
Abstract
Purpose of Review
Over the past 2 decades, our understanding of the role of genetics in the development and progression of aortic disease has grown considerably. This review serves as a foundational text for the reader to gain a thorough understanding of the basics of this emerging field while also displaying the latest breakthroughs which have had, and will continue to have, important impacts on clinical decision-making and patient outcomes. This review focuses on aneurysmal disease, arguably the richest subgroup of aortic diseases for genetic research.
Recent Findings
Thoracic aortic aneurysm and dissection (TAAD) is driven primarily by genetic factors. Genetic variants which drive TAAD tend to occur in genes involved in the structure and function of the aortic wall, specifically the extracellular matrix, TGF-β pathway, and smooth muscle cell contractile units. At least 37 gene variants (pathological or of unknown significance) have been found to be directly associated with the development of TAAD, independent of traditional cardiovascular (CV) risk factors such as smoking and hypertension. The standard suggestion for definitive treatment is prophylactic surgery at an aortic diameter of 5.0–5.5 cm, but several gene variants have been shown to dissect commonly at much smaller diameters (such as TGFβR1/2 or ACTA2), and so mutation-specific guidelines are leading to earlier surgery and better patient outcomes in these aggressive cases.
Abdominal aortic aneurysms (AAAs) also have a genetic component, but these are primarily driven by CV risk factors. Many gene variants have been found which are closely associated with these risk factors, such as those in the lipid metabolism pathway (SORT1). Four variants have been found through GWAS studies to directly influence AAAs independent of general risk factors, though the clinical implications are yet unclear.
Summary
New variants are discovered every year, and each creates a new avenue of research. As more data is collected on patients with each variant, such as the rate of growth, mode of inheritance, age at dissection, and diameter at dissection, we can craft more patient-specific guidelines for different subgroups of affected individuals, with the aim of achieving better long-term survival.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
There is no disease more conducive to clinical humility than aneurysm of the aorta
- William Osler
Introduction
The aorta is the largest artery in the human body and the site of many clinically challenging and significant diseases. According to CDC (Centers for Disease Control and Prevention) data from the USA, complications from aortic aneurysms caused 9923 deaths in 2018 alone [1]. Unfortunately for both patients and clinicians, aortic aneurysms are notoriously difficult to diagnose and treat: for example, the most common presenting symptom in thoracic aneurysms is death [2•], and in those who present at a hospital with a life-threatening dissection or rupture, 80% of cases are ultimately fatal [3•]. Clearly, therefore, a robust system for identifying affected patients prior to catastrophic consequences and for offering them preventive medical or surgical therapy is needed. Currently, the only routine screening protocol recommended by the US Preventative Task Force for aortic aneurysms is a single abdominal ultrasound scan offered to men aged 65–75 with a smoking history. Critically, this excludes women (42% of deaths in 2018 [1]), thoracic aneurysms (10% of deaths [1]), men younger than 65 (19.3% of deaths [1]), and never-smokers (13% of cases in males aged 65 + in one Swedish study [4]).
Furthermore, it is becoming increasingly clear in the modern era of precision medicine that no two aneurysms are alike: some arise without the typical risk factors being present, some grow faster than others, and some dissect at significantly shorter diameters. A one-size-fits-all approach for dealing with aneurysms, such as a simple 5.5 cm diameter for surgical intervention, is therefore being seen as increasingly inappropriate. Genomic studies have helped us to categorise these behaviours into distinct subsets caused by different genetic variants, enabling us to provide more personalised care to patients if we can determine which variants, if any, they carry. Knowing this information facilitates the screening of relatives and identification of those at particularly high risk of developing aneurysmal disease (i.e. those with the same genetic variant), even if the phenotype is not yet present.
The two main types of aortic aneurysm—thoracic and abdominal—are quite distinct from each other with regards to their genetic drivers and therefore should be considered separately.
Thoracic Aortic Aneurysm and Dissection (TAAD)—Overview
TAAD, typically thought of affecting the ascending aorta and aortic arch up to the ligamentum arteriosum, is arguably the disease which offers the richest substrate for genetic research. It is estimated to affect roughly 1% of the population [5••], but such numbers are difficult to calculate because only 5% exhibit clinical signs prior to dissection or rupture [2•]. Furthermore, many cases are misdiagnosed in acute care settings as a myocardial infarction (MI) [6], meaning that the mortality in Stanford type A dissections prior to hospital admission is likely higher than the 50% figure often quoted [5••].
It has been known for over 20 years that TAAD has a genetic component, confirmed by family pedigree analysis by Biddinger et al. (1997) and Coady et al. (1999) [7, 8], amongst others. Five percent of TAAD is called “syndromic” because other organ systems beyond the aorta are involved, as in Marfan Syndrome, Loeys-Dietz Syndrome, Ehlers-Danlos Syndrome, all autosomal dominant, and arterial tortuosity syndrome, which is autosomal recessive. These syndromic cases are easier to diagnose because there are cardiac and extracardiac manifestations of disease, particularly in the musculoskeletal and ocular systems. However, 95% of patients are “non-syndromic” (NSTAAD)2•, meaning these helpful clinical signs are not present. Genetic analysis, therefore, is likely to be particularly useful in this group for identifying affected patients and screening relatives.
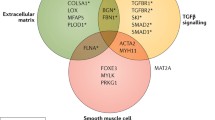
Since the advent of genomic sequencing, genotype–phenotype association studies have helped identify at least 37 gene variants [9••] (either known pathogenic variants or variants of unknown significance whose exact deleterious effects are not yet well established) which encompass 30% of familial non-syndromic TAAD, i.e. disease affecting more than a single family member [2•]. These variants almost exclusively affect proteins which constitute or regulate the function of the aortic wall, specifically those involved in the TGF-β signalling pathway, extracellular matrix production and maintenance, and vascular smooth muscle cell (SMC) contractile units, such as ACTA2, MYLK, and TGFBR2 [2•]. There is much more research to be done in this field as the causal gene variants in roughly 70% of familial cases are still yet to be identified.
Just under 80% of NSTAAD cases [2•] have no reported family history and are therefore termed “sporadic”. This number is high in part because relatives are not routinely screened by imaging modalities, instead relying on the presence of signs and symptoms. Robertson et al. (2016) showed that 27% of sporadic cases did actually have relatives who carried one of the known NSTAAD gene variants [10]. Furthermore, roughly 10% of those with an early sporadic dissection have a variant in a syndromic or familial NSTAAD gene, again affecting aortic wall proteins, and 28% of cases have a variant of unknown significance [6]. Some sporadic patients without affected relatives in previous generations may become founders of a new inherited aortic disease lineage, as suggested by some studies which have identified specific genetic variants in such patients [11, 12].
