
Abstract
The functional integrity of the kidney depends on normal development as well as on physiological cell turnover. Apoptosis induction is essential for these mechanisms. Multiple mechanisms are unleashed during obstructive nephropathy, one of the most complex being programmed cell death that leads to renal tubular atrophy and tubular loss. This review will focus on the interaction of nitric oxide and Hsp70 and on the regulation of renal antiapoptotic and protective oxidative stress responses.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
The incidence of chronic kidney disease continues to climb, not only in the United States but also worldwide (Campese and Park 2007). Inflammation of the tubulointerstitial compartment leading to fibrosis is a major factor in the progressive loss of renal function in patients with a wide variety of kidney diseases. About 80% of total kidney volume is composed of tubular epithelial cells and cells within the interstitial space. Most of the nonepithelial cells are associated with the rich vascular network of the kidney. Unlike most forms of chronic renal disease that often take months to establish fibrotic lesions, obstructive nephropathy induced by complete, continuous ureteral obstruction is an exceptionally aggressive form of tubulointerstitial fibrosis (Yang and Liu 2003). It is a complex renal disorder that begins with hydrodynamic and hemodynamic responses, leading to cellular changes in all renal compartments, and finally to interstitial fibrosis and tubular atrophy (Chevalier 2006). Renal fibrogenesis initiates at a very early stage after ureteral obstruction and progresses rapidly. The incidence of unilateral ureteral obstruction is reported as 1/1,000 in adults, and its incidence in children is of grave concern. Congenital obstructive nephropathy is a major cause of chronic renal failure in infancy (Chevalier 1999). Programmed cell death leads to renal tubular atrophy and tubular loss in neonatal unilateral ureteral obstruction (UUO; Chevalier et al. 1996).
Moreover, the severity of the apoptotic response to unilateral ureteral obstruction is far greater in the neonatal than in the adult rat, a factor that may contribute to the impaired growth of the obstructed kidney (Cachat et al. 2003). Rapid diagnosis and initiation of the treatment are vital to preserve function and/or to slow down renal injury.
Lately, genetically modified animals have been increasingly used to study the development of obstructive nephropathy. These animals have shown the complexity of apoptosis and tubulointerstitial fibrosis development involving a large number of closely related molecules functionally. In addition, the recent development of mechanical stretch in cultured epithelial cells that mimics renal tubular distention has led to the discovery of unexpected and contradictory roles of principal apoptosis modulating factors (Bascands and Schanstra 2005).
Apoptosis represents an efficient cellular suicide pathway with characteristics of death in individual cells, induced by both physiological and pathological stimuli with phagocytosis by adjacent cells but without an inflammatory response (Kerr et al. 1972).
At present, it is known that several renal pathologies are a cause and/or consequence of alterations in the mechanism of apoptosis regulation (Kasinath et al. 2006).
Many and varied mediators modulate the apoptotic signs, favoring or inhibiting them. Because of the significant role of apoptosis in the pathogenesis of renal cellular injury resulting from urinary tract obstruction, factors regulating the renal apoptotic response have been studied (Chevalier 2006). Stretching of the renal tubular cells by increased hydrostatic pressure provides a powerful mechanical stimulus to apoptosis in the obstructed kidney (Nguyen et al. 2000; Manucha et al. 2007). Ischemia is another stimulus to apoptosis, and UUO induces a sharp reduction in renal blood flow as well as impairment of autoregulation of renal blood flow (Chevalier and Thornhill 1995). Moreover, reactive oxygen species (ROS) are known to reduce the threshold at which tissues undergo apoptosis (Kayanoki et al. 1996), and reactive oxygen species are significantly increased in the chronically obstructed kidney (Kawada et al. 1999).
The neonatal obstructed kidney may be particularly susceptible to the generation of reactive oxygen species because endogenous renal antioxidant enzymes, including superoxide dismutase, are suppressed in the neonate (Gupta et al. 1999).
Among the modulators that have been recently studied, a relationship has been observed between nitric oxide (NO; Ito et al. 2005) and the chaperone protein Hsp70 (Vallés et al. 2003). Lately, there has been special interest in the interaction and regulation of these two apoptotic modulating factors in obstructive nephropathy.
2 NO as a bifunctional regulator of apoptosis
The ubiquitous distribution of the nitric oxide synthases and the remarkable diffusibility and diverse chemical reactivity of NO in biological systems make this molecule unique among the regulators of apoptosis. NO could be considered as a bifunctional regulator of apoptosis (Kim et al. 1999). The cytotoxic capacity of NO has been confirmed in numerous systems using diverse cell targets. NO cytotoxicity produced by NOS2 as well as by NOS1 has been the topic of intense study (Mannick et al. 1994).
