
Abstract
Essential fatty acids and their metabolites (γ-linolenic acid [GLA], dihomo-GLA, arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid; prostaglandin E1; prostacyclin [PGI2]; PGI3; lipoxins; resolvins; protectins; maresins; and nitrolipids) prevent platelet aggregation, produce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, inhibit platelet activation, possess peroxisome proliferator-activated receptor-γ ligand activity, and release nitric oxide. Thus, they lower blood pressure, are anti-arrhythmic and anti-inflammatory in nature, reduce low-density lipoprotein cholesterol, ameliorate the adverse actions of homocysteine, activate telomerase, and have cytoprotective properties—actions that prevent atherosclerosis and cardiovascular disease. Because coronary heart disease (CHD) and atherosclerosis are low-grade systemic inflammatory conditions, it is likely that reduced formation of lipoxins, resolvins, protectins, maresins, and nitrolipids plays a significant role in the pathogenesis of CHD. Hence, development of stable synthetic analogues of lipoxins, resolvins, protectins, and maresins may form a new therapeutic approach to CHD and other low-grade systemic inflammatory conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dyslipidemia, diabetes mellitus, hypertension, and obesity are important risk factors for coronary heart disease (CHD). Smoking cessation, β-blockers, anti-platelet agents, angiotensin-converting enzyme (ACE) inhibitors, and lipid-lowering agents such as statins reduce the risk of vascular events to a moderate but important degree [1]. But these medicines are not without significant side effects.
We and others observed that in CHD, hypertension, diabetes mellitus, hyperlipidemias, and obesity, essential fatty acid (EFA) metabolism is abnormal such that plasma phospholipid concentrations of arachidonic acid (AA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) are low [2–6, 7••]. Despite the fact that deficiency of polyunsaturated fatty acids (PUFAs) is present in CHD, hypertension, diabetes, hyperlipidemias, and obesity, and increased intake of PUFAs is of benefit in these diseases, exact mechanism(s) of their protective action remained unexplained.
Low-Grade Systemic Inflammation Occurs in CHD
CHD is a low-grade systemic inflammatory condition because these subjects have enhanced plasma levels of reactive oxygen species (ROS), C-reactive protein (CRP), interleukin (IL)-6, and tumor necrosis factor (TNF)-α, and low circulating levels of endothelial nitric oxide (NO) and various PUFAs [8•]. Increased ROS decreases anti-oxidant content of the cells/tissues. This leads to an imbalance between the pro- and anti-oxidant molecules that favor tissue damage in CHD [8•]. Hence, it is important that adequate cytoprotective and anti-oxidant defenses are available in the cells/tissues to protect them against excess pro-oxidants.
Balance Between Pro- and Anti-Oxidant Defenses in CHD
Hs-CRP, lipoprotein-associated phospholipase A2 (Lp-PLA2), proinflammatory cytokines, ROS, and myeloperoxidase (MPO) have potent cytotoxic actions and are increased in patients with CHD and atherosclerosis. To protect themselves from these cytotoxic molecules, cells/tissues should have adequate amounts of superoxide dismutase, vitamin E, catalase, and glutathione group of anti-oxidants. But these are not always sufficient to protect tissues from the cytotoxic action of ROS and MPO. In this context, lipid-soluble molecules such as lipoxins (LXs), resolvins, protectins, maresins, and nitrolipids that are derived from the ubiquitous PUFAs deserve special attention. LXs, resolvins, protectins, and maresins possess cytoprotective and cardioprotective actions, by virtue of their ability to inhibit production of IL-6 and TNF-α, suppress free radical generation, and enhance tissue repair [7••, 8•]. When EPA, DHA, AA, and aspirin are administered in adequate amounts, LXs, resolvins, protectins, maresins, and nitrolipids are formed that prevent myocardial damage. This explains the beneficial actions of EPA/DHA/AA and aspirin in both primary and secondary prevention of CHD.
This implies that deficiency of PUFAs seen in CHD leads to reduced formation of NO, LXs, resolvins, protectins, maresins, and nitrolipids that would initiate atherosclerosis and CHD and/or worsen the existing disease. Based on this evidence, it is predicted that plasma levels of hs-CRP, ROS, MPO, Lp-PLA2, and LP will be increased, whereas those of LXs, resolvins, protectins, maresins, and nitrolipids are decreased in patients with CHD. Furthermore, a balance between these pro- and anti-inflammatory molecules may aid in predicting prognosis of CHD [9••]. For instance, subjects with low levels of LXs, resolvins, protectins, maresins, and nitrolipids may have poor outcome and higher incidence of cardiac failure, arrhythmias, and recurrence of myocardial infarction. Statins and thiazolidinediones enhance the formation of myocardial 15-epi-LXA4 and EFAs mediate some of their actions, which could explain the ability of statins and glitazones to prevent CHD [10–13]. This implies that those who have low levels of EFAs and their metabolites could be resistant to the beneficial action of statins and glitazones; such subjects will benefit from supplementation of AA/EPA/DHA.