Elefteriades et al. (2015) further showed that certain other diseases, such as a bovine aortic arch, bicuspid aortic valve (BAV), temporal arteritis, renal cysts, and aortic coarctation, are associated with TAAD and may aid in raising suspicion of the diagnosis [13]. BAV is perhaps the most important, as it represents the most common congenital heart defect, affecting 1–2% of the population [14]. Aortic dilatation is estimated to occur 40–50% of patients with BAV [15] and dissection in 5% of cases [16].
Knowing which genetic variant is carried by a patient can affect management. Many variants lead to dissections under the 5.0–5.5 cm diameter range that is normally used as the threshold for surgery, so early intervention (usually curative) is warranted (Fig. 1). Furthermore, some variants are associated with a more rapidly progressive disease phenotype, as tends to be the case with familial-variant patients who present at an earlier age than those with sporadic-variants (58.2 vs 65.7) and have aneurysms that grow at faster rates (0.21 vs 0.16 cm/year) [17]. These factors may be relevant when deciding the frequency of follow-up scans, and the timing of the operation as the surgical threshold may be met sooner. Standard beta-blocker therapy to slow growth (widely practiced, but questionably effective) and avoidance of isometric exercises and contact sports, which are known triggers of dissection, would continue to be of use. A summary of different proposed surgical guidelines is shown in Fig. 1.

Surgical guidelines for patients carrying variants in specific genes involved in the development of TAAD based on current expert opinion. Those in red are the most recently discovered [9••]
TAAD—the Affected Pathways
An understanding of aortic structure and function is integral to elucidation of genetic pathways of disease. The aorta is an elastic tube which expands during systole, storing blood, and recoils in diastole, pushing stored blood out to the peripheries (the Windkessel effect)[18]. This is a tightly coordinated function, relying on elasticity and contractile strength, which regulates blood pressure and peripheral perfusion during the cardiac cycle [18]. A disruption of any component of the wall architecture can lead to significant weakening, rendering the aorta susceptible to aneurysmal dilatation or dissection, especially in face of concomitant hypertension.
The aortic wall itself is comprised of three layers:
-
1.
Tunica intima: endothelial cells on a basement membrane
-
2.
Tunica media: wavy layers of elastin and collagen embedding smooth muscle cells (SMCs)
-
3.
Tunica adventitia: collagen and fibroblasts
The region of interest for TAAD is the tunica media (TM), specifically the regulation of SMC production and activity. The TM consists of over 50 alternating layers of SMCs and elastin lamellae, with microfibril projections from the lamellae attaching obliquely to SMCs via focal adhesions which enable transmission of mechanical forces through the wall [19], like microscopic “oars”. The oblique direction reverses in each layer, thus minimising the forces on individual SMCs [20]. Loss of the SMCs, destruction of the elastin fibres, and deposition of proteoglycans and glycosaminoglycans in the remaining spaces in the extracellular matrix, a process called cystic medial necrosis, are what ultimately compromises the strength and elasticity of this layer and leads to TAAD [21].
Smooth Muscle Cell Contractile Units
SMC contractile units consist of the contractile proteins, which sense mechanical stress from pulsatile blood flow and contract and relax appropriately, as well as the many kinases which regulate intracellular function. As shown by Karimi and Milewicz (2016), components of this unit link both to the cytoplasmic and nuclear membranes, so mechanical stimuli have the ability to directly affect gene transcription and metabolic activity [22]. Variation in these systems, therefore, compromise the structural integrity of the vessel, its general metabolic health, and its ability to repair itself in response to insults—all key drivers in aneurysm formation.
TGF-β Pathway
Transforming growth factor β (TGF-β) is a cytokine which is vital in the preservation of SMC function and the development of blood vessels. It binds to TGFBR2 (TGF-β receptor 2) on the surface of SMCs, which then recruits and phosphorylates TGFBR1. This in turn phosphorylates SMAD2/3, which complex with SMAD4 and translocate to the nucleus to affect gene transcription, for example, upregulating production of contractile proteins such as alpha actin and smooth muscle myosin heavy chains [23]. TGF-β is also needed for to stimulate the differentiation of neural crest cells, from which much of the thoracic aorta is derived, into SMCs [24••]. Nonsense and frameshift variants, which can cause haploinsufficiency in which one allele is non-functional, and missense mutations, which disrupt protein function, cause a loss of function of TGF-β, thus impairing proper differentiation and function of SMCs [24••]. Having fewer functioning contractile units precludes proper mechanosensation and force generation, rendering the aortic wall more susceptible to damage at high pressures [24••]. Murine models reveal that early downregulation of TGF-β results in more significant aortic disease, likely due to compromised organogenesis. Paradoxically, however, variants expected to decrease TGF-β signalling actually increase it, perhaps due to disinhibition of a non-canonical TGF-β pathway which was previously slowed by negative feedback [25•].
Extracellular Matrix
The extracellular matrix (ECM) is a complex structure existing between cells of the aortic wall which significantly contributes to the aorta’s passive mechanical properties. It is synthesised by SMCs and fibroblasts and is comprised of elastin, involved in elasticity and recoil; collagen, involved in tensile strength and regulation of cell proliferation through interaction with integrins; and a variety of glycoproteins [26•]. The precise make-up of the ECM is itself dynamic, shifting in response to metabolic and mechanical stressors, partly under the regulation of proteases such as ADAMs, a disintegrin and metalloproteinase, and MMPs (matrix metalloproteinases)27 which degrade existing proteins to make room for newly synthesised proteins. Dysfunction of the ECM can contribute to aneurysm formation either through defects in the constituent proteins themselves or in the remodelling pathways. For example, variants in COL3A1 (type 3 procollagen; Vascular Ehlers-Danlos Syndrome) and FBN1 (ECM glycoprotein fibrilin-1; Marfan’s Syndrome) are both associated with an increased risk of TAAD, while polymorphisms in MMP3 and MMP13 have been associated with an increased risk of AAA [28].
