
Abstract
X-linked agammaglobulinemia (XLA) is a primary immunodeficiency caused by mutations in Bruton’s tyrosine kinase (BTK), and is characterized by markedly decreased numbers of blood B cells and an absence of all immunoglobulin isotypes. We performed whole exome sequencing in a male pediatric patient with dysgammaglobulinemia with IgA deficiency. Genetic analysis revealed a BTK missense mutation (Thr316Ala). To investigate whether a BTK mutation underlay this antibody deficiency with marked decrease of IgA in this patient, we performed functional analyses of B cells and phagocytes, and molecular analyses of somatic hypermutation and class switch recombination. The BTK missense mutation resulted in B cells with reduced BTK and high IgM expression. Equal proportions of CD19low and CD19normal fractions were observed, and both included naïve and memory B cells. Calcium influx and phospholipase Cγ2 phosphorylation upon IgM stimulation were marginally impaired in CD19low, but not in CD19+ B cells. Similar to XLA patients, IgA transcripts showed low SHM levels, whereas IgG transcripts were hardly affected. Our analyses suggest that the BTK mutation likely underlies the disease in this case, and that hypomorphic BTK mutations can result in normal circulating B cell numbers, but specifically impair IgA responses.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
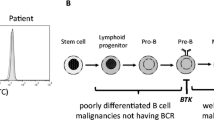
X-linked agammaglobulinemia (XLA) is a human immunodeficiency disease caused by mutations of the Bruton’s tyrosine kinase (BTK) gene. XLA is characterized by a profound deficiency of B lymphocytes and a decrease of all classes of immunoglobulin (Ig), because BTK mutations impair the differentiation of pro-B cells beyond the earlier pre-B cell stage [1–3].
BTK is a key molecule in pre-B cell activation and differentiation, and has multiple roles in the signaling, survival, and proliferation of B cells [4]. BTK is involved in the signaling pathways of various cell types through its association with the B cell antigen receptor (BCR) and Toll-like receptors (TLRs). After BCR ligation by antigen, phosphorylated B cell linker protein binds to BTK and phospholipase C (PLC)γ2 via the SH2 domains of BTK and PLCγ2. Activated BTK directly phosphorylates PLCγ2 at tyrosine residue 759, after which phosphorylated PLCγ2 causes calcium release and activation of canonical nuclear factor κB (NF-κB) signaling [4–7]. BTK is also a key signaling molecule of phagocytes; and recent studies have demonstrated the excessive production of reactive oxygen species (ROS) and increased apoptosis in human BTK-deficient neutrophils when stimulated via TLRs [8].
Over 600 different mutations in BTK have been reported in patients with XLA [9], but the genotype/phenotype correlation is unclear, and some missense mutations are associated with milder or atypical disease [4, 10]. The pathogenicity of a mutation in the BTK gene is less clear when BTK variants are found in antibody-deficient patients who have peripheral B cell percentages that are higher than expected in XLA (>2 % of lymphocytes) [11]. The few residual B cells from XLA patients can produce IgE upon in vitro stimulation with anti-CD40 and interleukin (IL)-4 [12]. Moreover, BTK-deficient B cells can generate low, but detectable levels of specific antibodies upon in vivo immunization and often produce self-reactive and poly-reactive antibodies [12–14]. It is still unknown, however, whether XLA B cells can undergo class switch recombination (CSR) to IgA, undergo somatic hypermutation (SHM), or have normal immunoglobulin receptor diversity.
We performed whole exome sequencing of a 3-year-old boy with dysgammaglobulinemia with a deficiency of IgA and identified a BTK missense mutation. This mutation has already been reported in a 31-year-old male with a reduced number of peripheral B cells (8 % at 12 years of age, and from 1.98 to 2.8 % at 28 years of age), dysgammaglobulinemia (normal serum IgG, low IgA and IgM), a history of recurrent infections, and an atypical clinical phenotype characterized by sclerosing cholangitis and chronic obstructive pancreatitis [15]. However, it is still unclear whether the mutation is causative of disease [16, 17], since expression of BTK and BTK phosphorylation was normal in this previous individual and no other biochemical analyses were performed.
In the present study, we showed the BTK (T316A) mutation underlies the decrease in the patient with dysgammaglobulinemia and examined B cell and phagocyte functions, as well as analyses of complementarity-determining region (CDR) length and SHM frequency in IgA and IgG transcripts to investigate the role of this mutation in IgA deficiency.
