of the nuclear strain as a function of applied strain in both the direction of stretch (X) and the transverse direction (Y), showing a reduced propagation of strain to the nucleus in NIH 3T3 fibroblasts expressing DN-KASH to disrupt the LINC complex. (c) Quantification of the overall cell strain in both dimensions as a function of applied membrane strain showing approximately 1:1 correspondence, indicating the cell-membrane interface is well adhered not appreciably changing in response to the applied strain. (d) Table of quantified strain ratios determined by the least squares fit lines in (b) and (c).
The nuclear deformation was quantified by using an automated routine in ImageJ. The nuclei were segmented using an intensity threshold and then fitted to an ellipse and then measuring the major and minor axes of that ellipse at each strain step. Simple width and height measurements of the cell body were used to quantify the cell strain. The applied membrane strain was estimated from rotation of the worm gears, with each full rotation corresponding to 4.75% strain. As seen in Fig. 4b, the magnitude of strain imparted to the nucleus per unit of applied membrane strain is lower for DN-KASH cells compared to the control cells, as expected to be the case since DN-KASH results in the disruption of the LINC complex. There is significantly higher scatter in the deformation behavior of the DN-KASH cells which we attribute to variable expression of the DN-KASH protein. As DN-KASH competes with endogenously expressed nesprin proteins, variation in the expression level can lead to variable levels of nuclei connectivity if every KASH binding site is not able to be saturated with the dominant negative. Shown in Fig. 4c, the deformation of the PDMS membrane is approximately equal to the cell deformation. This indicates that the cell is not detaching from the substrate in response to the applied strain. Quantifying the strain ratios (slopes of the linear least squares fit lines, shown in Fig. 4d) in the applied strain direction X, we can see that in control cells strain applied to the cell body is transferred to nuclear strain at a rate of approximately 73%, whereas in DN-KASH cells this rate of transfer decreases to 48%.
In general it is expected that the nucleus should deform at a lower rate than the cell body, as the nucleus has been shown to be much stiffer than the cell as a whole. Guilack et al.9 measured a threefold increase in the stiffness of the nucleus compared to the cell body using micropipette aspiration. They also showed that the mechanical response of both the nucleus and cell body are viscoelastic in nature, reaching a steady state after approximately 1 min. As the time required to track and image the cells in this study is greater than 1 min, the viscous contribution to the measured strains are expected to be negligible.
One proposed mechanism for mechanotransduction is that changes in the nuclear morphology changes the location of genes relative to the nuclear periphery, which in turn alter the gene expression.22 Work by Alam et al.1 showed that LINC complex disruption (through SUN1L) did not change the nuclear shape response to passive stimulation (substrate stiffness). Therefore, they concluded that LINC complex disruption likely does not affect mechanosensitive gene expression by altering the mechanosensitivity of the nucleus shape. In contrast, our present results using DN-KASH show that the mechanosensitivity of the nucleus shape is dependent on LINC complex connectivity under active mechanical stimulation (ie. the nucleus deforms 50% more with an intact LINC complex under the same applied strain) and therefore this mechanism for altered gene expression should not be disregarded for certain physiological loading conditions such as stretch.
Biaxial Stretch
To determine if load is transmitted to the LINC complex via nesprin-2G under biaxial loading, confluent monolayers of MDCK II cells transfected with the tension sensor plasmids were subjected to biaxial stretch. At each increment of applied membrane strain, fluorescence images were acquired of the nesprin-2G C25V localization (Fig. 5a). These fluorescence images were then masked as shown in Fig. 5b, where only nuclei which displayed clear localization of the nesprin-2G C25V sensor at the nuclear membrane and which remain within the field of view at all strain steps were included in the analysis. The FRET efficiency for areas within these masked regions was then determined using the sensorFRET approach5 (Fig. 5c). In this way, 121 individual cells distributed between 19 fields of view and 3 independent trials were characterized over 4 strain steps the results of which are shown in Fig. 5d. As lipofectamine based plasmid transfection can result in a wide range of expression levels, one concern is that overexpression will dilute the signal from loaded tension sensors with the signal from unloaded sensors in the cytoplasm once all the nesprin binding sites are saturated. The masking procedure, which only includes the perimeter of the nucleus, reduces this effect by excluding the majority of the cytoplasm from the analysis. If overexpression is appreciably diluting the signal, we would expect that increased fluorescence intensity (proportional to expression level) would result in increased FRET efficiency, which is not observed as shown in supplementary Fig. S3.