TAAD Genes
Fibrillin 1 (FBN1)
Fibrillin 1 is a large glycoprotein found in the extracellular matrix which polymerises to form the microfibrils that connect elastin lamellae to SMCs, and so it facilitates the transmission of mechanical forces across the aortic wall. FBN1 associates with TGF-β, latency-associated protein (LAP), and latency-associated TGF-β binding protein, to form the large latent complex (LLC) [29], which is responsible for sequestering TGF-β in the walls of the aorta and thus regulating the degree to which it influences gene transcription, as previously described. Should FBN1 be dysfunctional, there will be weaker connections between the layers of the tunica media, additionally leading to abnormal function of mechanosensation and gene regulation via SMC contractile units, and there will be inappropriate activation of the TGF-β pathway due to inadequate sequestration. Both of these consequences increase the risk of TAAD.
FBN1 is one of the most commonly disrupted genes in both syndromic and non-syndromic TAAD. As early as 1991, missense variants in FBN1 were shown to cause Marfan’s Syndrome (MFS)30. This condition, present in 2–3/100,000 individuals in the population [31], is the most common form of syndromic TAAD, with clinical features including joint-laxity (positive Steinberg sign), skeletal overgrowth, and vertebral column deformity [15]. There is also an increased incidence of aortic root dilatation and type A dissections.
There are over 2000 known unique variants of FBN1, accounting for 3% of familial TAAD [32] and 4% of TAAD without MFS [33]. The majority of these causative variants are of the missense type, which are autosomal dominant and have a high penetrance but variable expressivity between families and within the same family. The mechanism of disease is usually haploinsufficiency, resulting in reduced functional fibrillin-1 levels, or a dominant negative interference, whereby the dysfunctional protein interferes with the action of the normal protein [22]. The haploinsufficiency variant curiously has a 2.4 × higher death rate from cardiovascular complications than the dominant negative variant, but overall mortality is the same [34]. It is also more sensitive to the putative aortic growth-slowing effects of losartan [35].
Treatment guideline: prophylactic surgery at a diameter of 4.5–5cm [23], with earlier intervention recommended if the aorta is growing rapidly (> 0.5 cm/year)36. Clinicians are also referred to the Ghent criteria for diagnosis of MFS [37].
TGFβR 1/2
Variants in TGFβR 1/2 cause Loeys-Dietz Syndrome (LDS) types 1 and 2, respectively. LDS is a MFS-like disease, originally described in 2005, which is typified by signs such as congenital heart disease, cleft palate, bifid uvula, craniosynostosis, hypertelorism, and developmental delay [38]. The affected proteins are serine/threonine kinases which transfer the signal from TGF-β to intracellular SMAD proteins and thence to the nucleus for gene regulation. Disease-causing variants reduce the activity of the intracellular kinase domain [15] and therefore disrupt the pathway and weaken the aortic wall as previously described, resulting in syndromic TAAD. However, cases of NSTAAD have also been identified [14].
Disease progression is slightly different for the two genes: TGFβR1 mutations tend to present with aortic complications earlier in life (31.4 years vs 45.6 years average) [39]; have a higher incidence of type A dissections [39] and lower incidence of type B dissections; and have a 5 × lower risk of requiring surgery [40], though the overall mortality seems to be similar between the two [41]. Patients with Loeys-Dietz tend to have similar prognoses to MFS patients after diagnosis and medical management. Of note, there is also variation between the sexes: men with TGFβR1 variants have a greater risk for ascending aortic disease, whereas women have more disease in other arteries, but the aortic risks are similar for both sexes with TGFβR2 mutations [41].
Treatment guideline: prophylactic surgery at a diameter of 4–4.2 cm. This is still currently a controversial recommendation, with some arguing that this is too aggressive if good medical treatment is given [42] or that it is better to use a more holistic threshold encompassing sex, specific mutation, systemic features, and so on [41]. An example of the latter would be a 4.5 cm threshold, lowered to 4.0 cm in females with a low body surface area and a severe symptomatic TGFBR2 mutation [41].
SMAD3/SMAD2
Variants in SMAD3 cause Loeys-Dietz type 3 (aneurysm-osteoarthritis syndrome), an autosomal dominant disease characterised by craniofacial, skeletal, and cutaneous abnormalities with early onset osteoarthritis and other joint abnormalities [43], as well as aortic aneurysms [24••]. Most variants (61% missense, 23% frameshift [44•]) result in a loss of function of the MH2 [45] which is responsible for the oligomerisation of SMAD3/4 in the TGF-β pathway, thus producing a similar effect to TGFβR1/2 mutations. They comprise 2% of all familial TAAD [46], with roughly 60–70% of individuals having some form of aneurysm or dissection [43] and a median age of death in the mid-50s [47]. SMAD2 also complexes with SMAD3 when phosphorylated [48], and SMAD2-MH2 domain mutations produce similar effects to those mutations in SMAD3, though this has been less extensively studied.
Treatment guideline: prophylactic surgery at a diameter of 4–4.2 cm as with TGFβR1/2 for SMAD3 and a diameter of 5.0–5.5 cm for SMAD2.
TGF-β2/3
These isoforms of TGF-β may be affected directly by a pathogenic variant, resulting in Loeys-Dietz Syndrome types 4 and 5, respectively. The syndromic manifestations are similar to the other LDS types: marfanoid skeleton, bifid uvula, thin skin, and delayed wound healing [24••, 49]. Variants cause a direct rise in TGF-β levels, mimicking the effects of the other LDS types. There have been well-documented cases of both frameshift and nonsense variants in the gene [50].
Treatment guideline: prophylactic surgery at a diameter of 4.5–5.0 cm for TGF-β2 and a diameter of 5.0–5.5 cm for TGF- β3. The clinician may also consider yearly ultrasound scans of the aortic root and side branches with TGF-β3 variants as these are known to also cause aneurysms of the cerebral, iliac, and subclavian arteries [49].
ACTA2
ACTA2 is an SMC-specific alpha-actin which polymerises to form thin filaments. Together with myosin heavy chains, these filaments mediate contraction and relaxation of the vascular smooth muscle. Pathogenic variants are mostly missense dominant negative variants that can affect all four subdomains. They account for 12–21% of all familial TAAD cases, the most of any one gene [6]. Disrupted proteins cannot polymerise normally nor can ATP hydrolysis, which mediates contraction, occur [51]. The subsequent inability to adapt to vascular wall stress during pulsatile blood flow leads to aneurysms and dissections. Furthermore, SMC units become histologically disorganised and fragmented, and aortic tissues inappropriately accumulate proteoglycans [51]. Such pathologies also increase the risk for early onset coronary artery disease and ischaemic strokes (< 55 years in men/ < 60 years in women) [51], and one specific de novo missense variant (R179H) has even been shown to cause multisystemic smooth muscle dysfunction, characterised by pulmonary hypertension, hypotonic bladder, patent ductus arteriosus, congenital mydriasis, and hypoperistalsis [52].
