
Abstract
Acute myeloid leukemia (AML) cells are highly dependent on oxidative phosphorylation and the mitochondrial dynamics regulated by fusion-related genes MFN1, MFN2, and OPA1 and fission-related genes DNM1L and MFF. An analysis of previously published gene expression datasets showed that high expression of MFF was significantly associated with poor prognosis in patients with AML. Based on this finding, we investigated the impact of mitochondrial dynamics in AML. Transduction of shRNA against fission-related genes, DNM1L and MFF, inhibited growth and increased the mitochondrial area in AML cell lines. Extracellular flux analysis showed that deletion of mitochondrial dynamic regulators reduced mitochondrial respiration without significantly affecting glycolysis, except in shDNM1L-transfected cells. Immunodeficient NOG mice transplanted with DNM1L- or MFF-knockdown AML cells survived significantly longer than controls. Treatment of AML cell lines with Mdivi-1, which inhibits the DRP1 encoded by DNM1L, inhibited cell proliferation and oxidative phosphorylation. Our results show that mitochondrial dynamics play an important role in AML, and provide novel biological insights. The inhibition of mitochondrial dynamics induces unique mitochondrial alterations, which may be explored as a potential therapeutic target in AML.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous group of neoplastic disorders characterized by clonal proliferation of primitive stem or progenitor hematopoietic cells [1]. The standard therapy for AML involves induction and consolidation chemotherapy with cytarabine and anthracyclines [2]. Despite intensive chemotherapy, the overall cure rate is low, particularly in patients with relapsed or refractory disease, highlighting the need for novel therapeutic approaches for AML.
Recently, cancer metabolism has emerged as a potential therapeutic target for many types of cancers, including leukemia. Mitochondria are crucial bioenergetic hubs that control metabolic pathways, including the TCA cycle, fatty acid oxidation, electron transport chain, and oxidative phosphorylation (OXPHOS), and play a key role in metabolic reprogramming involved in tumorigenesis, growth, and resistance to therapeutic agents in cancers [3, 4]. AML cells have unique mitochondrial alterations that represent a targetable therapeutic vulnerability [5, 6]. Mitochondria are highly dynamic organelles that continually fuse and divide (referred to as ‘mitochondrial dynamics’) in response to diverse stimuli [7, 8]. Mitochondrial fusion is regulated by three large GTPases: mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy gene 1 (OPA1). Mitochondrial fission, which conflicts with mitochondrial fusion, is primarily regulated by dynamin-related protein 1 (DRP1) encoded by the DNM1L gene. DRP1 is recruited to the outer mitochondrial membrane by several mitochondria-bound proteins, including mitochondrial fission factor (MFF) [9]. AML cells are highly dependent on OXPHOS and require the tight regulation of mitochondrial dynamics by fusion- and fission-related genes [10]. Therefore, it is possible that impairment of mitochondrial plasticity due to aberrant fission or fusion contributes to the metabolic vulnerability of AML cells. Recently, several studies have suggested that targeting mitochondrial dynamics, particularly mitochondrial fusion, could improve the therapeutic response in hematologic malignancies [11, 12]. However, the mechanism through which mitochondrial fission affects AML biology remains unclear. In this study, we investigated the impact of mitochondrial dynamics on AML cells and found that aberrant fission affected cell proliferation and OXPHOS. Treatment of AML cell lines with mitochondrial division inhibitor-1 (Mdivi-1) [13, 14], which inhibits DRP1, reduces cell proliferation and OXPHOS in AML cell lines. Our results show that mitochondrial fission plays an important role in AML and provide novel biological insights. The inhibition of mitochondrial fission induces unique mitochondrial alterations, leading to a potential therapeutic target for AML.