The capacity of NO to induce apoptosis was first appreciated by Albina et al. (1993), who showed that NO caused apoptosis in macrophages. Since then, several cell types have been shown to undergo apoptosis in response to NO.
The proapoptotic effect seems to be independent of cGMP accumulation, except in vascular smooth muscle cells. Apoptosis by NO can be the result of DNA damage with a previous p53 protoncogene induction that produces cell cycle arrest by means of p21. Nevertheless, p53 can help in the repair of injured cells by p27 induction with a subsequent decrease in pRb phosphorylation (Freedman and Folkman 2004). Caspases, a family of cysteine proteases, are involved in apoptosis induction (Sola et al. 2004). The cellular redox state contributes to the complexity of this system. Cytotoxicity as the result of the interaction of NO with superoxide to yield peroxynitrite (ONOO) inflicts cellular injury through oxidation of many biological molecules. Furthermore, ONOO has also been implicated in the inactivation of Mn and Fe superoxide dismutase (Ischiropoulos et al. 1992).
Since 1994, however, information about NO and its interference with the apoptotic machinery has appeared in the literature. First for cGMP-dependent protein kinase activation and later for caspases inhibition (Nagai-Kusuhara et al. 2007); this mechanism was described in many tissue types but still not verified in renal tissue. In addition to the indirect inhibition of caspases activity, NO might modulate caspase expression and activity in a direct way due to its capacity for redox modifications by S-nitrosylation (Kim et al. 1997).
For caspase 3, cysteine 163 is essential and susceptible to redox modification by NO. The direct and/or indirect NO-mediated inactivation of caspases has been shown to reduce Bcl-2 cleavage and increase its concentration. Via this indirect mechanism, therefore, NO may also prevent cytochrome c release, inhibiting thereby activation of the dangerous caspases (Kim et al. 1998).
NO, a multifunctional mediator, has been shown to be antiapoptotic and antifibrotic in UUO (Hegarty et al. 2001). Urinary nitric oxide metabolites increased after relief of UUO compared with baseline. In vitro studies in stretched epithelial cells and in vivo studies in obstructed kidney of inducible nitric oxide synthase (iNOS) −/− mice have provided support for an antiapoptotic role for NO (Miyajima et al. 2001). Recently, using a nitric oxide biosynthetic precursor (L-arginine), it was demonstrated that renal damage, including apoptosis and fibrosis, was significantly improved by L-arginine treatment, suggesting that increased NO availability could be beneficial in the setting of UUO relief (Ito et al. 2005).
We have demonstrated decreased endogenous NO and lower iNOS expression at mRNA and protein levels in obstruction, regulated by the mitochondrial signal pathway, through the increased proapoptotic ratio Bax/Bcl-2 and subsequent caspase 3 activity (Manucha and Vallés 2008; Figs. 1 and 2). As a result, NO produces resistance to obstruction-induced cell death by the mitochondrial apoptotic pathway.

iNOS expression at mRNA, protein levels and endogenous NO generation in kidney cortex after 5 and 14 days of UUO. A Representative gel of iNOS mRNA in control and obstructed kidney cortexes after 5 days of obstruction, and in control and obstructed kidney for 14 days. Housekeeping gene β-actin expression is shown in the line underneath, in the same order as the densitometry bars. Graphical representation of iNOS/β-actin mRNA ratio showed an increased expression of iNOS isoform in obstructed cortex (OC) vs control cortex (CC) **p < 0.01 after 5 days of obstruction. Decreased iNOS expression from 14 days OC vs 5 days OC was demonstrated **p < 0.01. Results are means ± SEM of six independent observations. B Representative western blot and densitometric analysis of iNOS protein levels from kidney cortexes after 5 days of obstruction and following 14 days of obstruction. Immunoblots were quantified for iNOS expression. The relative amount of iNOS protein was determined after normalization of the level of iNOS protein of the appropriate control: 1 and was shown in histograms beneath the corresponding blots. Sharp decreased in iNOS protein levels from kidneys obstructed for 14 days compared to CC ***p < 0.001. Slight increase of iNOS protein levels in OC compared to CC after 5 days of obstruction: *p < 0.05. Results are means ± SEM of six separate experiments. C Measurement of nitrite generated (nmol NO2 generated/100 μL homogenate). Homogenates of renal cortex from obstructed 14 days vs CC, *p < 0.05. Following 5 days of obstruction, OC vs CC *p < 0.05. (Manucha and Vallés 2008)