Thoracic aortic disease is highly penetrant in individuals with disease causing ACTA2 variants, with one study finding 48% of cases presenting with an aortic event, of whom 88% presented with a thoracic aortic dissection [53]. Dissections present early (median age 35.5), and over 75% of patients will have some aortic event by the age of 85 [53].
Treatment guideline: prophylactic surgery at 4.5–5.0 cm. Up to 1/3 of patients may dissect below 5 cm, many of whom will be pregnant women in the peripartum period [53]; extra care should be taken to counsel such patients. Furthermore, murine knockout models suggest that the deleterious effects of ACTA2 are not significant under conditions of normal blood pressure [54], so it is important that the patient’s blood pressure be well controlled medically.
MYH11
MYH11 encodes smooth muscle myosin heavy chains (SM-MHC), each of which joins with two regulatory light chains and two essential light chains to form thick filaments. These work in tandem with thin filaments (actin/ACTA2) to control SMC contraction [24••]. Causal variants may account for up to 1% of familial TAAD, with the phenotype of increased pulse wave velocity and decreased aortic compliance [55]. However, as with ACTA2, mouse models suggest that MYH11 variants may only contribute to TAAD if there is concomitant hypertension or some other epigenetic factor [54]. Furthermore, data from the Netherlands suggests that variants in MYH11 may not even track in kindreds with TAAD, indicating that these variants may enhance aortic disease, but have a limited ability to cause it outright [56]. Data which seem to show a direct link between MYH11 variants and TAAD are inconclusive; for example, a 16p13.1 duplication (a genomic region containing MYH11) was found to increase the risk of sporadic TAAD eight-fold, but it is not clear that it is MYH11 specifically within this region that is producing the effect [14].
Treatment guideline: prophylactic surgery at 4.5–5.0 cm.
MYLK
Ca2 + /calmodulin (CaM)-dependent myosin light chain kinase (MLCK, encoded by the MYLK gene) phosphorylates regulatory light chains (RLCs) in vascular smooth muscle, in turn increasing the activity of myosin II ATPase, which stimulates contraction [24••]. There are two well-known haploinsufficient variants—one truncation and other disrupting the calmodulin binding sequence—which can reduce MLCK activity by up to 85% [57]. Half-knockout murine models suggest that a mere 50% reduction in activity results in a 40% reduction in RLC phosphorylation/aortic contraction, and while vessels can typically handle such a reduction in contraction capacity, the aorta experiences the highest pressures and therefore exhibits pathology first [58]. As there have been few documented and studied cases of MYLK variants, further clinical phenotyping is difficult. Research is also made more difficult by the observation that functional MLCK variant assays do not correlate well with their ability to cause disease; variants with trivial reductions in kinase activity can still be pathogenic [59•].
Treatment guideline: because MYLK mutations can cause dissection in the absence of aortic dilatation [59•], a size threshold for surgery is not wholly appropriate, but can provisionally be taken as 4.5–5.0 cm. Alternative thresholds have been proposed; for example, Wallace et al. suggest an age threshold of roughly 40 years for those with a haploinsufficiency variant since very few type A dissections occur before this age [59•]. This is in line with common practices for hereditary cancers such as ovarian cancer (e.g. BRCA1 mutation)[59•].
PRKG1
PRKG1 encodes type 1 cGMP-dependent protein kinase (PKG-1). In vascular smooth muscle cells, PKG-1α (the major isoform present) is activated by NO (nitric oxide) and in turn activates guanylyl cyclase, which then produces cGMP. cGMP activates the regulatory myosin-binding subunit of the enzymes that dephosphorylate RLC [22]. PKG-1 therefore opposes the action of MLCK, stimulating SMC relaxation. As pathogenic variants of MYLK are loss-of-function, it might be expected that pathogenic variants of PRKG1 to be gain-of-function, and this is precisely what is seen. For example, the Arg177Gln mutation deactivates the inhibitory domain and makes PKG-1 constitutively active, resulting in excessive SMC relaxation [60]. Over 60% of affected patients may develop an aortic dissection at a mean age of just over 30 years [60].
Treatment guideline: prophylactic surgery at 4.5–5.0 cm.
COL3A1
COL3A1 encodes type 3 procollagen, which is secreted and cleaved by procollagen proteases to be assembled as part of the extracellular matrix [61]. Variants are generally autosomal dominant with near 100% penetrance, roughly 2/3 of which are missense variants causing substitution of glycine for another amino acid in the Gly-X–Y repeating unit. Another 1/3 are exon-skip variants that cause splicing errors [62]. Given that collagen is a homotrimer, only ~ 1/8 molecules produced are normal if one of the alleles is defective (i.e. 0.5 [3•]), as with these variants [63, 64]. Contrastingly, haploinsufficiency variants have 50% of the normal collagen—these make up the remaining observed variants [62]. The associated phenotype is Vascular Ehlers-Danlos Syndrome (EDS), characterised by translucent skin, arterial/intestinal/uterine fragility, bruising, and specific facial features [65]. Arterial rupture accounts for most deaths in vascular EDS.
Treatment guideline: prophylactic surgery at ≤ 5.0 cm.
FOXE3
Mouse models show that forkhead transcription factor (FOXE3) variants lead to a decrease in the ability of SMCs to withstand the stress-induced apoptosis that can occur at high blood pressures and to a reduction in the number of SMCs populating the aortic wall during organogenesis. The main evidence for the role of FOXE3 in TAAD comes from one European family with 8 cases of acute dissection at a median age of 45.6 years. The likely pathogenic variants are of the missense type affecting the C-terminus of the DNA binding domain, the same domain affected by variants which cause certain ocular diseases, as are commonly seen in the other connective tissue disorders mentioned here [66].
Treatment guideline: prophylactic surgery at 5.0–5.5 cm. Since there are limited studies into the pathogenic variants of FOXE3, it is not possible to derive a more specific treatment guideline.