Materials and methods
Cell lines, reagents, and antibodies
U937, THP-1, K562, HEL, NB4, MOLM1, MOLM13, MOLM14, MV4-11, and UCSD/AML1 cells were cultured in RPMI1640 supplemented with 10% FCS. MV4-11 was purchased from ATCC (Manassas, VA, USA). THP-1, U937, and K562 were purchased from JCRB Cell Bank (Japanese Collection of Research Bioresources Cell Bank). MOLM13 and MOLM14 cells were gifts from Dr. Yusuke Furukawa (Jichi Medical University, Japan). Human bone marrow CD34 progenitor cells (Lonza K.K, Kanagawa, Japan) were cultured in X-vivoTM20 supplemented with SCF to a final concentration of 25 ng/mL, TPO to 50 ng/mL, and FLT-3 to a concentration of 50 ng/mL. The proliferation of leukemia cells was measured using a Cell Counting Kit-8 (CCK-8, Dojindo, Kumamoto, Japan). Stock solutions of Mdivi-1 (Selleckchem, Houston, TX, USA) and the selective OPA1 inhibitor, MYLS22 (Selleckchem) were prepared in dimethyl sulfoxide (DMSO), and serial dilutions were prepared in the culture medium prior to each experiment. The final DMSO concentration was less than 0.2% in all experiments. Cells in the logarithmic growth phase (1 × 104 cells/mL) were treated with different chemical concentrations.
MTT assay
Cell viability was evaluated by CCK-8 assay. Cells were plated in 96-well clear-bottom plates at a cell density of 104 cells/well and incubated at 37 °C for 1–5 days. Before the measurement, 10 µL of CCK-8 was added to each well. After 2.5 h of incubation, luminescence was measured at 460 nm using a microplate reader.
Quantitative real-time PCR
RNA was isolated from leukemia cells using the RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany). cDNA was synthesized using random primers and SuperScript VILO (Thermo Fisher Scientific, Waltham, MA). Quantitative PCR was performed using SYBR qPCR Mix (TOYOBO, Osaka, Japan) and Step One Plus (Thermo Fisher Scientific).
GEO data analysis
The gene expression profiles of 12,417 [15] were downloaded from the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database [16].
Lentiviral transduction of AML cells with shRNA
pLKO.1 puro was a gift from Dr. Bob-Weinberg (Addgene plasmid #8453). Control shRNA (scramble) was a gift from Dr. David Sabatini (Addgene Plasmid #1864). The shRNA oligonucleotides are provided in the Supplemental Table 1. pMD2.G-VSV-G and psPAX2-Gag-Pol plasmids were purchased from Addgene. On day 2 post-transduction, cells were selected by exposure to puromycin (1 μg/mL; Sigma-Aldrich) for 4 days. Knockdown efficiency was analyzed by measuring mRNA levels using quantitative PCR on day 4.
Transmission electron microscopy
Transmission electron microscopy (TEM) was performed, as previously reported [17], on THP-1 cells transduced with shRNA vectors targeting mitochondrial dynamics-related genes to investigate whether the inhibition of the regulators of mitochondrial dynamics affects mitochondrial morphology. Quantification of the mitochondrial volume was performed by first drawing the contour of the cross-section of each mitochondrion and then calculating the enclosed area using ImageJ.
Flow cytometry
ROS levels were detected using CellROX Deep Red Reagent (Invitrogen) or MitoSOX (Invitrogen). MitoTracker (Invitrogen) was used to assess mitochondrial mass. Mitophagy was measured using a mitophagy detection kit (Dojindo). The purpose of these methods was to determine whether the dysfunction of mitochondrial dynamics was linked to oxidative stress.
Apoptosis assay
Apoptosis assays were performed using the APC Annexin V Apoptosis Detection Kit (BioLegend, San Diego, USA) according to the manufacturer's protocol. Leukemia cells were incubated with 5 µL of Annexin-V and 10 µL of propidium iodide (PI) in 100 µL Annexin V Binding Buffer for 15 min at room temperature in the dark.