Mitochondrial apoptotic pathway induction after 14 days of kidney obstruction; A Induction of mRNA expression for Bcl-2 and Bax and the ratio of mRNA Bax/mRNA Bcl-2 in kidney cortexes after UUO for 5 and 14 days mRNA for Bcl-2 and Bax were measured by reverse transcription polymerase chain reaction. Histograms show the relative concentration of mRNAs for Bcl-2 and Bax to β-actin mRNA. Cortexes obstructed for 14 days compared with CC *p < 0.05. Data represent the means ± SEM of six independent experiments. B Western Blot analysis for 32 kDa pro-caspase 3 protein and caspase 3 activity in obstructed and control kidney cortexes. Upper Panel: Total protein (50 μg) was extracted and equal amounts of protein were loaded and separated by molecular weight on 12% SDS-PAGE. Blot represents one out of six separate experiments. Lower Panel: Caspase 3 activity was assessed by level of Ac-DEVD-AMC cleavage release of fluorescence AMC tag. Activity is expressed as pmol AMC/min/μg protein. Cortexes obstructed for 14 days compared with CC **p < 0.01. Caspase 3 activity and pro-caspase 3 protein assay data were obtained from the same six independent samples. (Manucha and Vallés 2008)
3 Heat shock-induced cell protection from apoptosis induction
Heat shock protein (HSP) Hsp70 has been reported to protect various cells and tissues from ischemic damage. Acting as molecular chaperones, HSPs play essential roles in mediating protein folding, assembly, transport, and degradation (reviewed in Balch et al. 2008; Hartl 1996; Morimoto et al. 1994). In cells exposed to hyperthermia, the induced synthesis of these proteins helps to prevent protein denaturation and aggregation and assists in the refolding or removal of damaged proteins (Stokoe et al. 1992). Induction of HSPs protects cells not only from damage due to heat but also from damage due to oxidative injury and cytokine-mediated cytotoxicity. Whether HSPs protect cells by blocking protein denaturation in general or whether a specific heat-sensitive target is protected is not known. The role of Hsp70 in blocking the apoptotic process has also been examined. Two decades ago, the participation of HSPs in apoptosis modulation was established for the first time (Franceschi 1989). The apoptotic cascade initiation is in part regulated by protein-protein interactions between death-promoting (Bax, Bad, and Bcl-xs) and inhibiting (Bcl-2, Bcl-xL, and Mcl-1) members of the Bcl-2 family (Nuñez and Clarke 1994; Reed 1994; Werner 1996).
Bcl-2-expressing cells resist apoptosis initiated by a number of physiological and stressful conditions including hyperthermia (Tsujimoto 1989). In prior heat stress in ATP-depleted renal tubular cells, the interaction between Hsp70 and Bcl-2 may be responsible, at least in part, for the protection afforded by Hsp70 against ATP depletion injury (Wang et al. 1999).
Given its localization within mitochondria and its role in preventing cytochrome c release, preservation of Bcl-2 by Hsp70 could account for the protection of epithelial cells (Borkan et al. 1993). Nevertheless, it has been proposed that HSPs also act by means of a mechanism independent of Bcl-2, intervening at several points to halt progression of the apoptotic cascade (Strasser and Anderson 1995). Previous studies have indicated that at least some of the antiapoptotic activity of Hsp70 can be attributed to its ability to suppress the activity of JUN-kinase (Kumar and Tatu 2003; Gabai et al. 1997). Activation of stress-activated protein kinase SAPK/c-Jun N-terminal kinase (JNK) has been strongly inhibited in cells in which Hsp70 was induced to a high level, indicating that Hsp70 blocks apoptosis by inhibiting signaling events upstream of SAPK/JNK activation. Hsp70 also inhibits apoptosis events at some point downstream of SAPK/JNK activation (caspase 3-mediated). Alternatively, Hsp70 may act by preventing cell death by interfering with the ability of cytochrome c and Apaf-1 to recruit pro-caspase 9. In this case, Hsp70 suppresses apoptosis by directly associating with Apaf-1 and blocking the assembly of a functional apoptosome (Beere et al. 2000).
4 Interaction between Nitric Oxide and Hsp70
A novel alternative antiapoptotic mechanism for NO is the induction of Hsp32 (heme oxygenase) and Hsp70, by means of NO-mediated modification in intracellular antioxidants levels (Mosser et al. 1997).