LOX
Lysyl oxidase (LOX) cross-links collagens and elastin by catalysing the formation of aldehydes from lysine residues in the precursors of these two proteins. This stabilises the collagen fibrils and creates strength and elasticity in the aortic wall [67]. A number of missense variants have been discovered which lead to either haploinsufficiency or a reduction in activity due to changes in the catalytic domain. These lead to abnormal aortic development with decreased/fragmented elastic fibres and fusiform enlargements of the ascending aorta/aortic root [67]. LOX can also influence TAAD development by binding and inactivating free TGF-β via its amine oxidase activity [68].
Treatment guideline: insufficient evidence is available to make a firm conclusion on recommended treatments, so the default diameter threshold of 5.0–5.5 cm applies.
Other Genes
There are many other genes which have good evidence for being involved in TAAD formation but are not discussed herein. Some of the main genes are summarised in Table 1:
*Autosomal recessive inheritance. The remainder are autosomal dominant. The treatment guideline for the above variants is surgical repair at 5–5.5 cm.
Abdominal Aortic Aneurysms (AAAs)—Overview
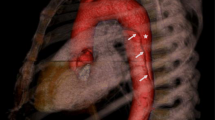
An AAA can be defined as a permanent localised dilatation of the abdominal aorta above a diameter of 3.0cm [69]. Like in TAAD, the aortic wall in AAAs exhibits destruction of elastin/collagen structures and a loss of SMCs, but it also shows significant inflammation and neovascularisation [70]. Although there is some overlap between the two diseases—up to 28% of those with an AAA also have a TAA [71]—TAA and AAA have been shown to segregate independently through family lineages [69], i.e. a family history of TAA has a minimal effect on the risk for developing AAA and vice versa. This could be because the two vessel segments have different embryological origin: abdominal SMCs are derived from mesoderm, while thoracic SMCs are derived from neural crest cells [72]. Furthermore, unlike TAAs, it is not yet possible using variants alone to categorise AAAs into distinct groups with different prognoses. A visual comparison of the two types of aortic aneurysm is shown in Fig. 2.

The ligamentum arteriosum, the fibrous remnant of the ductus arteriosus, naturally divides the aorta into two regions with distinct characteristics of aneurysmal disease. Figure illustration by Rob Flewell. PA = pulmonary artery
Nevertheless, twin studies show that the heritability (i.e. the degree to which variability of a trait in the population is attributable to genetic factors) of AAA is up to 70% [73], and roughly 20% of patients have an affected relative, compared to as low as 2% in controls [74]. A positive family history therefore at least doubles one’s risk of developing an AAA, with a greater risk of inheritance in males [75]. An analysis of six genome wide association studies (GWAS) involving over 10,000 patients only identified nine potential gene loci that are significantly associated with AAAs, five of which are not specific to AAA but rather contribute to arteriopathies in general [76••].
AAA Genes
Cell Growth
Some genes associated with AAAs regulate the growth of cells within the aortic wall. One such example is CDKN2BAS1/ANRIL (rs1075727 [76••], rs10757278 SNPs [77]); this locus encodes an antisense RNA product that affects the expression of CDKN2A/B. These cyclin-dependent kinases mediate vascular tissue proliferation and inflammation, so a defect in this pathway can lead to several arterial pathologies, including AAA and coronary artery disease (“pan-atherosclerosis”)78. Another important gene is DAB2IP (rs7025486), which encodes DAB2-interacting protein, an inhibitor of cell proliferation and survival [79]. A disease-causing variant would facilitate aortic wall degeneration.
Inflammation
The relationship between vascular inflammation and aneurysmal disease is well established and is exemplified by the link between IL6R (interleukin 6 receptor) and AAAs [80]. IL6 binds its receptor, which then dimerises with gp-130 to form a complex that activates various intracellular kinases and the transcription factor STAT3. Upregulation of multiple components of this pathway is seen in AAA [81]. Another important example is ILRN (encodes IL1 receptor antagonist). IL-1 is a cytokine which triggers multiple inflammatory pathways by binding its receptor. It provides a target for drug therapeutics in inflammatory diseases such as rheumatoid arthritis. The endogenous receptor antagonist IL1Ra mimics the action of these drugs, and variants which upregulate the antagonist perhaps paradoxically increase the risk of AAA [82].
Lipid Metabolism
It is not surprising that atherosclerosis-mediated weakening of the abdominal aortic wall is influenced heavily by lipid metabolism genes. Sortilin 1 (SORT1 gene), for example, is a protein which interacts with PCSK9 acidic sphingomyelinase and apolipoprotein B100 in hepatocytes and thus regulates plasma LDL cholesterol levels [83]. Sortilin 1 is also present in vascular smooth muscle, where it regulates calcification [84]. Variants therefore confer a risk for arteriosclerosis and dyslipidaemia, with broad implications for cardiovascular disease. LDLR (LDL-receptor) variants are the commonest cause of familial hypercholesterolaemia and can also lead to a rise in plasma LDL and widespread risk for atherosclerosis [85]. Lipoprotein A, empirically atherogenic via an unclear mechanism [86], and lipoprotein-related protein 5 (LRP5), involved in bone, glucose, and lipid metabolism [87], are other examples.
AAA-Specific Variants
A meta-analysis of 6 GWAS studies from 2017 [76••] found four genetic loci that are strongly associated with AAA but are not associated with general cardiovascular risk factors or disease of other arteries:
-
1.
1q32.3: near the promotor of SMYD2, a protein involved in myofibril organisation. SMYD2 methylates Hsp90 and then forms a complex with it and the sarcomere protein Titin to contribute to sarcomere stability in skeletal and cardiac muscle [88]. It also methylates IL6/TNFα genes to inhibit NF-κB/ERK signalling [89]. Lower levels in AAA smooth muscle may contribute to the disease.
-
2.
13q12.11: LINC00540. This 3’-non-coding RNA region has no known function, but it appears to be linked to FGF9 (increased in AAA). FGF9 is a regulator of vascular smooth muscle/endothelial cell apoptosis [24••, 90]. It therefore impacts on angiogenesis, a key feature in AAA aetiology.
-
3.
20q13.12: near PCIFI/MMP9/ZNF335. The strongest effect of this variant is on PLTP, a cholesterol transport protein which, when overexpressed in mice, causes plaque lesions to rapidly increase in size and reduce their collagen content, thereby making them less stable. This is likely a VLDL-related mechanism. It may also contribute to the formation of plaques [91].
-
4.