Cell cycle analysis
Leukemia cells were cultured in complete medium containing 10 μM BrdU at 37 °C for 60 min. And then fixed and stained with 7AAD and an allophycocyanin-conjugated (APC) anti-BrdU antibody using the BrdU Flow Kit (BD Pharmingen, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions.
Measurement of ECAR and OCRs
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using an XFp analyzer and Seahorse Mito stress test kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s protocol. The detailed procedure has been published previously [18].
Retroviral BM transduction assays
pMIG-FLAG-MLL-AF9 was a gift from Daisuke Nakada (Addgene; plasmid #71,443). G3T-hi cells (Takara Bio, Shiga, Japan) were transiently transfected with MSCV vectors and the pCL-Eco retrovirus packaging vector using polyethylenimine (PEI). C57BL6J mice were injected with 5-fluorouracil (150 mg/kg; Kyowa Kirin, Japan) and after 4 days, the BM cells were harvested and incubated for 24 h in X-Vivo 15 (Lonza, Allendale, NJ) supplemented with 50 ng/mL SCF, 50 ng/mL TPO, 10 ng/mL IL-3, and 10 ng/mL IL-6 (Peprotech, Rocky Hill, NJ, USA). After incubation, cells in plates coated with RetroNectin (Clontech, Mountain View, CA) were spin-infected (490 g for 45 min at 20 °C) with retroviral supernatant supplemented with polybrene (8 μg/mL). Fifty thousand BM cells were transplanted into the lethally irradiated C57BL/6 mice.
Colony-forming assay
Leukemia cells were plated in MethoCult M3234 medium (STEMCELL Technologies) supplemented with 20 ng/mL SCF, 10 ng/mL GM-CSF, 10 ng/mL IL-3, and 10 ng/mL IL-6, according to the manufacturer's instructions.
RNA sequencing (RNA-seq)
We performed RNA-seq on THP-1 cells transduced with shScr, shDNM1L, or shMFF to understand how the inhibition of fission-related genes suppresses leukemia growth and assess the associated changes in gene expression. For the RNA-seq experiments, poly(A)-selected libraries were prepared using the NEBNext Poly (A) mRNA Magnetic Isolation Module (NEBE7490) and NEBNext Ultra RNA Library Prep Kit for Illumina (E7530). All libraries were sequenced on an Illumina NovaSeq 6000 system using 2 × 150 bp paired-end sequencing. DESeq2 was used for data processing, normalization, and differential expression analysis.
AML xenograft tumors
Female NOG mice (8-week-old) received tail vein injection of THP-1 cells (2 × 106) suspended in 100 µL X-VIVO. From day 14 after BMT, NOG mice were injected daily with 2.5 mg/kg mdivi-1 (Selleckchem), or DMSO vehicle intraperitoneally for 3–5 weeks (5 days a week). All procedures were approved by the Animal Care and Use Committee of the University of Miyazaki.
Statistical analysis
Statistical analysis was performed using the unpaired Student t- or log-rank test, as appropriate. Calculations were performed using GraphPad Prism 8. Group data are expressed as the mean ± standard deviation. Values of P < 0.05 were considered statistically significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Results
Mitochondrial dynamics is crucial for AML biology
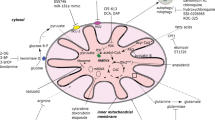
The mitochondria constantly undergo dynamic fission and fusion to maintain their function and integrity (Fig. 1A). We analyzed the relevance of the expression of the regulators of mitochondrial dynamics for the survival of patients with AML in previously published gene expression data from GEO datasets. High expression of MFF in AML was significantly associated with a poor prognosis (Fig. 1B, C). However, the expression of DNM1L, OPA1, MFN1, and MFN2 were not associated with AML prognosis. The expression levels of mitochondrial dynamics-related genes were confirmed in human CD34 progenitor cells and various AML cell lines (Fig. 1D). There were no genes consistently highly expressed in AML cell lines, despite a higher expression of mitochondrial dynamics-related genes in THP-1 compared to that in the CD34+cells. There was no difference in the mitochondrial size between CD34+cells and THP-1 (Supplementary Fig. 1A). Next, we knocked down mitochondrial fission- or fusion-related genes in THP-1 and MOLM13 cells (Supplementary Fig. 1B). Transduction of shRNA against the fission-related genes DNM1L or MFF induced growth inhibition relative to the shScramble (shScr) control in both cell lines (Fig. 1E). Although knockdown of the fusion-related genes OPA1, MFN1, and MFN2 did not show growth inhibition common to both AML cell lines, depletion of MFN1 or OPA1 suppressed either one of the AML cell lines (Supplementary Fig. 1C). These results suggest that mitochondrial dynamics-related genes contribute to AML cell proliferation.