Metabolism of EFAs
EFAs, which are needed for survival, cannot be synthesized in the body and hence have to be obtained in our diet; thus, they are essential [6–9••]. There are two types of naturally occurring EFAs in the body: the omega (n)-6 series derived from linoleic acid (LA; 18:2), and the n-3 series derived from α-linolenic acid (ALA; 18:3). Both n-6 and n-3 series are metabolized by the same set of enzymes to their respective long-chain metabolites.
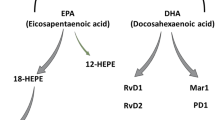
LA is converted to γ-linolenic acid (GLA; 18:3, n-6) by the action of the enzyme Δ6 desaturase (d-6-d), and GLA is elongated to form dihomo-GLA (DGLA; 20:3, n-6), the precursor of the 1 series of prostaglandins (PGs). DGLA can also be converted to AA (20:4, n-6) by the action of the enzyme Δ5 desaturase (d-5-d). AA forms the precursor of the 2 series of PGs, thromboxanes, and the 4 series of leukotrienes (LTs). ALA is converted to EPA (20:5, n-3) by d-6-d and d-5-d. EPA forms the precursor of the 3 series of PGs and the 5 series of LTs (Fig. 1). AA and EPA give rise to their respective hydroxy acids, which, in turn, are converted to respective LTs. In addition, AA, EPA, and DHA form precursor to anti-inflammatory compounds LXs, resolvins, protectins (neuroprotectin D1 is one such compound derived from DHA), and maresins [6–9••, 12–14••]. PGs, LTs, LXs, and resolvins are highly active, modulate inflammation, and are involved in several physiologic and pathologic processes. Although the terms EFAs and PUFAs are used interchangeably, it should be understood that all EFAs are PUFAs but all PUFAs are not EFAs. Only LA and ALA qualify to be EFAs, whereas GLA, DGLA, AA, EPA, and DHA are PUFAs. LA, GLA, DGLA, AA, ALA, EPA, and DHA are also called LCPUFAs (long-chain PUFAs). In general, many authors use the terms EFAs and PUFAs interchangeably. This convention is followed in the present discussion.

Schematic showing the metabolism of essential fatty acids, their action on various enzymes, and factors that account for their beneficial actions in coronary heart disease. AA—arachidonic acid; ACE—angiotensin-converting enzyme; CETP—cholesteryl ester transfer protein; DHA—docosahexaenoic acid; eNO—endothelial nitric oxide; EPA—eicosapentaenoic acid; FXR—farnesoid X receptor; LT—leukotriene; LXR—liver X receptor; NF—nuclear factor; PG—prostaglandin; PPAR—peroxisome proliferator—activated receptor; RAR-RXR—retinoic acid receptor-retinoid X receptor; ROS—reactive oxygen species; SREBP—sterol regulatory element-binding protein; TXA3—thromboxane A3; UCP—uncoupling protein
Factors Influencing the Metabolism of EFAs
Dietary LA and ALA compete with one another for the same set of enzymes, and Δ6 and Δ5 desaturases prefer n-3 to n-6. Oleic acid (OA; n-9) that is not an EFA is also metabolized by the same desaturases. But, in view of the preference of these enzymes to LA and ALA, under normal physiologic conditions, the metabolites of n-9 are formed only in trivial amounts. Hence, presence of significant amounts of 20:3 n-9, a metabolite of OA, in the cells and plasma indicates EFA deficiency.