The same trend was observed in all three experiments, where the FRET efficiency decreases upon 1 turn of applied stretch, remains approximately constant for the second turn, and then returns to its initial value upon unloading. However, due to the relatively small changes in FRET efficiency being measured (1%) and high variability, the differences between loading steps do not reach the threshold for statistical significance (T-Test p = 0.10 for Initial to 1 Turn). In order to observe morphological changes in the nucleus as a function of biaxial loading, confluent monolayers of MDCK II cells with live cell staining of lamin (Lamin-Chromobody®: Chromotek GmbH, Munich, Germany) were also subjected to biaxial stretch. As shown in Fig. 5e, the nucleus deforms and then upon unloading, the nuclear area decreases further than the initial condition and adopts a blebby morphology with wrinkles in the nuclear envelope.
One possible explanation consistient with all of these observations (i.e. the small absolute change in FRET efficiency observed, after the initial strain FRET efficiency is insensitive to further loading, and the change in nuclear morphology upon unloading) is that the actin cytoskeleton, which mechanically bridges nesprin-2G C25V to the focal adhesions attached to the PDMS substrate, is permanently deforming at high strains. This permanent deformation could be realized passively, through actin fiber failure, or active remodeling of the cytoskeleton by the cell.

(a) Representative fluorescence intensity images of MDCK II cells transfected with the nesprin-2G C25V tension sensor. (b) Image showing the masks (red) applied to the intensity image to include only the nuclear perimeter. (c) Pixel-wise FRET efficiency values for each of the masked regions shown in (b). (d) Average (+/-SEM) FRET efficiency of all 121 cells at each strain step. (e) Representative images of live cell staining of lamin before, during and after stretch, showing shrinkage and increase of wrinkles upon unloading.
To further investigate this possibility, live cell staining of actin (Actin-Chromobody®: Chromotek GmbH, Munich, Germany) in a monolayers of MDCK II cells was used to monitor morphological changes in the actin organization upon loading. Changes in the actin morphology can be observed when comparing the same cells before, during, and after stretch as shown in Fig. 6a. However, this evidence should be considered qualitative, as small changes in focal height and the time delay between straining and imaging could significantly impact the quantification. High speed and high resolution 3D imaging would be required for systematic quantification of these changes which exceeds the instrumentation capabilities of the microscope used in this study. Similar to what was observed in the lamin staining, only small changes in actin localization can be observed upon deforming the membrane, whereas more dramatic changes are seen upon unloading. With areas showing prominent changes highlighted by red circles in Fig. 6a, the width of the cortical actin network appears to narrow in some regions while dispersing in others, which would also suggest permanent changes in the cytoskeleton due to even a single cycle of applied stretch. Although quantification of this reorganization was not possible with the current data set, similar instances of changes in actin localization were observed in 3 independent experiments.
In addition to qualitative observations of the actin morphology, the live cell staining of actin also provides an opportunity to quantify the strain applied on a cell by cell basis. Using the actin localized at the cell cortex to define the boundary of each cell, the cell areas of 90 individual cells were manually masked and used to determine the cell strain at each loading step, as shown in Fig. 6b. Lower cell strains compared to the membrane strain could indicate issues such as delamination of the monolayer, separation of cell-cell junctions, or that the cell monolayer is sufficiently stiff to constrain the deformation of the membrane. For our experiments, the cell area strain per turn of the worm gears is not significantly different from the membrane behavior (T Test p = 0.61), therefore the influence of these issues, if present, is not appreciable compared to the other sources of experimental variability.