Materials and methods
Exome and sequencing analysis of BTK
All human exons were captured by SeqCap EZ Exome SR Version 3.0 (Roche NimbleGen, Inc., Madison, US), and paired-end sequencing of captured fragments was performed on an Illumina HiSeq 1000 deep sequencing instrument. DNA/RNA extraction, cDNA synthesis, cell isolation, and validation study information are described in detail in the supplementary methods section. This study design was approved by the local scientific ethics committee, and informed consent was obtained from all subjects.
Flow cytometry
Peripheral blood mononuclear cells (PBMCs) were stained with fluorescein-conjugated antibodies and analyzed by Fortessa flow cytometry (Beckton Dickenson, Franklin Lakes, US) or FACS Calibur (Beckton Dickenson, Franklin Lakes, US) and FlowJo software (TreeStar Inc, Ashland, US). Additional antibody information is described in detail in the supplementary methods section.
Phosphorylation assay
A total of 1 × 106 cells/100 μl PBMCs were incubated in RPMI 1640 and left untreated or stimulated with human CD40 Ligand (2 μg; Miltenyi Biotec, Bergisch Gladbach, Germany) for 15 min or goat F(ab’)2 anti-human IgM UNLB (10 μg; Southern Biotech, Birmingham, US) for 5 min at 37 °C [18]. Cells were then fixed and permeabilized using the BD Phosflow kit and stained with anti-NF-κB p65 (pS529)-Alexa Fluor 488 and anti-PLCγ2 (pY759)-Alexa Fluor 647 (all from BD Pharmingen Inc., San Diego, US).
Calcium flux
PBMCs were loaded with 2 μM Fluo4AM (Life Technologies, Carlsbad, CA, US) in RPMI 1640 supplemented with 5 % heat-inactivated fetal bovine serum, incubated for 45 min at 37 °C, and stimulated with goat F(ab’)2 anti-human IgM (5.0 μg/ml; Southern Biotech, Bergisch Gladbach, Germany) for 8 min and ionomycin (8 μg/ml; Life technologies, Carlsbad, US) for the last minute, as previously described [19]. The results were analyzed by kinetic plots using FlowJo software (Tree Star Inc., Ashland, US).
Mutation analysis in IgA and IgG transcripts
IgA and IgG transcripts were amplified from PBMCs cDNA using primers in the leader sequence of the immunoglobulin heavy chain variable region (IGHV) in combination with an immunoglobulin heavy alpha (IGHA) or an immunoglobulin heavy gamma (IGHG) consensus reverse primer. All PCR products were subcloned and then sequenced, as previously described [20, 21]. The mutation frequencies within IGHV and the immunoglobulin heavy chain (IGH)-CDR3 sizes were determined from each unique clone. Statistical analyses were performed with the Mann–Whitney test. P values <0.05 were considered to be statistically significant.
Results
Patient and laboratory data
The patient is a 3-year-old male born to the first child of non-consanguineous parents, who suffered from perianal abscesses, recurrent acute otitis media, recurrent respiratory tract infections, and a skin eruption with fever, which was diagnosed pathologically as leukocytoclastic vasculitis. He did not present with lymphadenopathy, autoimmune disease, or gastrointestinal infection.
Diagnostic workup was started at 10 months of age, and the patient had normal serum IgG and IgG subclass (IgG 1335, IgG1 963, IgG2 337, IgG3 151, IgG4 < 3.0 mg/dl) and low IgM (from 14 to 49 mg/dl), but undetectable serum IgA levels <4 mg/dl (Supplementary Table 1), and occasional neutropenia induced by infection. Specific IgG levels against previous vaccinations (measles and rubella) were normal, as were T cell receptor excision circles (TRECs) and kappa-deleting recombination excision circles (KRECs) numbers. Since recurrent bacterial infections were not controlled by prophylactic use of antibiotics, we provisionally put him on intravenous immunoglobulin therapy at 2 years of age. This decreased the frequency of bacterial infections.