The mechanism by which NO stimulates the expression of Hsp70 may involve the interaction of NO with thiol-containing molecules. Ample evidence exists to support the view that NO readily oxidizes low molecular weight thiols, forming S-nitrosothiols and disulfide. Among cellular low molecular weight thiols, glutathione is the most abundant as well as being one of the intracellular targets of NO. NO can oxidize intracellular reduced glutathione and thereby change the antioxidant levels within the cell, resulting in oxidative or nitrosative stress. This action stimulates the induction of heat shock proteins Hsp32 (heme oxygenase) and Hsp70, which protect cells from apoptotic cell death induced by tumor necrosis factor (TNF) plus actinomycin D. (Kanner et al. 1991) and by oxidative or nitrosative stress (Harbrecht et al. 1994).
Pretreatment of hepatocytes with NO has been shown to alter the redox state accompanied by oxidation of glutathione (GSH) and by formation of S-nitrosoglutathione. A GSH-oxidizing agent (diamide) and a GSH alkylating agent (N-ethylmaleimide) both induced Hsp70 mRNA but a GSH synthesis inhibitor (buthionine sulfoximine) did not; this suggests that NO induces Hsp70 expression through GSH oxidation (Kim et al. 1997). The above-mentioned induction may occur via the activation of heat shock factor 1 (Xu et al. 1997). The accumulation of misfolded proteins causes the mobilization of the HSPs resulting in the free pool of Hsp70, and the subsequent removal of the negative regulatory influence on HSF activation, during heat shock or other stresses. The released HSF is phosphorylated and assembles into trimers, acquires DNA binding activity, and leads to elevated Hsp70 mRNA transcripts. During NO stimulation, multiple and complex pathophysiological changes occur in vascular smooth muscle cells, including protein damage or modifications due to the cytotoxic effect of NO (Lipton et al. 1993).
Thus, NO- and heat shock-induced Hsp70 production share many similarities in the activation of Hsf1 and in the regulation of Hsp70 gene expression.
The molecular mechanism underlying the antiapoptotic effects of NO-mediated HSP expression may be associated with two possibilities (Harbrecht et al. 1992). The first is the direct suppression of apoptotic signal transduction involving the inhibition of caspase family protease activation. The second involves the chaperon-mediated import of precursor proteins into mitochondria by HSPs. This action controls mitochondrial function and membrane permeability, thereby preventing the release of cytochrome c that is required for further activation of caspases.
A discussion of the relationship between Hsp70 and apoptosis induction during obstructive nephropathy was first given by Chan et al. (2001). Other results indicated that Hsp70 could modulate the apoptosis cascade during renal obstruction (Dmitrieva and Burg 2005; Manucha et al. 2005; Van de Water et al. 2006).
Recently, we have reported that nitric oxide prevents obstruction-induced cell death by means of the mitochondrial apoptotic pathway, through the induction of heat shock protein 70 (Manucha and Valles 2008). Our results showed that the apoptotic effect created by lower nitric oxide decreased Hsp70 expression. It was associated with the direct induction of the apoptotic signal transduction involving the activation of caspase 3 by decreasing stabilization of Bcl-2 (Fig. 3).

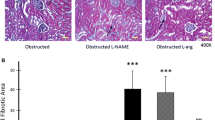
In vivo effect of L-Arginine and L-NAME treatment on Hsp70 protein expression in neonatal rats Representative Western blot and densitometry of Hsp70 in control and 14 days obstructed renal cortex homogenate from rats pretreated with L-Arginine or L-NAME. After L-Arginine-pretreatment for 14 days, a significant increase of inducible Hsp70 protein levels was noted in obstructed and control cortex homogenates related to cortex tissue of non-treated rats, ***p < 0.01 and **p < 0.001, respectively. Absence of Hsp70 expression was demonstrated after L-NAME treatment. Blot represents one out of four separate experiments. (Manucha and Vallés 2008)
Of the factors regulating the renal apoptotic response in renal injury that result from urinary tract obstruction, reactive oxygen species are known to reduce the threshold of tissues to undergo cell death (Kayanoki et al. 1996).
The neonatal obstructed kidney may be particularly susceptible to the generation of reactive oxygen species because endogenous renal antioxidant enzymes, including superoxide dismutase, are suppressed in the neonate (Gupta et al. 1999).
Overproduction of ROS has been identified as a key component of apoptotic pathways involving activation of endogenous endonucleases (Fernandez et al. 1995) and direct DNA fragmentation (Mertens et al. 1995).
Under normal physiological conditions, a balance between superoxide and nitric oxide exists in vivo. NO and superoxide react together at a diffusion-controlled rate to yield peroxynitrite (ONOO−), which inflicts cellular injury through oxidation of many biological molecules. Furthermore, ONOO− has been implicated in the inactivation of Mn and Fe superoxide dismutase (Ischiropoulos et al. 1992).