(a) Live MDCK II cells expressing Actin-Chromobody label were tracked at 3 loading steps (initial, 37% strain, and unloaded), showing permanent reorganization of the actin cytoskeleton due to stretch. Areas showing large changes in morphology are denoted by red circles. (b) Comparison of membrane area strain (quantified from fiduciary marks) to cell area strain (quantified based on the cortical actin) as a function of applied stretch.
Conclusions
In this work we developed 3D printable stretching devices for applying both uniaxial and biaxial mechanical stimuli to cells in situ. We have made the design of these devices freely available in the supplemental material for other researchers to use. In order to demonstrate the usefulness of this experimental capability in probing the biophysics of cells, we showed that external mechanical forces are propagated to the nucleus through the actin cytoskeleton and LINC complex proteins. Under both uniaxial and biaxial stretch applied to MDCK cells, FRET based tension sensors integrated into mini-nesprin-2G showed a decrease in FRET (corresponding to increased force) in response to applied stretch. Additionally, under uniaxial deformation of NIH 3T3 cells, we were able to show that for cells treated with DN-KASH (to disrupt the LINC complex) approximately 48% of the strain imparted to the cell is transferred to the nucleus, in contrast to untreated control cells in which the nucleus strains approximately 73% of the applied cell strain. As high biaxial strains resulted permanent nuclear shape changes (increased blebbiness) as well as a relative decrease in force (although still higher than the initial condition) which are both indicative of cytoskeletal deformation, failure, or remodeling, imaging of the actin cytoskeleton in MDCK cells before, during, and after deforming the substrate was performed and showed qualitative changes in the actin morphology in response to deformation.
Methods
Stretcher Characterization
The uniaxial stress strain behavior was determined using a 1 cm wide strip of PDMS membrane attached to the uniaxial stretcher on one end to apply displacement while the other end was fixed to a lab scale to measure the load. Imaging of fiduciary marks during deformation using a fixtured camera was used to measure the strain. For the biaxial characterization Abaqus FEA software was used to determine the membrane area under a biaxial stress state. Additionally, 3 independent measurments of the strain (measured via permanent marker fiduciary marks) vs. worm gear rotation behavior was also determined in order to quantify the uncertainty of the strain application.
Biaxial Stretcher Preparation
Cruciform substrates with the dimensions shown in S10 were cut from PDMS sheets from Specialty Manufacturing Inc. (Class VI Silicone, Gloss, 12”×12”, 0.005” thickness, 40 durometer) using a scalpel. Care was taken to cut the curved inside corners of the cruciform in a single continuous motion in order to prevent irregularities in the edge that can act as stress concentrations and cause premature failure of the membrane. The membrane is centered on the stretching apparatus with the slots for the clamping bars oriented towards the stretcher base. The clamping bars are installed to hold the membrane in place and the timing pulleys are turned 360° to remove any slack in the membrane. The top surface of the membrane is then exposed to UV for 30 min and the well is attached using petroleum jelly. The well is then covered with 30 μg/mL fibernectin for 30 min to promote cell adhesion.
Cell Culture
MDCK II and NIH 3T3 mouse fibroblasts were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal calf serum and 500 μg/mL of Penicillin–Streptomycin antibiotics. Cells were maintained in an atmospherically controlled incubator at 37 °C and 5% CO2 atmosphere. Media were changed every other day.
Cell Fluorescent Labeling
MDCK II cells stably expressing nesprin-2G T40V tension sensor were generated by Lipofectamine 2000 (Thermo Fisher) transfection followed by G418 selection. Transient transfection of nesprin-2G C25V was achieved in MDCK II cells using Lipofectamine 2000 as well. DN-KASH expressing NIH 3T3 cells were developed by lentiviral infection of DN KASH (in the indicuble pinducer20 vector, addgene plasmid number 44012) and expressed using 1 μg/ml doxycycline. MDCK II cells expressing Actin-Chromobody and Lamin-Chromobody labeling (ChromoTek®: Munich, Germany) were a gift from Teemu Ihalainen (University of Tampere, Finland). For the uniaxial strain quantification experiments, the cell body was labeled with CellTracker Green CMFDA (Thermo Fisher) which was diluted to a final working concentration of 20 μM in DMSO then media, the solution was placed on the cells for 15 minutes at 37 °C, while the nucleus was labeled with Hoescht which at a final working concentration of 1.62 μM in media and was placed on the cells for 5 minutes.