Mitochondrial dynamics is crucial for acute myeloid leukemia (AML) biology. A Simplified model of mitochondrial fusion and fission in human cells. Mitochondrial fusion combines two mitochondria together, whereas fission separates one into two. Fusion is coordinated by the MFN1, MFN2, and OPA1. Fission is regulated by DNM1L and MFF. B, C Kaplan–Meier survival curves analyzed using PrognoScan: GSE12417-GPL570 showed that high expression of MFF predicted reduced overall survival in AML. D Expression level of mitochondrial dynamics related genes in human CD34 cells and AML cell lines. Expression was normalized against the corresponding level of β-actin. E Proliferation of THP-1 and MOLM13 cells transduced with shScr, shDNM1L, or shMFF, respectively
Inhibition of mitochondrial dynamics disrupts mitochondrial oxidative metabolism
TEM images revealed that the cross-sections of the mitochondria demonstrated a significantly larger area in THP-1 cells with fission-related genes knockdown (Fig. 2A–D). In contrast, inhibition of the mitochondrial fusion genes did not change the mitochondrial size except for shMFN1#2 transfected cells which significantly increased mitochondrial size (Fig. 2D). As shown in Fig. 2E–G, the ROS production in DRP1 or MFF depletion cells was comparable to that in the control cells. In contrast, MFN1, MFN2, and OPA1 depletion significantly increased ROS production compared to that in the control (Fig. 2E–G). The loss of MFF induced significant mitophagy defects in AML cells (Fig. 2H). To evaluate the anti-leukemic mechanism mediated by mitochondrial dynamics inhibition, we investigated the cell cycle distribution and apoptosis in AML cells. As a result, the cells in G0/G1 phase increased markedly only with OPA1 depletion (Supplemental Fig. 2). Neither inhibition of mitochondrial fission nor fusion induced apoptosis in AML cells (Supplemental Fig. 3). We further analyzed the OCR and ECAR using a Seahorse XF analyzer to assess mitochondrial respiration and glycolysis. The results demonstrated reduced mitochondrial respiration by the deletion of mitochondrial dynamics regulators without significantly affecting glycolysis, except in shDNM1L transfected cells, which showed a significant metabolic shift toward glycolysis (Fig. 2I, J).