In contrast, NO may protect cells from reactive oxygen intermediate (ROI)-mediated cytotoxicity by scavenging superoxide anions which are implicated in toxicity through the formation of hydrogen peroxide or hydroxyl radical (Bautista and Spitzer 1994). Nitric oxide has been shown to inhibit superoxide anion generation. The mechanism for such inhibition is thought to be due to the inactivation of nicotinamide adenine dinucleotide phosphate-oxidase due to the scavenging effects of NO on superoxide (Clancy et al. 1992).
High levels of NO exposure induce protective stress responses, stimulating Hsp70 expression (Kim et al. 1997).
Induction of HSPs protects cells not only from damage due to heat but also from damage due to oxidative injury and cytokine-mediated cytotoxicity. Recently, we demonstrated that after 24 h of unilateral ureteral obstruction, protection against tubulointerstitial fibrosis by losartan, independent from changes in blood pressure, includes decreased oxidative stress linked to upregulation of Hsp70 expression (Manucha et al. 2005).
Previously, it has been shown that both ROI production and lipid peroxidation are inhibited by NO donor-induced Hsp70 expression. Furthermore, only cells overexpressing Hsp70 were found to be protected from both ROI- and TNF-induced cytotoxicity. Overexpression of Hsp27 only protected from exogenous ROI exposure but not from TNF cytotoxicity (Jäättelä et al. 1992; Jäättelä and Wissing 1993).
Recent data have shown that HSPs may protect from TNF toxicity by inhibiting the action of ROI on mitochondrial membrane potential (Cossarizza et al. 1995).
As has been discussed by Billiar et al., inhibition of TNFα toxicity by an NO donor pretreatment could occur through the inhibition of ROI production in mitochondria, preventing ROI-mediated alterations in mitochondrial membrane potential (Kim et al. 1997). This could prevent cytochrome c release, which is involved in apoptosis through activation of cysteine protease (Liu et al. 1996). Since Hsp70 is not a mitochondrial protein, however, it is unlikely that Hsp70 acts directly as a mitochondrial antioxidant. Hsp70 may instead block signal transduction to the mitochondria, resulting in the inhibition of mitochondrial ROI production either by inhibiting second lipid messenger(s) to the mitochondria (Jacquier-Sarlin et al. 1994) or by preventing the interaction between the death domain of TNFα receptor and signal molecule(s) (Hsu et al. 1995). Alternatively, it is possible that Hsp70 may enhance the chaperon-mediated import of precursor proteins into the mitochondria which control mitochondrial function and lead to decreased ROS formation (Harkness et al. 1994). On this matter, in a recent chapter of book, we present some of the factors identified along with proposed interactions (Fig. 4).

The principal proposed mechanisms of induction and inhibition of apoptosis in the kidney Pro-apoptotic effects: (1) accumulation of the tumor suppressor protein p53; (2) peroxynitrite formation; (3) caspase and pro-caspase activation. Anti-apoptotic effects: (1) redox state modulation; (2) inhibition of Cytochrome C release; (3) caspase activity inhibition; (4) cytoprotective stress proteins and cGMP-dependent protein kinase induction. (Vallés and Manucha 2008)
Taken together, our data demonstrate that the effect of the interaction of NO with Hsp70 is a result of the capacity of both to prevent the activation of the mitochondrial apoptotic pathway in neonatal early kidney obstruction. Induction of Hsp70 protects cells not only from damage due to apoptosis induction but also from damage due to oxidative injury. These findings demonstrate that NO can induce cytoprotection in early obstructed kidney cortex tubular epithelial cells through the stimulation of Hsp70 expression.
In conclusion, the accumulated data suggest that relevant levels of nitric oxide may contribute to apoptotic pathway suppression by the upregulation of Hsp70 and that interaction is an early line of defense for protecting cells from death. The induction of Hsp70 expression precedes conventional markers of renal injury, protecting cells not only from damage due to apoptosis induction but also from damage due to oxidative injury. Further studies will continue to elucidate the regulatory events of apoptosis induction in obstructive nephropathy.