21q22.2: ERG. This transcriptional regulator is involved in VEGF-mediated vascular development and endothelial cell activation. As previously mentioned, angiogenesis is a key step in the development of AAAs. It also mediates vascular inflammation via mechanisms similar to cIAP-2 and IL-8 [92].
Future Work
Sequencing Techniques
Much of the data in TAAD comes from whole exome sequencing (WES) of affected patients. This technique is ideal for identifying pathogenic variants in coding regions or canonical splice sites and allows us to compile a “genetic dictionary” of variants, mechanisms, prognoses, and treatment guidelines to aid with the management of aneurysmal diseases [5••]. As sequencing and testing capabilities are expanded and made more widely available, the number of disease-associated genetic variants will likely increase. Further genotype-guided prospective study will facilitate better understanding of the prognosis of each variant and more personalised treatment.
Screening in TAAD
Screening programs are mostly based on expert opinion, and there is very little official advice or evidence available about what to do if a rare genetic variant is found in a patient, how frequently to monitor the associated aneurysm, and when it is appropriate to routinely monitor extra-aortic arteries [3•]. Screening of second- and third-degree relatives of an affected individual is also not routinely recommended, though data suggest that almost a quarter of second-degree relatives and 15% of third-degree relatives will also be affected [3•]. Furthermore, there is a dearth of data on the quality of life and psychological effects of a proposed TAAD screening program. Given the low penetrance and heterogeneity of many TAAD variants, extensive testing of low-risk individuals could lead to substantial false positive and negative results, causing unnecessary worry and/or overtreatment.
Beyond traditional screening methods such as echocardiograms and computed tomography scans, there are many new techniques being developed which offer a unique insight into a patient’s aneurysm status. For example, a Belgian group is pioneering the use of FDG-PET/CT (fluorodeoxyglucose positron emission tomography/computed tomography) to monitor disease progression by measuring the degree of FDG uptake by the aortic wall; uptake is correlated with a higher number of adventitial inflammatory cells and a lower number of medial SMCs [93]. Some groups are in the process of developing blood tests to diagnose and monitor what may be radiologically silent aneurysms through unique patterns of RNA expression in the blood. One such signature has already been shown to identify aneurysms with 80% accuracy while also differentiating between ascending/descending and familial/sporadic aneurysms [94]. Micro RNAs (miRNA), which can be studied using microarrays of aortic tissue, like miR-574-5p can also differentiate between aneurysm patients and controls [95••]. Combined with the ever-expanding catalogue of genetic variants and their associated clinical phenotypes, such blood tests offer an exciting opportunity to greatly improve both diagnosis and management of aneurysmal disease.
Gene-Environment Interactions
The field of epigenetics studies the ways in which the environment can affect gene expression through DNA methylation, histone acetylation, and miRNA expression. Changes in DNA methylation in particular have been implicated in vascular SMC apoptosis, inflammation, and degradation of the ECM [96]. Such methylation patterns could be induced by long-term exposure to certain risk factors such as smoking, and this could explain in part why many AAA genetic loci are strongly associated with these risk factors. Multiple GWASes have found that smoking intensity, quantified as cigarettes smoked per day, segregates with multiple loci such as CHRNA5, CHRNB3, and CHRNA6—all nicotinic receptors—as well as near CYP2A6 and CYP2B6, which are enzymes that metabolise nicotine [97]. Though these did not reach the genome-wide level of significance, a SNP in CHRNB3 (rs1451240) became significant when the “cigarettes per day” measure was substituted by FTCD (Fagerström Test for Cigarette Dependence), a measure of nicotine dependence [97]. Variant carriers may find it more difficult to extinguish the smoking habit, thus increasing smoke exposure long term. Sustained exploration of these ideas could prove useful in further understanding the aetiology of aortic aneurysmal diseases.
Conclusions
Not all aortic aneurysms are created equal. Some grow faster. Some rupture earlier. Some do not present with clear clinical signs, while others do. Understanding the genetics of this disease offers great opportunities not only to improve identification of affected patients, but also then to stratify them into groups based on their specific genetic variant, thus facilitating more personalised care based on that specific group’s prognosis. Cataloguing the different variants and studying the nature of the aneurysms they produce, along with other aforementioned research streams, will be vital in the ongoing advancement of aneurysm care.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Underlying Cause of Death, 1999–2018 Request. https://wonder.cdc.gov/ucd-icd10.html. Accessed 30 Sep 2020.
Faggion Vinholo, T., Zafar, M. A., Ziganshin, B. A. & Elefteriades, J. A. Nonsyndromic thoracic aortic aneurysms and dissections-is screening possible? Semin Thorac Cardiovasc Surg 31, 628–634 (2019). A detailed analysis of how clinicians can translate knowledge of the prognoses of different mutations into actionable guidelines for screening relatives.
Mariscalco Giovanni, Debiec Radoslaw, Elefteriades John A., Samani Nilesh J., & Murphy Gavin J. Systematic review of studies that have evaluated screening tests in relatives of patients affected by nonsyndromic thoracic aortic disease. Journal of the American Heart Association 7, e009302 (2018). A systematic review looking at the prevalence of specific gene mutations in first, second, and third degree relatives. This has important implications for deciding who to screen.
Svensjö S, et al. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation. 2011;124:1118–23.
Brownstein, A. J., Ziganshin, B. A. & Elefteriades, J. A. Human aortic aneurysm genomic dictionary: is it possible? Indian J Thorac Cardiovasc Surg 35, 57–66 (2019). A discussion on the possibility of creating a genomic “dictionary”- a catalogue of different mutations with different clinical sequelae and guidelines. Features more detail about the characteristics of different mutations.
Ostberg, N. P., Zafar, M. A., Ziganshin, B. A. & Elefteriades, J. A. The Genetics of Thoracic Aortic Aneurysms and Dissection: A Clinical Perspective. Biomolecules 10, (2020).
Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–11.
Coady MA. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134:361.
Faggion Vinholo, T. et al. Genes Associated with thoracic aortic aneurysm and dissection: 2019 update and clinical implications. Aorta (Stamford) 7, 99–107 (2019). An annual update on our group's ongoing work to sequence patients at our center for new/existing variants.
Robertson EN, et al. Familial non-syndromal thoracic aortic aneurysms and dissections — incidence and family screening outcomes. Int J Cardiol. 2016;220:43–51.