Inhibition of mitochondrial dynamics disrupts mitochondrial oxidative metabolism. Representative transmission electron microscope (TEM) images showing the morphology of mitochondria in THP-1 transduced with shScr (A), shDNM1L (B), or shMFF (C), respectively. D Quantification of the mitochondrial cross-section area from the TEM images of THP-1. Each dot represents a single mitochondrion. Flow-cytometry analysis of Mitotracker (E), CellROX (F), MitoSOX (G), and Mitophagy (H). Each symbol represents an individual sample. Data are collected in at least four experiments per condition. I Energy metabolism was analyzed using the XF extracellular flux analyzer. Basal and maximum oxygen consumption rate (OCR) in mitochondria were much lower in THP-1 transduced with shRNA against mitochondrial dynamics-related genes. J The OCR/extracellular acidification rate (ECAR) ratio in THP-1 transduced with shRNA against mitochondrial dynamics-related genes. shDNM1L transfected cells showed a significant metabolic shift toward glycolysis compared to control cells. Data are represented by the mean ± SD. *P < 0.05; **P < 0.01; ****P < 0.001

Limiting mitochondrial fission suppresses KMT2A-MLLT3 leukemia growth and oxidative phosphorylation in ex vivo models. A Schematic outline of KMT2A-MLLT3 mouse model. KMT2A-MLLT3 cells transduced with shScr, shDnm1l or shMff were analyzed. B Leukemia cells infected with shRNA were cultured in methylcellulose medium. Graph shows the number of colonies and cell counts of KMT2A-MLLT3 cells. C The basal and maximum oxygen consumption rate (OCR) of DNM1L or MFF depleted cells were lower than those of control cells. D No difference was observed in OCR/ECAR ratio between DNM1L or MFF depleted and control cells. All data are expressed as the mean ± standard deviation. **P < 0.005 (Two-tailed t-test.). E Flow-cytometry analysis of CellROX in DNM1L or MFF depleted and control cells
Limiting mitochondrial fission suppresses KMT2A-MLLT3 leukemia growth and oxidative phosphorylation ex vivo
To examine whether mito-fission regulators influence energy metabolism in KMT2A-rearranged leukemia cells, we used a murine model in which AML was driven by the KMT2A-MLLT3 oncogene (Fig. 3A). KMT2A-MLLT3 leukemia cells from murine bone marrow were transduced with shRNA against Dnm1l or Mff and serial colony-forming assays were performed. We observed significant inhibition of cell proliferation and colony-forming ability in mice receiving DNMlL- or MFF-depleted AML cells (Fig. 3B). Measurements of energy metabolism showed reduced OCR in DNM1L- or MFF-deficient AML cells without a metabolic shift to the glycolytic system compared to that in the control cells (Fig. 3C, D). These results show that fission-related genes are important for energy production and cell proliferation in leukemia models of different species. To further evaluate the anti-leukemic mechanism mediated by mitochondrial fission inhibition, we measured the level of cellular ROS, apoptosis and cell cycle distribution by using flowcytometry. Knockdown of Dnm1L or Mff did not induce increased ROS, apoptosis or cell cycle arrest in ex vivo AML mouse models (Fig. 3E, Supplementary Fig. 4A, B). Therefore, anti-proliferative effect of inhibition of mitochondrial fission may be primarily due to the metabolic inhibition rather than the increased ROS production or apoptosis in AML cells.
Inhibition of mitochondrial fission regulates the transcriptome of leukemia cells
The volcano plot of RNA-seq data revealed that 65 genes were upregulated and 100 genes were downregulated by >twofold in shDNM1L compared to that in shScr (P < 0.05), and 77 genes were upregulated and 21 genes were downregulated by >twofold in shMFF compared to that in shScr (P < 0.05) (Fig. 4A, B; Supplemental Table 2). Three genes (FP236383.3, CHST2, and GAS6) were upregulated, and four genes (MTX1P1, KRT8, KIFC1, and XIRP1) were downregulated in the shDNM1L and shMFF cell lines (Fig. 4C). Gene set enrichment analysis was used to determine the extent to which the expression of metabolic target genes changed; however, no gene set common to the two groups was found (Supplemental Table 3). These results revealed that although shDNM1L and shMFF share common features, such as mitochondrial morphology, cell proliferation inhibition, and OXPHOS inhibition, they have different gene expression profiles.