References
Albina JE, Cui S, Mateo RB, Reichner JS (1993) Nitric oxide-mediated apoptosis in murine peritoneal macrophages. J Immunol 150:5080–5085
Balch WE, Morimoto RI, Dillin A, Nelly JW (2008) Adapting proteostasis for disease intervention. Science 319:916–919 DOI 10.1126/science.1141448
Bascands JL, Schanstra JP (2005) Obstructive nephropathy: insights from genetically engineered animals. Kidney Int 68:925–937 DOI 10.1111/j.1523-1755.2005.00486.x
Bautista AP, Spitzer JJ (1994) Inhibition of nitric oxide formation in vivo enhances superoxide release by the perfused liver. Am J Physiol 266:G783–G788
Beere HM, Wolf BB, Cain K, Tailor P, Morimoto RI, Cohen GM, Green DR (2000) Heat shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nature Cell Biology 2:469–475 DOI 10.1038/35019501
Borkan SC, Emami A, Schwartz JH (1993) Heat stress protein-associated cytoprotection in inner medullary collecting duct cells from rat kidney. Am J Physiol 265:F333–F341
Cachat F, Lange-Sperandio B, Chang AY, Kiley SC, Thornhill BA, Forbes M, Chevalier RL (2003) Ureteral obstruction in neonatal mice elicits segment-specific tubular cell responses leading to nephron loss. Kidney Int 63:564–575 DOI 10.1046/j.1523-1755.2003.00775.x
Campese VM, Park J (2007) HMG-CoA reductase inhibitors and the kidney. Kidney Int 71:1215–1222 DOI 10.1038/sj.ki.5002174
Chan W, Krieg RJ Jr, Ward K, Santos F Jr, Lin KC, Chan JC (2001) Progression after release of obstructive nephropathy. Pediatr Nephrol 16(3):238–244 DOI 10.1007/s004670000519
Chevalier RL, Chung KH, Smith CD, Ficenec M, Gomez RA (1996) Renal apoptosis and cluster in following ureteral obstruction: the role of maturation. J Urol 156:1474–1479 DOI 10.1016/S0022-5347(01)65633-7
Chevalier RL (1999) Molecular and cellular pathophysiology of obstructive nephropathy. Pediatr Nephrol 13:612–619 DOI 10.1007/s004670050756
Chevalier RL, Thornhill BA (1995) Ureteral obstruction in the neonatal rat: renal nerves modulate hemodynamic effects. Pediatr Nephrol 9:447–450 DOI 10.1007/BF00866725
Chevalier RL (2006) Obstructive nephropathy: towards biomarker discovery and gene therapy. Nat Clin Pract Nephrol 2(3):157–168 DOI 10.1038/ncpneph0098
Clancy RMJ, Leszcznska-Piziak J, Abramson SB (1992) Nitric oxide, and endothelial cell relaxation factor inhibits neutrophils superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest 90:1116–1121 DOI 10.1172/JCI115929
Cossarizza A, Cooper EL, Quaglino D, Salvioli S, Kalachnikova G, Franceschi C (1995) Mitochondrial mass and membrane potential in coelomocytes from the earthworm Eisenia foetida: studies with fluorescent probes in single intact cells. Biochem Biophys Res Commun 214(2):503–510 DOI 10.1006/bbrc.1995.2315
Dmitrieva NI, Burg MB (2005) Hypertonic stress response. Mutat Res 569(1–2):65–74 DOI 10.1016/j.mrfmmm.2004.06.053
Fernandez A, Kiefer J, Fosdick L, McConkey DJ (1995) Oxygen radical production and thiol depletion are required for Ca(2)-mediated endogenous endonuclease activation in apoptotic thymocytes. J Immunol 155:5133–5139
Franceschi C (1989) Cell proliferation, cell death and aging. Aging (Milano) 1:3–15
Freedman DA, Folkman J (2004) Maintenance of G1 checkpoint controls in telomerase immortalized endothelial cells. Cell Cycle 6:811–816
Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY (1997) Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem 272:18033–18037 DOI 10.1074/jbc.272.29.18033
Gupta A, Nigam D, Shukla GS, Agarwal AK (1999) Profile of reactive oxygen species generation and antioxidative mechanisms in the maturing rat kidney. J Appl Toxicol 19:55–59 DOI 10.1002/(SICI)1099–1263(199901/02)19:1<55::AID-JAT538>3.0.CO;2-K
Harbrecht BG, Billiar TR, Stadler J, Demetris AJ, Ochoa J, Curran RD, Simmons RL (1992) Inhibition of nitric oxide synthesis during endotoxemia promotes intrahepatic thrombosis and an oxygen radical-mediated hepatic injury. J Leukoc Biol 52:390–394
Harbrecht BG, Stadler J, Demetris AJ, Simmons RL, Billiar TR (1994) Nitric oxide and prostaglandins interact to prevent hepatic damage during murine endotoxemia. Am J Physiol 266:G1004–G1010