We carried out detailed immunophenotyping of the patient’s B lymphocytes. The percentage of B cells within the lymphocyte gate at 1 year of age was 14.2 %; however, this gradually decreased with age to 1.4–5.7 % at 3 years of age (Fig. 1a and Supplementary Fig. 1). CD24++ CD38++ transitional B cells were increased at the age of 1 year; however, these were normal at 3 years of age compared to age-matched controls (Fig. 1b and Supplementary Fig. 1) [22]. The frequencies of IgD-CD27+ memory B cells at 1 year of age were reduced. Intriguingly, however, the frequencies of CD27+ memory B cells and IgG+ B cells were normal. In contrast, surface IgA+ B cells were not detected (Fig. 1c and Supplementary Fig. 1). CD19+ B cells consisted of two fractions: CD19low and CD19normal (Fig. 1a). Because previous observations found that decreased CD19 expression was associated with impaired BCR signaling or immature B cells [23, 24], we compared the expression of other surface markers between the two fractions. Expression of CD19 was low in the majority of CD21low B cells (Fig. 1a). Transitional B cells were hardly detected in CD19low B cells (Fig. 1b). Surface IgM expression was high on both CD19low and CD19normal cells compared to that in healthy donors (HD) (Fig. 1d).

Patient (3-year-old) peripheral B cell subpopulation. a Lymphocyte CD19, CD20, and CD21 expression. b Total CD19+ and CD19low cells CD24 and CD38 expression. c Total CD19+ and CD19low cells CD27 and IgD expression, and total CD19+ cells IgG and IgA expression. d IgM expression in healthy donor (dotted line) and patient CD19+ cells (CD19normal, solid; CD19low, dotted lines)
Whole exome sequencing reveals a missense mutation in BTK (Thr316Ala)
To identify the causative gene defect in the patient with dysgammaglobulinemia with marked decrease of IgA, we performed whole exome sequencing. We excluded mutations in dbSNP database version 137 and selected coding, splice site, and non-synonymous mutations (Supplementary Fig. 2). This analysis identified possible mutations in BTK. Sanger sequencing of BTK confirmed the presence of a missense substitution in exon 11 (NM_000061.2:c.946A >G) of the patient, causing a change from threonine to alanine in position 316 of the SH2 domain (NP_000052.1:p.Thr316Ala). This missense substitution was located in a highly conserved site and was classified as a “deleterious mutation” by the effect predictor SIFT and as “probably damaging with a score of 1.000” by Polyphen 2 [25]. The same mutation was not found in exome sequencing on 26 patients with common variable immunodeficiency at our institute. This BTK (T316A) mutation has previously been reported in a 31-year-old male with atypical XLA that exhibited low IgA and normal IgG [15]. We observed no B cells with spontaneous reversion to normal BTK in the purified B cell fraction of the present patient (data not shown). The patient’s parents did not harbor the same mutation, suggesting that it was de novo (Fig. 2a). BTK expression in monocytes was reduced in the patient when compared with that of control subjects (difference of mean fluorescence intensity (∆MFI):HD, 1.985; patient, 0.996) (Fig. 2b).

Genetic analysis of BTK. a Sanger sequencing of BTK exon 11 in the patient and his parents. b Flow cytometric analysis of BTK expression in the patient. Dotted line, healthy donor; solid line, patient; gray, isotype control. Values represent ∆MFI. One representative dataset out of two independent experiments is shown
In addition to the BTK mutation, the patient and his mother carried a heterozygous missense substitution in TNFRSF13B (tumor necrosis factor receptor superfamily, member 13B (TNFRSF13B), encoding transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI)); NM_012452.2:c.637G > A, NP_036584.1:p.Ala213Thr (Supplementary Fig. 3a). This mutation is listed in dbSNP (rs140914723) and does not appear to contribute to dysgammaglobulimenia because the immunoglobulin levels of the patient’s mother were normal (serum IgG, 927 mg/dl; IgM, 132 mg/dl; IgA, 112 mg/dl), and TACI expression on memory B cells of the patient was normal (Supplementary Fig. 3b). We found no mutations in other genes than BTK and TNFRSF13B that could explain the dysgammaglobulimenia (data not shown).
Normal BCR signaling in CD19+ B cells carrying the T316A BTK mutation
BTK signals to downstream pathways of the BCR. Therefore, we determined whether signaling in B cells harboring the mutated BTK was affected. To examine BCR signaling, we measured calcium flux and phosphorylation of PLCγ2 upon BCR cross-linking. The calcium influx in CD19+ cells from the patient was normal even upon suboptimal stimulation with anti-IgM F(ab’)2 (Fig. 3a). However, the calcium influx in CD19low cells was slightly low and delayed upon suboptimal stimulation (Fig. 3b). The BCR-mediated PLCγ2 phosphorylation of CD19+ cells was normal compared to B cells from controls [∆MFI: controls, 5.2 ± 2.2; patient, 6.0 ± 0.1 (mean ± SD)] (Fig. 3c). PLCγ2 activation in CD19low cells was marginally low compared to that in CD19normal cells (Supplementary Fig. 4a).