Volcano plot showing the log2FC up- and downregulated genes for shDNM1L vs. shScr (A) and shMFF vs. shScr transfected cells (B). Upregulated and downregulated genes that are common between shDNM1L and shMFF are shown in red and blue, respectively. C Venn diagram showing the up- and downregulated gene overlap between shDNM1L and shMFF. D Kaplan–Meier curves of mice survival were established for AML cell lines with knockdown of DNM1L and MFF, engrafted in NOG mice. Significance was determined using log-rank tests (**P < 0.01)
Disruption of mitochondrial fission contributes to prolonged survival in AML mice model
Next, we examined the effects of mitochondrial fission inhibition in vivo using NOG mice. Mice were transplanted with THP-1 cells transduced with shScr, shDNM1L, or shMFF, observed for survival. Mice transplanted with DRP1- and MFF-deleted cells showed significantly increased survival compared to that with controls (Fig. 4D). These results demonstrate that the inhibition of fission-related genes can suppress leukemia cell growth in an AML xenograft model.
Mdivi-1 disrupts AML proliferation via oxidative metabolism inhibition in vitro
To determine whether pharmacological inhibition of mitochondrial dynamics inhibits OXPHOS, we treated AML cell lines with Mdivi-1 or MYLS22, selective inhibitors of OPA1. Mdivi-1 had a more reduced cell viability and anti-proliferative effect at lower concentrations than MYLS22 (Fig. 5A–C). The addition of Mdivi-1 suppressed OCR in the AML cell lines THP-1 and MOLM13 in a concentration-dependent manner (Fig. 5D). These results showed that Mdivi-1 reduced OCR and inhibited proliferation, similar to DNML1L knockdown. To evaluate the effect of Mdivi-1 in vivo, we treated NOG mice transplanted with THP-1 with Mdivi-1 or vehicle, however, there was no difference in survival between the two groups (Fig. 5E, F).

Mdivi-1 disrupts AML proliferation via oxidative metabolism inhibition in vitro. The in vitro half maximal inhibitory concentration (IC50) of Mdivi-1 (A) or MYSL22 (B) for the growth of AML cell lines was calculated for each cell line. C Proliferation of THP-1, MOLM13, and K562 incubated with vehicle or 10–50 μM of Mdivi-1, respectively. D The basal OCR and maximal respiratory capacity in Mdivi-1 treated cells were significantly lower than those of vehicle-treated cells. E Schema of Mdivi-1 treatment in NOG mice transplanted with THP-1. F Survival of NOG mice treated with vehicle (n = 6) or Mdivi-1 (n = 5). Treatment was started 2 weeks after BMT. ns not significant
Discussion
Strategies for targeting the mitochondria in cancer treatment are based on the concept that cancer cells critically depend on alterations in their cellular metabolism [3]. As exemplified by the aerobic glycolysis proposed by Warburg [19], cancer cells reprogram their metabolism in response to the tumor microenvironment. Mitochondria can reprogram their size, shape, and localization in response to metabolic demands and are involved in various biological processes, such as the cell cycle, stress response, and cell death [20,21,22]. Recent evidence indicates that mitochondrial dynamics could also contribute to the tumorigenesis of many cancers, including myeloid disorders [21,22,23,24]. Mitochondrial fragmentation and increased DNM1L expression have been detected in several cancer cell lines and in patient samples. Furthermore, silencing of DNM1L in several cancer models inhibited tumor growth and improved survival outcomes [25,26,27]. In contrast, analyses of OPA1 RNA-seq data showed significant mRNA overexpression in a wide range of tumor types, and OPA1 inhibition reduced breast cancer cell proliferation, migration, and invasion in the in vitro and in vivo models [28]. Mitochondrial fission and fusion are crucial for maintaining cancer cell proliferation and survival depending on the situation, indicating that this relationship is not simple.