Of several factors that influence the activities of desaturases and elongases, saturated fats, cholesterol, trans-fatty acids, alcohol, adrenaline, and glucocorticoids inhibit Δ6 and Δ5 desaturases. Pyridoxine, zinc, and magnesium are necessary co-factors for normal Δ6 desaturase activity. Insulin activates Δ6 desaturase, whereas diabetics have reduced Δ6 desaturase activity. The activity of Δ6 desaturase decreases with age. Oncogenic viruses and radiation inhibit Δ6 desaturase. Total fasting, protein deficiency, and glucose-rich diets reduce the activity of Δ6 desaturase. A fat-free diet and partial caloric restriction enhances Δ6 desaturase. Activities of Δ6 and Δ5 desaturases are decreased in diabetes mellitus, hypertension, hyperlipidemia, and metabolic syndrome. Trans- and saturated fatty acids, and cholesterol, interfere with EFA metabolism and promote inflammation, atherosclerosis, and CHD. This implies that trans-fats, saturated fats, and cholesterol have proinflammatory actions, whereas EFAs and PUFAs possess anti-inflammatory properties. The ability of trans-fats, saturated fats, and cholesterol to interfere with the formation of AA, EPA, and DHA from dietary LA and ALA could lead to decreased formation of LXs, resolvins, PGI2 (prostacyclin), PGI3, and other beneficial eicosanoids that prevent platelet aggregation, and leukocyte chemotaxis and activation. LXs, resolvins, protectins, and maresins decrease the formation of proinflammatory cytokines and produce vasodilatation, events that prevent or arrest atherosclerosis. In contrast, trans-fats, saturated fats, and cholesterol may directly activate leukocytes, induce the generation of free radicals, and enhance the production and release of proinflammatory cytokines that facilitate atherosclerosis. Trans-fats, saturated fats, and cholesterol may directly activate leukocytes and macrophages to enhance their ability to produce free radicals and proinflammatory cytokines. It is possible that trans-fats, saturated fats, and cholesterol may inhibit the formation of LXs, resolvins, protectins, maresins, PGI2, and PGI3. Thus, EFAs, especially EPA and DHA, are cytoprotective to endothelial cells, whereas trans-fats, saturated fats, and cholesterol produce endothelial dysfunction. AA, EPA, and DHA augment NO generation from endothelial cells and thus prevent endothelial dysfunction. In contrast, trans-fats, saturated fats, and cholesterol produce endothelial dysfunction and thus inhibit endothelial NO (eNO) production. Furthermore, NO quenches superoxide anion and thus prevents the cytotoxic action of superoxide anion and protects endothelial cells from free radical-induced damage. This implies that endothelial cells need adequate amounts of AA, EPA, and DHA so that they can generate physiologic amounts of eNO not only to prevent pathologic platelet aggregation and atherosclerosis but also to protect themselves from the cytotoxic actions of free radicals [2–4, 7••–9••].
NO reacts with PUFAs to yield their respective nitroalkene derivatives that can be detected in plasma. These nitroalkene derivatives, termed nitrolipids, produce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, inhibit platelet activation, and show anti-atherosclerotic properties [2–4, 7••–9••, 15•].
PUFAs Modulate HMG-CoA Reductase and ACE Enzymes, and Possess Anti-arrhythmic, Anti-hypertensive, Anti-atherosclerotic, Anti-inflammatory, Cytoprotective, and Cardioprotective Actions
PUFAs inhibit HMG-CoA reductase enzyme similar to statins and hence are useful in the treatment of hyperlipidemias [11]. In fact, statins and PUFAs have many overlap actions that suggest that PUFAs mediate many, if not all, actions of statins [11], and this could be one mechanism by which they lower cholesterol levels. Recent studies revealed that statins augment concentrations of LXs in the heart [12, 13], lending support to this concept. Furthermore, when a combination of statins and PUFAs was administered, a synergistic beneficial effect was seen in patients with combined hyperlipemia [16].
Several studies suggested that PUFAs have modulatory influence on renin secretion and action, yet at times are independent of both renin secretion and PG formation [17, 18]. Because the anti-hypertensive actions of PUFAs seem to be independent of formation of PGs, it is likely that fatty acids themselves are able to bring about this action. Alternatively, LXs, resolvins, protectins, and maresins formed from various PUFAs possibly have anti-hypertensive action by inhibiting the ACE activity. But this proposal needs to be verified and confirmed.