Harkness TAA, Nargang FE, van der Klei I, Neupert W, Lill R (1994) A crucial role of the mitochondrial protein import receptor MOM19 for the biogenesis of mitochondria. J Cell Biol 124:637–648 DOI 10.1083/jcb.124.5.637
Hartl FU (1996) Molecular chaperones in cellular protein folding. Nature 381:571–580 DOI 10.1038/381571a0
Hegarty NJ, Young LS, Kirwan CN, O'Neill AJ, Bouchier-Hayes DM, Sweeney P, Watson RW, Fitzpatrick JM (2001) Nitric oxide in unilateral ureteral obstruction: effect on regional renal blood flow. Kidney Int 59:1059–1065 DOI 10.1046/j.1523-1755.2001.0590031059.x
Hsu H, Xiong J, Goeddel DV (1995) The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81:495–504 DOI 10.1016/0092-8674(95)90070-5
Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS (1992) Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 298(2):431–437 DOI 10.1016/0003-9861(92)90431-U
Ito K, Chen J, Seshan SV et al (2005) Dietary arginine supplementation attenuates renal damage after relief of unilateral ureteral obstruction in rats. Kidney Int 68(2):515–528 DOI 10.1111/j.1523–1755.2005.00429.x
Jäättelä M, Wissing D, Bauer PA, Li GC (1992) Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J 11:3507–3512
Jäättelä M, Wissing D (1993) Heat-shock proteins protect cells from monocyte cytotoxicity: possible mechanism of self-protection. J Exp Med 177(1):231–236 DOI 10.1084/jem.177.1.231
Jacquier-Sarlin MR, Fuller K, Dinh-Xuan AT, Richard MJ, Polla BS (1994) Protective effects of Hsp70 in inflammation. Experientia (Basel) 50:1031–1038
Kanner J, Harel S, Granit R (1991) Nitric oxide as an antioxidant. Arch. Biochem. Biophys 289:130–136 DOI 10.1016/0003-9861(91)90452-O
Kasinath BS, Mariappan MM, Sataranatarajan K, Lee MJ, Feliers D (2006) mRNA translation: unexplored territory in renal science. J Am Soc Nephrol 12:3281–3292 DOI 10.1681/ASN.2006050488
Kawada N, Moriyama T, Ando A, Fukunaga M, Miyata T, Kurokawa K, Imai E, Hori M (1999) Increased oxidative stress in mouse kidneys with unilateral ureteral obstruction. Kidney Int 56:1004–1013 DOI 10.1046/j.1523-1755.1999.00612.x
Kayanoki Y, Fujii J, Islam KN, Suzuki K, Kawata S, Matsuzawa Y, Taniguchi N (1996) The protective role of glutathione peroxidase in apoptosis induced by reactive oxygen species. J Biochem (Tokyo) 119:817–822
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer 4:239–257
Kim YM, de Vera ME, Watkins SS, Billiar TR (1997) Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem 272:1402–1411 DOI 10.1074/jbc.272.2.1402
Kim Y, Bombeck CH, Billiar TR (1999) Nitric oxide as a bifunctional regulator of apoptosis. Circulation Research 84:253–256
Kim YM, Kim TH, Seol DW, Talanian RV, Billiar TR (1998) Nitric oxide suppression of apoptosis occurs in association with an inhibition of Bcl-2 cleavage and cytochrome c release. J Biol Chem 273:31437–31441 DOI 10.1074/jbc.273.47.31437
Kim YM, Talanian RV, Billiar TR (1997) Nitric oxide inhibits apoptosis by preventing increases in caspase-3-like activity via two distinct mechanisms. J Biol Chem 272:31138–31148 DOI 10.1074/jbc.272.49.31138
Kumar Y, Tatu U (2003) Stress protein flux during recovery from simulated ischemia: induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics 3:513–526 DOI 10.1002/pmic.200390065
Lipton SA, Choi YB, Pan ZH et al (1993) A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364:626–632 DOI 10.1038/364626a0
Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86(1):147–157 DOI 10.1016/S0092-8674(00)80085-9
Mannick JB, Asano K, Izumi K, Kieff E, Stamler JS (1994) Nitric oxide produced by human B lymphocytes inhibits apoptosis and Epstein-Barr virus reactivation. Cell 79:1137–1146 DOI 10.1016/0092-8674(94)90005-1
Manucha W, Carrizo L, Ruete C, Molina H, Vallés P (2005) Angiotensin II type I antagonist on oxidative stress and heat shock protein 70 (Hsp70) expression in obstructive nephropathy. Cell Mol Biol 51(6):547–555