In hematological diseases, several studies clearly indicate that targeting mitochondrial dynamics could be a promising therapeutic strategy [11, 12, 17, 24, 29]. A recent study suggested that the genetic depletion of mitochondrial fusion genes or pharmacological inhibition of OPA1 (MYLS22) had a significant anti-leukemic activity in AML [11]. However, the biological significance of the mitochondrial dynamics in AML remains unclear. In the present study, we demonstrated the potential therapeutic targeting role of mitochondrial fission in AML. Using previously published gene expression datasets, we found that high expression of MFF was significantly associated with poor prognosis in patients with AML. To determine the role of mitochondrial fission in AML cells, we investigated the impact of genetic depletion or pharmacological inhibition of DRP1 and MFF. The inhibition of mitochondrial fission showed an anti-leukemic effect, with mitochondrial morphological changes and the suppression of OXPHOS. These findings differ from those previously reported by Larrue et al. [11], who reported that the depletion of mitochondrial fission had a limited impact on AML cells. The cause of this discrepancy is unknown; however, the results may vary depending on the type of cell line used. Intracellular and mitochondrial ROS production was unchanged with mitochondrial fission inhibition but was promoted upon mitochondrial fusion depletion. In HCC cell lines, DNM1L overexpression or MFN1 knockdown was associated with ROS overproduction, whereas DNM1L knockdown or MFN1 overexpression attenuated ROS generation [30]. Our results regarding the relationship between mitochondrial dynamics and ROS production are consistent with those of the previous study. In addition, genetic depletion of DNM1L and MFF did not induce cell cycle arrest and apoptosis. Therefore, the anti-proliferative effect of inhibition of mitochondrial fission may be primarily due to the metabolic inhibition rather than the increased ROS production, cell cycle arrest, or apoptosis in AML cells. Conversely, OPA1 inhibition increased ROS production and induced G1 arrest. This result was consistent with a previous report that suggested a significantly increased proportion of G1 with depletion of OPA1 in AML cell lines [11], and the anti-leukemic effect of OPA1 could be due to cell cycle arrest.
Additionally, knockdown of MFF induced suppressed mitophagy in vitro. In immortalized human fibroblasts, overexpression of MFF induced extensive mitochondrial fragmentation with the concomitant activation of mitophagy [31]. Thus, MFF depletion may induce dysregulated mitophagy, resulting in the accumulation of damaged mitochondria. A metabolic shift towards glycolysis was observed in only the silencing of DNM1L. In general, overexpression of DNM1L leads to mitochondrial fragmentation and a metabolic shift from OXPHOS toward glycolysis in cancer cells. However, when DRP1 is inhibited, lung cancer cells become more reliant on glycolysis by losing redox control and suppressing lactate utilization [32]. Thus, the role of DNM1L in mitochondrial division and metabolic alterations seems to vary depending on the cancer cell type.
Previous studies suggested that Mdivi-1, a inhibitor of DRP1 [14], decreased cancer cell proliferation by inducing mitochondrial fusion and altering oxygen consumption [25, 32]. In our study, pharmacological inhibition of DRP1 by Mdivi-1 showed anti-leukemic effects as well as genetic depletion of DRP1 in vitro. Although genetic depletion of mitochondrial fission inhibited OXPHOS and prolonged survival in a mouse leukemia model, Mdivi-1 as a single agent did not support its efficacy in vivo. However, it has also been suggested that the depletion of DRP1 may restore the sensitivity of AML cells to other chemotherapeutic drugs. DRP1 inhibition enhances venetoclax-induced mitochondrial apoptosis in TP53-mutated AML cells via BAX/BAK activation [33]. Although Mdivi-1 has off-target effects on the ETC, previous reports have revealed that Mdivi-1 treatment did not affect normal hematopoiesis and had little to no reported in vivo toxicity in mice [13, 24]. Thus, the inhibition of mitochondrial fission may contribute to overcoming drug resistance in certain subsets of leukemia.
In conclusion, our study revealed that the inhibition of mitochondrial fission by MFF or DRP1 depletion induced potent antileukemic effects in vitro, ex vivo, and in vivo through the inhibition of OXPHOS. The significant anti-leukemic activity observed with the DRP1 inhibitor, Mdivi-1, suggests that mitochondrial fission could represent a future therapeutic target in AML.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Khwaja A, Bjorkholm M, Gale RE, Levine RL, Jordan CT, Ehninger G, et al. Acute myeloid leukaemia. Nat Rev Dis Primers. 2016;2:16010.