B cell signaling. Calcium mobilization in patient and healthy donor CD19+ (a), and patient CD19normal and CD19low cells (b). Arrows indicate time points of stimulation. Flow cytometric analyses of PLCγ2 phosphorylation in CD19+ (c) and NF-κB p65 phosphorylation in CD19+ (d) stimulated with anti-IgM antibody or CD40 ligand. Solid lines, stimulated cells; dotted lines or gray lines, unstimulated cells. Values represent ∆MFI ± SD. Healthy volunteers (n = 4 or 5) and patients with CVID (n = 3 or 4) and IgA deficiency (n = 1 or 2) were enrolled as controls
Defective IgA production upon in vitro CD40 ligand stimulation
To evaluate CD40 signaling, we analyzed the phosphorylation of NF-κB p65 at S529 in patient B cells stimulated with recombinant CD40 ligand and found it to be normal [∆MFI: controls, 7.1 ± 5.1; patient, 4.1 ± 1.2 (mean ± SD)] (Fig. 3d). To study whether this signal induced IgG and IgA production, we purified B cells (patient) and naïve B cells (healthy donors), cultured them in the presence of anti-CD40 and IL-21 for 12 days, and measured supernatant IgG and IgA levels by ELISA. B cells harboring the BTK (T316A) mutation produced comparable levels of IgG to control subjects; however, the production of IgA was significantly impaired in mutant B cells (Supplementary Fig. 4b). Thus, despite seemingly normal CD40 signaling, BTK mutant B cells were unable to induce class switching and production of IgA.
Strongly reduced SHM levels in IgA transcripts of the patient and classical XLA patients
B cells from the patient produced IgG both in vitro and in vivo, and the patient showed a normal IgG response to vaccination against measles and rubella. To further investigate the quality of the patient’s antibodies, we analyzed SHM frequencies in IGHV genes and the IgH-CDR3 sizes of IgG and IgA transcripts. SHM frequencies of IgG transcripts in B cells with BTK (T316A) were slightly, but significantly, reduced as compared to those in age-matched healthy volunteers and IgA-deficient patients (P < 0.01). A similar decrease was observed in classical XLA patients; however, this was much less severe than what was previously observed for CD19-deficient patients (Fig. 4a). SHM frequencies in IgA transcripts of the patient were lower than in controls and IgA-deficient patients (P < 0.001), and were similar to classical XLA patients and CD19-deficient patients. (Figure 4a). The median IgH-CDR3 sizes in patient IgA transcripts were significantly longer than in controls (P = 0.003) (Fig. 4b); however, there were no significant differences in patient and control CDR3 sizes in IgG transcripts (P = 0.36) (Fig. 4b).

Somatic hypermutation and BCR repertoire analyses. a IGHV mutation frequencies of IgA and IgG transcripts. b IGH-CDR3 sizes of IgA and IgG transcripts. Dashed lines represent centroblast median values as previously reported [23]. Gray dots, unique sequences; red lines, median values. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001 (Mann–Whitney test). The values in parentheses indicate the total number of analyzed sequences in each subset. The total number of clones from IgA transcripts that we were able to generate from six classical BTK-deficient patients (<0.5 % B cells) was only fourteen. We also analyzed age-matched healthy volunteers (n = 4) as controls, IgA-deficient patients (n = 2), and CD19-deficient patients (n = 7)
Thus, both in our patient and in classical XLA patients, the mutation levels and the selection mechanism for short IGH-CDR3 regions are specifically impaired in IgA transcripts. This indicates that the observed anomaly in class switched transcripts is not peculiar to the BTK (T316A) mutation, but is generally observed in classical BTK mutations.
Neutrophils with BTK (T316A) exhibit excessive ROS production
To further investigate whether the BTK mutation leads to a functional impairment in innate immune cells [8, 26, 27], we analyzed the function of neutrophils. In a luminol assay, neutrophils with BTK (T316A) produced excessive ROS compared to normal neutrophils (Supplementary Fig. 5).