Manucha W, Carrizo L, Ruete C, Vallés P (2007) Apoptosis induction is associated with decreased NHE1 expression in neonatal unilateral ureteral obstruction. BJU Int 1:191–198 DOI 10.1111/j.1464-410X.2007.06840.x
Manucha W, Vallés P (2008) Cytoprotective role of nitric oxide associated with Hsp70 expression in neonatal obstructive nephropathy. Nitric Oxide 18(3):204–215
Mertens JJ, Gibson NW, Lau SS, Monks TJ (1995) Reactive oxygen species and DNA damage in 2-bromo-(glutathion-Syl) hydroquinone-mediated cytotoxicity. Arch Biochem Biophys 320:51–58 DOI 10.1006/abbi.1995.1341
Miyajima A, Chen J, Poppas DP, Darracott Vaughan Jr ED, Felsen D (2001) Role of nitric oxide in renal tubular apoptosis of unilateral ureteral obstruction. Kidney Int 59:1290–1303 DOI 10.1046/j.1523-1755.2001.0590041290.x
Morimoto RI, Tissieres A, Georgopoulos C (1994) The biology of heat shock proteins and molecular chaperones. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B (1997) Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis. Mol Cell Biol 17:5317–5327
Nagai-Kusuhara A, Nakamura M, Mukuno H, Kanamori A, Negi A, Seigel GM (2007) cAMP responsive element binding protein mediates a cGMP/protein kinase G-dependent antiapoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp Eye Res 84:152–162 DOI 10.1016/j.exer.2006.09.010
Nguyen HT, Bride SH, Badawy AB et al (2000) Heparin-binding EGF-like growth factor is up-regulated in the obstructed kidney in a cell- and region-specific manner and acts to inhibit apoptosis. Am J Pathol 156:889–898
Nuñez G, Clarke MF (1994) The Bcl-2 family of proteins: regulators of cell death and survival. Trends Cell Biol 4:399–403 DOI 10.1016/0962-8924(94)90053-1
Reed JC (1994) Bcl-2 and the regulation of programmed cell death. J. Cell Biol 124:1–6 DOI 10.1083/jcb.124.1.1
Sola A, Alfaro V, Vinas JL, Hotter G (2004) Exogenous adenosine enhances caspase-3 activity in warm renal ischaemia. Pflugers Arch 447(4):387–391 DOI 10.1007/s00424-003-1197-6
Stokoe D, Engel K, Campbell DG, Cohen P, Gaestel M (1992) Identification of MAPKAP kinase 2 as a major enzyme responsible for the phosphorylation of the small mammalian heat shock proteins. FEBS Lett 313:307–313 DOI 10.1016/0014-5793(92)81216-9
Strasser A, Anderson RL (1995) Bcl-2 and thermotolerance cooperate in cell survival. Cell Growth Differ 6:799–805
Tsujimoto Y (1989) Stress-resistance conferred by high level of Bcl-2 protein in human B lymphoblastoid cell. Oncogene 4:1331–1336
Vallés P, Jorro F, Carrizo L, Manucha W, Oliva J, Cuello-Carrión FD, Ciocca DR (2003) Heat shock proteins Hsp27 and Hsp70 in unilateral obstructed kidneys. Pediatr Nephrol 18(6):527–535
Vallés P, Manucha W (2008) Nitric oxide in the kidney: physiological roles and regulation. In: Gimenez MS, Gomez NM (eds) Advances in chemistry and biology of nitric oxide. Research Signpost, Kerala, pp 1–19
Van de Water B, de Graauw M, Le Dévédec S, Alderliesten M (2006) Cellular stress responses and molecular mechanisms of nephrotoxicity. Toxicol Lett 162(1):83–93 DOI 10.1016/j.toxlet.2005.10.014
Wang Y, Knowiton AA, Christensen TG (1999) Prior heat stress inhibits apoptosis in adenosine triphosphate-depleted renal tubular cells. Kidney Int 55:2224–2235 DOI 10.1046/j.1523-1755.1999.00476.x
Werner MH (1996) Stopping death cold. Structure 4:879–883 DOI 10.1016/S0969-2126(96)00094-9
Xu Q, Hu Y, Kleindienst R, Wick G (1997) Nitric oxide induces heat-shock protein 70 expression in vascular smooth muscle cells via activation of heat shock factor 1. J Clin Invest 100:1089–1097 DOI 10.1172/JCI119619
Yang J, Liu Y (2003) Delayed administration of hepatocyte growth factor reduces renal fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 284:F349–F357
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Manucha, W., Vallés, P. Hsp70/nitric oxide relationship in apoptotic modulation during obstructive nephropathy. Cell Stress and Chaperones 13, 413–420 (2008). https://doi.org/10.1007/s12192-008-0050-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12192-008-0050-4