Guo D, et al. Genetic variants in LRP1 and ULK4 are associated with acute aortic dissections. Am J Hum Genet. 2016;99:762–9.
Prakash S, LeMaire SA, Bray M, Milewicz DM, Belmont JW. Large deletions and uniparental disomy detected by SNP arrays in adults with thoracic aortic aneurysms and dissections. Am J Med Genet A. 2010;152A:2399–405.
Elefteriades, J. A., Sang, A., Kuzmik, G. & Hornick, M. Guilt by association: paradigm for detecting a silent killer (thoracic aortic aneurysm). Open Heart 2, e000169 (2015).
Luyckx, I., Loeys, B. L., & Curriculum topic: disease of the aorta and trauma to the aorta and heart. The genetic architecture of non-syndromic thoracic aortic aneurysm. Heart 101, 1678–1684 (2015).
Isselbacher, E. M., Lino Cardenas, C. L. & Lindsay, M. E. Hereditary influence in thoracic aortic aneurysm and dissection. Circulation 133, 2516–2528 (2016).
Pomianowski, P. & Elefteriades, J. A. The genetics and genomics of thoracic aortic disease. Annals of Cardiothoracic Surgery 2, 271–279–279 (2013).
Albornoz G, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg. 2006;82:1400–5.
Cocciolone AJ, et al. Elastin, arterial mechanics, and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018;315:H189–205.
Davis, E. C. Smooth muscle cell to elastic lamina connections in developing mouse aorta. Role in aortic medial organization. Lab Invest 68, 89–99 (1993).
Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344:477–9.
Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. 2013;184:907–24.
Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta and how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol. 2016;32:26–34.
Andelfinger G, Loeys B, Dietz H. A decade of discovery in the genetic understanding of thoracic aortic disease. Can J Cardiol. 2016;32:13–25.
Pinard, A., Jones, G. T. & Milewicz, D. M. Genetics of thoracic and abdominal aortic diseases: aneurysms, dissections, and ruptures. Circ Res 124, 588–606 (2019). An in-depth look at the pathogenesis several variants by the Milewicz group, including some not covered in this chapter.
Takeda, N. et al. TGF-β signaling-related genes and thoracic aortic aneurysms and dissections. International Journal of Molecular Sciences 19, 2125 (2018). A great resource for studying the precise molecular mechanisms of the TGF-β pathway in the clinical context of thoracic aortic aneurysms.
Jana, S., Hu, M., Shen, M. & Kassiri, Z. Extracellular matrix, regional heterogeneity of the aorta, and aortic aneurysm. Experimental & Molecular Medicine 51, 1–15 (2019). Contains much more detail about the structure of the aortic wall, along with a review of different ways of modelling it in the context of aortic aneurysms and of the different ECM genes involved in aneurysm formation.
Wang X, Khalil RA. Matrix metalloproteinases, vascular remodeling, and vascular disease. Adv Pharmacol. 2018;81:241–330.
Rabkin, S. W. Chapter Seven - The role matrix metalloproteinases in the production of aortic aneurysm. in Progress in Molecular Biology and Translational Science (ed. Khalil, R. A.) vol. 147 239–265 (Academic Press, 2017).
Verstraeten A, Alaerts M, Laer LV, Loeys B. Marfan syndrome and related disorders: 25 years of gene discovery. Hum Mutat. 2016;37:524–31.
Dietz HC, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–9.
Judge DP, Dietz HC. Marfan’s syndrome. The Lancet. 2005;366:1965–76.
Regalado ES, et al. Pathogenic FBN1 variants in familial thoracic aortic aneurysms and dissections. Clin Genet. 2016;89:719–23.
Tan L, et al. FBN1 mutations largely contribute to sporadic non-syndromic aortic dissection. Hum Mol Genet. 2017;26:4814–22.
Franken R, et al. Genotype impacts survival in Marfan syndrome. Eur Heart J. 2016;37:3285–90.
Franken Romy et al. Beneficial outcome of losartan therapy depends on type of FBN1 mutation in Marfan syndrome. Circulation: Cardiovascular Genetics 8, 383–388 (2015).
Hiratzka LF, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. J Am Coll Cardiol. 2010;55:e27–129.
Loeys BL, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–85.
Loeys BL, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81.
Tran-Fadulu V, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGFBR1 or TGFBR2 mutations. J Med Genet. 2009;46:607–13.
Teixidó-Tura G, et al. Heterogeneity of aortic disease severity in patients with Loeys-Dietz syndrome. Heart. 2016;102:626–32.
Jondeau G, et al. International Registry of Patients Carrying TGFBR1 or TGFBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016;9:548–58.
Milewicz D, et al. Precision medical and surgical management for thoracic aortic aneurysms and acute aortic dissections based on the causative mutant gene. J Cardiovasc Surg (Torino). 2016;57:172–7.
van de Laar IMBH, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–6.
Schepers, D. et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum Mutat 39, 621–634 (2018). This paper contains more details of known mutations in TGFB2/3 and SMAD2/3 which cause Loeys-Dietz Syndrome. It is a great exposition into the way in which basic science integrates well with clinical practice in the field of aortic disease.
Wischmeijer A, et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. Am J Med Genet A. 2013;161A:1028–35.
Regalado ES, et al. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res. 2011;109:680–6.
van der Linde D, et al. Aggressive cardiovascular phenotype of aneurysms-osteoarthritis syndrome caused by pathogenic SMAD3 Variants. J Am Coll Cardiol. 2012;60:397–403.
Micha D, et al. SMAD2 Mutations are associated with arterial aneurysms and dissections. Hum Mutat. 2015;36:1145–9.
Bertoli-Avella AM, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65:1324–36.
Boileau C, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–21.
Guo D-C, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–93.
Milewicz DM, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010;152A:2437–43.
Regalado ES, et al. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet. 2015;8:457–64.
Bellini C, Wang S, Milewicz DM, Humphrey JD. Myh11(R247C/R247C) mutations increase thoracic aorta vulnerability to intramural damage despite a general biomechanical adaptivity. J Biomech. 2015;48:113–21.
Zhu L, et al. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–9.
Harakalova M, et al. Incomplete segregation of MYH11 variants with thoracic aortic aneurysms and dissections and patent ductus arteriosus. Eur J Hum Genet. 2013;21:487–93.
Wang L, et al. Mutations in myosin light chain kinase cause familial aortic dissections. The American Journal of Human Genetics. 2010;87:701–7.