Discussion
Whole exome analysis of a 3-year-old male with dysgammaglobulinemia with IgA deficiency unexpectedly revealed a BTK missense mutation (Thr316Ala). The numbers of peripheral blood B cells, total and specific serum IgG levels, and KRECs levels of the patient at diagnosis were normal. We nevertheless confirmed that this mutation was disease causing, because this de novo mutation was located in a highly conserved site; no other mutation found in whole exome analysis could explain the antibody deficiency; the percentage of peripheral B cells was gradually decreased to around 1 %; BTK expression in B cells was low; and neutrophils exhibited augmented ROS production. Our patient was similar to the previously reported patient with T316A in clinical phenotype and immunological data. Both patients exhibited recurrent bacterial infections; the frequency of B cells gradually decreased with age; and they showed normal IgG and low IgA. Although the previous reports did not show the abnormalities in the expression of BTK, we detected a low expression of BTK. Moreover, similar to classical XLA patients, the B cells had high surface IgM levels and carried similarly low levels of SHM in IgA transcripts compared to IgG transcripts. Impairment in antibody maturation and diversity may have been involved in his susceptibility to infection which was controlled by intravenous immunoglobulin supplementation.
Circulating B cells in XLA patients in previous reports were largely CD38high CD24high transitional B cells and were IgMhigh, CD19low, and CD21low [24, 28, 29]. Expression of IgM is usually down-regulated as B cells mature [30]. High IgM expression of B cells from XLA patients was reported to reflect the immature stage of B cell differentiation [31]. In contrast, all the B cells with BTK (T316A), including naïve and memory B cells, showed high IgM expression. Unusually high level of IgM expression in mature B cells was observed in CD19- or CD45-deficient mice, and it is speculated that this may be a reflection of impairment of constitutive BCR signaling [32, 33]. These cumulative data suggest that all B cells in our patient have some defects in in vivo BCR signaling.
Interestingly, B cells with the BTK (T316A) mutation consisted of CD19normal and CD19low subsets, in which CD19low B cells showed marginally reduced calcium influx and PLCγ2 response. This observation was not peculiar to our BTK deficiency, since CD19low B cells from other B cell deficiency patients also exhibited impaired BCR-mediated response (data not shown). Expression of CD19 is affected by CD21 and CD81 that form a signaling complex together with CD19 [23, 34]. Decreased expression of CD19 has also been seen in B cells from a patient with Igβ deficiency and with B cell linker protein deficiency; however, the factors that regulate CD19 expression are not well understood [24]. The reason for the presence of CD19normal/CD21normal and CD19low/CD21low populations in BTK (T316A) B cells is unknown.
CD40 signaling is critical in both CSR and SHM [4]. CD40-mediated NF-κB p65 phosphorylation was normal; and induction of activation-induced cytidine deaminase (AID) was observed in B cells with BTK (T316A) after stimulation with an anti-CD40 antibody and IL-21 (data not shown). The involvement of BTK in CD40 signaling is still controversial [35]. Several reports suggested that BTK was activated upon CD40 stimulation and was required for the synergy between CD40 and BCR [36–38]. Although anti-CD40-induced NF-κB phosphorylation was normal in the patient, we could not exclude the possibility that CD40 signaling was defective in the B cells, because B cells failed to produce IgA upon stimulation not only with an anti-CD40 antibody and IL-21, but also with an anti-CD40 antibody and IL-10 or IL-4 (data not shown).
CSR to IgA was severely affected, and SHM and CDR3 sizes of IgA transcripts were abnormal in the B cells with BTK (T316A). This agrees with a previous study that reported normal IgG but low IgA in a patient with the same T316A mutation [15], and reduced IgA levels in an XLA patient with a splice-site mutation in intron 10 of BTK [39]. Interestingly, we found that SHM frequencies of IGHV genes in Cα transcripts were more severely affected than Cγ transcripts in classical XLA patients as well, unlike in IgA-deficient patients. Cumulatively, these results indicate that BTK function is particularly important in isotype switch to IgA. What would be a potential explanation for the specific defects in IgA in BTK deficiency? Signaling through TACI or transforming growth factor-β1receptor is important for CSR to IgA [40]. BTK may be involved in these signaling pathways. One possibility is that BTK is involved in the TACI signaling pathway. TACI signaling is essential for AID expression and IgA secretion in response to a proliferation-inducing ligand (APRIL) and B cell-activating factor (BAFF) [41], but the mechanism by which TACI triggers CSR to IgA is unclear [40, 42]. Previous reports showed that PBMCs from patients with a TACI deficiency were able to produce IgG, but not IgA when stimulated with anti-CD40 and IL-4 or IL-10 [43, 44]. These data raised the possibility that TACI signaling was impaired in our patient. Further experiments are necessary to address this issue using functional assays such as stimulation with APRIL, BAFF, and TLRs agonists.