Döhner H, Wei AH, Appelbaum FR, Craddock C, DiNardo CD, Dombret H, et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood. 2022;140:1345–77.
Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46.
Sainero-Alcolado L, Liaño-Pons J, Ruiz-Pérez MV, Arsenian-Henriksson M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. 2022;29:1304–17.
Skrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–88.
Panina SB, Pei J, Kirienko NV. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021;9:17.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–84.
Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. 2020;21:204–24.
Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24:659–67.
Hayashi Y, Harada H. Mitochondrial dynamics as a pathobiological mediator of clonal myeloid disorders. Cancer Sci. 2023;114:2722–8.
Larrue C, Mouche S, Lin S, Simonetta F, Scheidegger NK, Poulain L, et al. Mitochondrial fusion is a therapeutic vulnerability of acute myeloid leukemia. Leukemia. 2023;37:765–75.
Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 2019;9:890–909.
Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The putative Drp1 inhibitor mdivi-1 Is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583-594.e6.
Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204.
Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–201.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41:D991–5.
Pei S, Minhajuddin M, Adane B, Khan N, Stevens BM, Mack SC, et al. AMPK/FIS1-mediated mitophagy is required for self-renewal of human AML stem cells. Cell Stem Cell. 2018;23:86-100.e6.
Saito Y, Kinoshita M, Yamada A, Kawano S, Liu HS, Kamimura S, et al. Mannose and phosphomannose isomerase regulate energy metabolism under glucose starvation in leukemia. Cancer Sci. 2021;112:4944–56.
Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–70.
Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5.
Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–51.
Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–83.
Chen H, Chan DC. Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab. 2017;26:39–48.
Aoyagi Y, Hayashi Y, Harada Y, Choi K, Matsunuma N, Sadato D, et al. Mitochondrial fragmentation triggers ineffective hematopoiesis in myelodysplastic syndromes. Cancer Discov. 2022;12:250–69.
Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. 2012;26:2175–86.
Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene. 2013;32:4814–24.
Nagdas S, Kashatus JA, Nascimento A, Hussain SS, Trainor RE, Pollock SR, et al. Drp1 promotes KRas-driven metabolic changes to drive pancreatic tumor growth. Cell Rep. 2019;28:1845-59.e5.
Zamberlan M, Boeckx A, Muller F, Vinelli F, Ek O, Vianello C, et al. Inhibition of the mitochondrial protein Opa1 curtails breast cancer growth. J Exp Clin Cancer Res. 2022;41:95.
Schimmer AD. Mitochondrial shapeshifting impacts AML stemness and differentiation. Cell Stem Cell. 2018;23:3–4.
Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFkB and TP53 pathways. Autophagy. 2016;12:999–1014.
Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget. 2012;3:798–810.
Hu M, Zhao Y, Cao Y, Tang Q, Feng Z, Ni J, et al. DRP1 promotes lactate utilization in KRAS-mutant non-small-cell lung cancer cells. Cancer Sci. 2020;111:3588–99.
Jang JE, Hwang DY, Eom JI, Cheong JW, Jeung HK, Cho H, et al. DRP1 inhibition enhances venetoclax-induced mitochondrial apoptosis in TP53-mutated acute myeloid leukemia cells through BAX/BAK activation. Cancers. 2023;15:745.
Acknowledgements
This work was supported in part by KAKENHI (19K1736), the Japan Leukemia Research Fund, and a Grant-in-Aid for Clinical Research from the Miyazaki University Hospital.
Funding
Japan Society for the Promotion of Science, 19K1736, Mariko Kinoshita, University of Miyazaki Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
About this article

Cite this article
Kinoshita, M., Saito, Y., Otani, K. et al. Mitochondrial dynamics as a potential therapeutic target in acute myeloid leukemia. Int J Hematol (2024). https://doi.org/10.1007/s12185-024-03843-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12185-024-03843-8