Gao N, et al. Signaling through myosin light chain kinase in smooth muscles. J Biol Chem. 2013;288:7596–605.
Wallace, S. E. et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genetics in Medicine 21, 144–151 (2019). An in-depth look at MYLK variants and a great example of how new variants should be studied clinically and in the laboratory.
Guo D, et al. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. The American Journal of Human Genetics. 2013;93:398–404.
Smith LB, et al. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res. 2011;90:182–90.
Shalhub S, et al. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J Vasc Surg. 2014;60:160–9.
Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–80.
Pyeritz, R. E. Ehlers–Danlos Syndrome (2009). https://doi.org/10.1056/NEJM200003093421009https://www.nejm.org/doi/pdf/10.1056/NEJM200003093421009.
Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32.
Kuang S-Q, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. 2016;126:948–61.
Guo D, et al. LOX Mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118:928–34.
Lindsay, M. E. & Dietz, H. C. The genetic basis of aortic aneurysm. Cold Spring Harb Perspect Med 4, a015909 (2014).
Kuivaniemi H, Ryer EJ, Elmore JR, Tromp G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev Cardiovasc Ther. 2015;13:975–87.
Kent, K. C. Clinical practice. Abdominal aortic aneurysms. N Engl J Med 371, 2101–2108 (2014).
Larsson E, et al. High frequency of thoracic aneurysms in patients with abdominal aortic aneurysms. Ann Surg. 2011;253:180–4.
Cheung C, Bernardo AS, Trotter MWB, Pedersen RA, Sinha S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin–dependent disease susceptibility. Nat Biotechnol. 2012;30:165–73.
Wahlgren, C. M., Larsson, E., Magnusson, P. K. E., Hultgren, R. & Swedenborg, J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J Vasc Surg 51, 3–7; discussion 7 (2010).
Johansen K, Koepsell T. Familial tendency for abdominal aortic aneurysms. JAMA. 1986;256:1934–6.
Kuivaniemi H, et al. Familial abdominal aortic aneurysms: collection of 233 multiplex families. J Vasc Surg. 2003;37:340–5.
Jones, G. T. et al. Meta-analysis of genome-wide association studies for abdominal aortic aneurysm identifies four new disease-specific risk loci. Circ Res 120, 341–353 (2017). The pivotal paper which discovered the new AAA-specific loci which are independent of traditional risk factor genes.
Helgadottir A, et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat Genet. 2008;40:217–24.
Kuivaniemi, H., Ryer, E., HR, Y. & Elmore, J. Genetic risk factors for abdominal aortic aneurysms. in 1–30 (2013).
Gretarsdottir S, et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat Genet. 2010;42:692–7.
Harrison SC, et al. Interleukin-6 receptor pathways in abdominal aortic aneurysm. Eur Heart J. 2013;34:3707–16.
Lindeman JHN, et al. Enhanced expression and activation of pro-inflammatory transcription factors distinguish aneurysmal from atherosclerotic aorta: IL-6- and IL-8-dominated inflammatory responses prevail in the human aneurysm. Clin Sci (Lond). 2008;114:687–97.
Interleukin 1 Genetics Consortium. Cardiometabolic effects of genetic upregulation of the interleukin 1 receptor antagonist: a Mendelian randomisation analysis. Lancet Diabetes Endocrinol 3, 243–253 (2015).
Kjolby, M. et al. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab 12, 213–223 (2010).
Zhang Z, Jiang W, Yang H, Lin Q, Qin X. The miR-182/SORT1 axis regulates vascular smooth muscle cell calcification in vitro and in vivo. Exp Cell Res. 2018;362:324–31.
Bradley DT, et al. A variant in LDLR is associated with abdominal aortic aneurysm. Circ Cardiovasc Genet. 2013;6:498–504.
Helgadottir A, et al. Apolipoprotein(a) genetic sequence variants associated with systemic atherosclerosis and coronary atherosclerotic burden but not with venous thromboembolism. J Am Coll Cardiol. 2012;60:722–9.
Galora S, et al. Low-density lipoprotein receptor-related protein 5 gene polymorphisms and genetic susceptibility to abdominal aortic aneurysm. J Vasc Surg. 2013;58:1062-1068.e1.
Du SJ, Tan X, Zhang J. SMYD proteins: key regulators in skeletal and cardiac muscle development and function. Anat Rec (Hoboken). 2014;297:1650–62.
Xu G, et al. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) production. J Biol Chem. 2015;290:5414–23.
Fibroblast Growth Factor-9 Activates c-kit progenitor cells and enhances angiogenesis in the infarcted diabetic heart. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4670684/. Accessed 05 Nov 2020.
Matthijs M, et al. Acute elevation of plasma PLTP activity strongly increases pre-existing atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:1277–82.
Dryden NH, et al. The transcription factor Erg controls endothelial cell quiescence by repressing activity of nuclear factor (NF)-κB p65. J Biol Chem. 2012;287:12331–42.
Courtois A, et al. 18F-FDG uptake assessed by PET/CT in abdominal aortic aneurysms is associated with cellular and molecular alterations prefacing wall deterioration and rupture. J Nucl Med. 2013;54:1740–7.
Wang, Y. et al. Gene expression signature in peripheral blood detects thoracic aortic aneurysm. PLoS One 2, (2007).
Boileau, A. et al. MiR-574–5p: A circulating marker of thoracic aortic aneurysm. Int J Mol Sci 20, (2019). This paper shows how genetic analysis of aortic aneurysms can help with diagnosis as well as clinical management, in this case by finding circulating miRNAs associated with the disease. As our genetic knowledge grows, so too will the capacity for such innovations to greatly improve clinical care.
Toghill BJ, et al. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis. 2015;241:121–9.
Rice JP, et al. CHRNB3 is more strongly associated with Fagerström test for cigarette dependence-based nicotine dependence than cigarettes per day: phenotype definition changes genome-wide association studies results. Addiction. 2012;107:2019–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
John Elefteriades is Principal of CoolSpine, serves on the Data and Safety Monitoring Board of Terumo, and consults for CryoLife. Asanish Kalyanasundaram declares no conflict of interest.
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Genetics
Rights and permissions
About this article

Cite this article
Kalyanasundaram, A., Elefteriades, J. The Genetics of Inheritable Aortic Diseases. Curr Cardiovasc Risk Rep 16, 13–24 (2022). https://doi.org/10.1007/s12170-022-00687-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12170-022-00687-x