In conclusion, we identified a BTK (T316A) mutation in a young boy with dysgammaglobulinemia with marked decrease in IgA, despite normal numbers of circulating B cells and serum IgG levels. Thus, mildly affected BTK function is sufficient for early B cell differentiation, but cannot fully support IgA responses.
Abbreviations
- AID:
-
Activation-induced cytidine deaminase
- APRIL:
-
A proliferation-inducing ligand
- BAFF:
-
B cell-activating factor
- BTK:
-
Bruton’s tyrosine kinase
- CDR:
-
Complementarity-determining region
- CSR:
-
Class switch recombination
- CVID:
-
Common variable immunodeficiency
- KRECs:
-
Kappa-deleting recombination excision circles
- NF-κB:
-
Nuclear factor kappa B
- PLCγ2:
-
Phospholipase Cγ2
- ROS:
-
Reactive oxygen species
- RSS-KDE:
-
Recombination signal sequence-kappa-deleting element
- SHM:
-
Somatic hypermutation
- TNFRSF13B:
-
Tumor necrosis factor receptor superfamily, member 13B
- TACI:
-
Transmembrane activator and calcium-modulator and cyclophilin ligand interactor
- TRECs:
-
T cell receptor excision circles
- XLA:
-
X-linked agammaglobulinemia
References
Mohiuddin MS, Abbott JK, Hubbard N, Torgerson TR, Ochs HD, Gelfand EW. Diagnosis and evaluation of primary panhypogammaglobulinemia: a molecular and genetic challenge. J Allergy Clin Immunol. 2013;131(6):1717–8.
Ochs HD, Smith CIE. X-linked agammaglobulinemia. A clinical and molecular analysis. Medicine. 1996;75(6):287–99.
Nomura K, Kanegane H, Karasuyama H, Tsukada S, Agematsu K, Murakami G, et al. Genetic defect in human X-linked agammaglobulinemia impedes a maturational evolution of pro-B cells into a later stage of pre-B cells in the B-cell differentiation pathway. Blood. 2000;96:610–7.
Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519–33.
Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2(12):945–56.
Kurosaki T, Hikida M. Tyrosine kinases and their substrates in B lymphocytes. Immunol Rev. 2009;228:132–48.
Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev. 2005;203:216–34.
Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, et al. The kinase BTK negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13(4):369–78.
Valiaho J, Smith CI, Vihinen M. BTK base: the mutation database for X-linked agammaglobulinemia. Hum Mutat. 2006;27(12):1209–17.
Teimourian S, Nasseri S, Pouladi N, Yeganeh M, Aghamohammadi A. Genotype-phenotype correlation in Bruton’s tyrosine kinase deficiency. J Pediatr Hematol Oncol. 2008;30:679–83.
Abbott JK, Ochs HD, Gelfand EW. Coding-region alterations in BTK do not universally cause X-linked agammaglobulinemia. J Allergy Clin Immunol. 2013;132(5):1246–8.
Nonoyama S, Tsukada S, Yamadori T, Miyawaki T, Jin YZ, Watanabe C, et al. Functional analysis of peripheral blood B cells in patients with X-linked agammaglobulinemia. J Immunol. 1998;161:3925–9.
Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J Exp Med. 2004;200(7):927–34.
Cunningham-Rundles C. Ponda PP. Molecular defects in T- and B-cell primary immunodeficiency diseases. Nat Rev Immunol. 2005;5(11):880–92.
Graziani S, Di Matteo G, Benini L, Di Cesare S, Chiriaco M, Chini L, et al. Identification of a BTK mutation in a dysgammaglobulinemic patient with reduced B cells: XLA diagnosis or not? Clin Immunol. 2008;128(3):322–8.
Fleisher TA, Notarangelo LD. What does it take to call it a pathogenic mutation? Clin Immunol. 2008;128(3):285–6.
Conley ME. Genetics of hypogammaglobulinemia: what do we really know? Curr Opin Immunol. 2009;21(5):466–71.
Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131(2):477 e1–485 e1.
Foerster C, Voelxen N, Rakhmanov M, Keller B, Gutenberger S, Goldacker S, et al. B cell receptor-mediated calcium signaling is impaired in B lymphocytes of type Ia patients with common variable immunodeficiency. J Immunol. 2010;184(12):7305–13.
Berkowska MA, Driessen GJ, Bikos V, Grosserichter-Wagener C, Stamatopoulos K, Cerutti A, et al. Human memory B cells originate from three distinct germinal center-dependent and -independent maturation pathways. Blood. 2011;118(8):2150–8.
Zelm MCv, Bartol SJW, Driessen GJ, Mascart F, Reisli I, Franco JL, et al. Human CD19- and CD40L-deficiencies impair antibody selection and differentially affect somatic hypermutation. J Allergy Clin Immunol. 2013 in press.
Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162(2):271–9.
Zelm MCv, Reisli I, Burg Mvd, Burg Mvd, Castaño D, Noesel CJMv, Tol MJDv, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–12.
Dobbs AK, Bosompem A, Coustan-Smith E, Tyerman G, Saulsbury FT, Conley ME. Agammaglobulinemia associated with BCR(-) B cells and enhanced expression of CD19. Blood. 2011;118(7):1828–37.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Mohamed AJ, Yu L, Backesjo C-M, Vargas L, Faryal R, Aints A, et al. Bruton’s tyrosine kinase (BTK): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev. 2009;288:58–73.
Doyle SL, Jefferies CA, Feighery C, O’Neill LA. Signaling by toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J Biol Chem. 2007;282(51):36953–60.
Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. 2009;27:199–227.
Anker R, Conley ME, Pollok BA, Pollok BA. Clonal diversity in the B cell repertoire of patients with X-linked agammaglobulinemia. J Exp Med. 1989;169:2109–19.
Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959–71.
Conley ME. B cells in patients with X-linked agammaglobulinemia. J Immunol. 1985;134(5):3070–4.
Benschop RJ, Cambier JC. B cell development: signal transduction by antigen receptors and their surrogates. Curr Opin Immunol. 1999;11:143–51.
Lam K-P, Kuhn R, Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–83.
van Zelm MC, Smet J, Adams B, Mascart F, Schandene L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Investig. 2010;120(4):1265–74.
Satterthwaite AB, Witte ON. The role of Bruton’s tyrosine kinase in B-cell development and function. Immunol Rev. 2000;175:120–7.
Brunner C, Kreth HW, Ochs HD, Schuster V. Unimpaired activation of c-Jun NH2-terminal kinase (JNK) 1 upon CD40 stimulation in B cells of patients with X-linked agammaglobulinemia. J Clin Immunol. 2002;22(4):244–51.
Bishop GA. The multifaceted roles of TRAFs in the regulation of B-cell function. Nat Rev Immunol. 2004;4(10):775–86.
Haxhinasto SA, Bishop GA. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J Biol Chem. 2004;279(4):2575–82.
Maekawa K, Yamada M, Okura Y, Sato Y, Yamada Y, Kawamura N, et al. X-linked agammaglobulinemia in a 10-year-old boy with a novel non-invariant splice-site mutation in BTK gene. Blood Cells Mol Dis. 2010;44(4):300–4.
Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8(6):421–34.
He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, et al. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nat Immunol. 2010;11(9):836–45.
Salzer U, Bacchelli C, Buckridge S, Pan-Hammarstrom Q, Jennings S, Lougaris V, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–76.
Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. 2007;120(5):1178–85.
Martinez-Gallo M, Radigan L, Almejun MB, Martinez-Pomar N, Matamoros N, Cunningham-Rundles C. TACI mutations and impaired B-cell function in subjects with CVID and healthy heterozygotes. J Allergy Clin Immunol. 2013;131(2):468–76.
Acknowledgments
The authors would like to thank Eri Kumaki, Miko Okamura, Shizuko Minegishi, Takashi Watanabe, Masaki Takazawa, and Atsushi Hijikata for technical assistance.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
About this article

Cite this article
Mitsuiki, N., Yang, X., Bartol, S.J.W. et al. Mutations in Bruton’s tyrosine kinase impair IgA responses. Int J Hematol 101, 305–313 (2015). https://doi.org/10.1007/s12185-015-1732-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-015-1732-1