Micropatterning
The center of the membrane was stamped with fibronectin rectangles 30 × 100 μm. The stamps used to stamp fibronectin rectangles were made with polydimethylsiloxane (PDMS). Stamps were coated with fibronectin and were pressed onto the prepared membrane to transfer the fibronectin to the stretcher substrate. Once stamped, the area was coated with 0.2% BSA-647 (Molecular Probes, Eugene, OR) to visualize the surrounding area of the rectangles. The stamped area was washed and treated with Pluronic F-127 to limit cell adhesion to only the fibronectin rectangles.
FRET Imaging
Images were acquired using a Zeiss LSM 710 confocal microscope using both 405 nm or 458 nm excitation wavelengths from an argon laser source. A 40 × water immersion objective lens (NA = 1.1) was used for all imaging. Live cells were imaged in spectral mode using a 32-channel spectral META detector to record spectra of each pixel spanning wavelengths from 416 to 718 nm. Images were captured in 16 bit mode, scanned bi-directionally, and averaged 4 times. For sensorFRET based efficiency imaging, spectral images at both 405 and 458 nm excitation wavelengths were acquired. The normalized emission shape of the mTFP and mVenus fluorophores as well as the calibration parameter \(\gamma\) (= 0.101) required for the sensorFRET analysis were experimentally determined from control cells expressing single fluorophores (see Arsenovic et al.5 for details). FRET index analysis was carried out using only 458nm excitation as described in Arsenovic et al.4
Cell Tracking
To track cells during the application of strain, a membrane marking is kept in the field of view in real-time while the membrane is being deformed. The stage is continually re-positioned and re-focused to compensate for transverse and axial drift from the focal plane. The magnitude of position changes required for a given strain step increases as the viewing area deviates from the center of the membrane. The typical time required for applying a 15% strain step is 1–2 min. Additionally, as multiple fields of view were imaged per strain step to collect data from multiple cells in a single experiment, there is an additional delay between steps due to moving to find each area of interest.
References
Alam, S. G. et al. The mammalian linc complex regulates genome transcriptional responses to substrate rigidity. Sci. Rep. 6:38063, 2016.
Alam, S., D. B. Lovett, R. B. Dickinson, K. J. Roux, and T. P. Lele. Nuclear forces and cell mechanosensing. Prog. Mol. Biol. Transl. Sci. 126:205–215, 2014. https://doi.org/10.1016/B978-0-12-394624-9.00008-7.
Arsenovic, P. T. & Conway, D. E. Using nesprin tension sensors to measure force on the linc complex. In The LINC Complex, 59–71 (Springer, 2018).
Arsenovic, P. et al. Nesprin-2G, a component of the nuclear LINC complex, is subject to myosin-dependent tension. Biophys. J. 110:34–43, 2016. https://doi.org/10.1016/j.bpj.2015.11.014.
Arsenovic, P. T., C. R. Mayer, and D. E. Conway. SensorFRET: a standardless approach to measuring pixel-based spectral bleed-through and FRET efficiency using spectral imaging. Sci. Rep. 7:15609, 2017. https://doi.org/10.1038/s41598-017-15411-8.
Baddam, S. et al. The desmosomal cadherin desmoglein-2 experiences mechanical tension as demonstrated by a fret-based tension biosensor expressed in living cells. Cells 7:66, 2018.
Brenner, M. D. et al. Spider silk peptide is a compact, linear nanospring ideal for intracellular tension sensing. Nano Lett. 16:2096–2102, 2016).
Elosegui-Artola, A. et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171:1397.e14–1410.e14, 2017. https://doi.org/10.1016/j.cell.2017.10.008.
Guilak, F., J. R. Tedrow, and R. Burgkart. Viscoelastic properties of the cell nucleus. Biochem. Biophys. Res. Commun. 269:781–786, 2000.
Ingber, D. E., and I. Tensegrity. Cell structure and hierarchical systems biology. J. Cell Sci. 116:1157–1173, 2003. https://doi.org/10.1242/jcs.00359.
Lee, A. et al. An equibiaxial strain system for cultured cells. Am. J. Physiol. Physiol. 271:C1400–C1408,1996.
Lombardi, M. L. et al. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 286:26743–26753, 2011. https://doi.org/10.1074/jbc.M111.233700.
Luxton, G. W. G., E. R. Gomes, E. S. Folker, E. Vintinner, and G. G. Gundersen. Linear arrays of nuclear envelope proteins harness retrograde actin flow for nuclear movement. Science (New York) 329:956–959, 2010. https://doi.org/10.1126/science.1189072.
Luxton, G. W. G., and D. A. Starr. KASHing up with the nucleus: novel functional roles of KASH proteins at the cytoplasmic surface of the nucleus. Curr. Opin. Cell Biol. 28:69–75, 2014. https://doi.org/10.1016/j.ceb.2014.03.002.
Maniotis, A. J., C. S. Chen, and D. E. Ingber. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. U.S.A. 94:849–854, 1997.
Mohan, A. et al. Spatial proliferation of epithelial cells is regulated by e-cadherin force. Biophys. J. 115:853–864, 2018.
Schürmann, S. et al. The isostretcher: an isotropic cell stretch device to study mechanical biosensor pathways in living cells. Biosens. Bioelectron. 81:363–372, 2016.
Shao, Y. et al. Uniaxial cell stretching device for live-cell imaging of mechanosensitive cellular functions. Rev. Sci. Instrum. 84:114304, 2013.
Tajik, A. et al. Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 15:1287, 2016.
Tseng, Q. et al. A new micropatterning method of soft substrates reveals that different tumorigenic signals can promote or reduce cell contraction levels. Lab Chip 11:2231–2240, 2011. https://doi.org/10.1039/c0lc00641f.
Ursekar, C. P. et al. Design and construction of an equibiaxial cell stretching system that is improved for biochemical analysis. PLoS ONE 9:e90665, 2014.
Wang, N., J. D. Tytell, and D. E. Ingber. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol.10, 75–82, 2009). https://doi.org/10.1038/nrm2594.
Acknowledgments
We wish to thank Teemu Ihalainen for providing reagents. We acknowledge support from NIH R35GM119617 and R03AR068096, NSF CMMI-1653299, and the Thomas F. and Kate Miller Jeffress Memorial Trust. Microscopy was performed at the VCU Nano Characterization Core Facility.
Author Contributions
CM and PA wrote the manuscript. CM, PA, KB and KD performed the experiments. CM designed the biaxial stretcher, PA and KB designed the uniaxial stretcher. DC led the project and provided input to the manuscript. All authors helped edit and revise the manuscript.
Data Availability
Any datasets generated during and/or analyzed during the study are available from the corresponding author on reasonable request.
Conflict of interest
Carl R. Mayer, Paul T. Arsenovic, Kranthidhar Bathula, Kevin B. Denis, and Daniel E. Conway declare that they have no conflicts of interest.
Ethical approval
No human studies were carried out by the authors for this article. No animal studies were carried out by the authors for this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Daniel Fletcher.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mayer, C.R., Arsenovic, P.T., Bathula, K. et al. Characterization of 3D Printed Stretching Devices for Imaging Force Transmission in Live-Cells. Cel. Mol. Bioeng. 12, 289–300 (2019). https://doi.org/10.1007/s12195-019-00579-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12195-019-00579-y