Previously, I showed that PUFAs inhibit leukocyte ACE activity [19]. Of all the fatty acids tested, EPA was the most effective (EPA > ALA > DHA > GLA > LA > AA), whereas AA was the least effective when their ability to inhibit purified ACE activity was tested. DHA and EPA were the most effective fatty acids in inhibiting the leukocyte ACE activity (EPA > DHA > ALA = AA > LA > GLA). On the other hand, PGs (PGE1, PGE2, PGI2, and PGF2α) and free radicals (superoxide anion, hydrogen peroxide, and hydroxyl radical) showed marginal (∼20%) inhibitory action on ACE activity. In contrast, NO showed powerful inhibitory action on ACE activity [19], whereas PUFAs enhanced eNO generation [20]. The effects of PUFAs on ACE activity and NO generation, and the inability of free radicals and PGs to suppress ACE activity, are interesting because there is a close interaction between platelets, leukocytes, and endothelial cells in the pathogenesis of CHD [8•]. Pro-atherosclerotic events such as hemodynamic forces, hyperlipidemia, hypertension, and smoking induce the expression of proinflammatory genes that initiate and accelerate atherosclerosis at the points of shear stress. These factors enhanced infiltration of intima by leukocytes and macrophages, induced low-level activation of nuclear factor-κB, elevated the expression of vascular cell adhesion molecule-1, intracellular adhesion molecule-1, IL-1, IL-6, and monocyte chemoattractant protein-1, and increased the production and release of free radicals and uncoupling protein expression in endothelial cells, platelets, and leukocytes in atherosclerosis-susceptible regions [21]. These events can be prevented, and the atherosclerosis process and onset of CHD can be arrested if there is adequate production of PGE1, PGI2, PGI3, LXs, resolvins, protectins, maresins, NO, and anti-inflammatory cytokines IL-4, IL-10, and transforming growth factor-β by endothelial cells. These protective molecules will be produced in adequate amounts provided there are sufficient stores of respective precursors such as PUFAs and L-arginine and their respective enzymes in the endothelial cells [8•]. These results suggest that when tissue concentrations of PUFAs are low, the activity of ACE will be high, resulting in increased formation of angiotensin-II and a simultaneous decrease in eNO.
AA, EPA, and DHA are converted in the presence of aspirin to epi-LXs, LXs, and resolvins that possess potent anti-inflammatory actions. Epi-LXs enhance the formation of eNO [3, 6, 8•, 9••, 22]. NO blocks the interaction between leukocytes and the vascular endothelium and stimulates the formation of PGI2, a potent vasodilator and platelet anti-aggregator, from AA [8•, 22]. This suggests that the beneficial actions of aspirin could be attributed not only to its ability to enhance the formation of PGI2 and suppress the synthesis of thromboxane A2, but also to the formation of epi-LXs and eNO. Thus, PUFAs regulate renin formation and action, inhibit angiotensin-II formation by its action on ACE activity, enhance eNO formation, and form precursors to beneficial biologically active molecules such as PGE1 (from DGLA), PGI2 (from AA), PGI3 (from EPA), LXs (from AA, EPA, and DHA), resolvins (from AA, EPA, and DHA), protectins (from DHA), maresins (from DHA), and 5,6-, 8,9-, 11,12-, and 14,15-EETs and hydroxyeicosatetraenoic acids (19- and 20-HETEs; from AA), and thus serve as endogenous regulators of vascular tone, platelet aggregation, and blood pressure [8•, 23••, 24••].
PUFAs Function as Endogenous Diuretics
Healthy volunteers administered EPA (3.9 g) and DHA (2.4 g) every day for 6 weeks showed significant increase in renal plasma flow and glomerular filtration rate, decrease in renal vascular resistance, and an increase in excretion of PGE3, with no change in blood pressure and heart rate [25]. A diet rich in evening primrose oil (a rich source of GLA and LA) and safflower oil decreased proteinuria, glomerular sclerosis, and tubular abnormalities in diabetic rats [26].
Spontaneously hypertensive rats showed lower systolic blood pressure when fed fish oil (EPA and DHA), evening primrose oil (a rich source of GLA), and fish oil plus evening primrose oil, suggesting that a combination of GLA, EPA, and DHA produces optimal beneficial actions with regard to renal indices and blood pressure [27], while fish oil prevented increase in blood pressure induced by high-salt diet in stroke-prone spontaneously hypertensive rats [28]. These results suggest that availability of optimal amounts of GLA and EPA/DHA reduces blood pressure and preserves renal function in diabetic and hypertensive rats. Thus, PUFAs may show actions similar to those observed with conventional, synthetic diuretics. These beneficial actions of PUFAs can be attributed to the formation of beneficial PGA, PGE3, PGI2, PGI3, LXs, resolvins, protectins, and maresins.
PUFAs Activate the Parasympathetic Nervous System
Autonomic function is an important factor that regulates heart rate, blood pressure, and cardiac rhythm. Vagal acetylcholine (ACh) and adrenergic norepinephrine and epinephrine regulate the autonomic function and thus the variations in heart rate variability (HRV) and baroreflex sensitivity. n-3 fatty acids reduce the risk of sudden death by preventing life-threatening cardiac arrhythmias by significantly increasing HRV [29]. A direct positive correlation was noted between the content of DHA in cell membranes and HRV index, suggesting an anti-arrhythmic effect of n-3 fatty acids [30]. Because increased parasympathetic tone is responsible for increase in ventricular fibrillation threshold and protects against ventricular arrhythmias, it is likely that EPA/DHA supplementation enhances parasympathetic tone. This is supported by the observation that EPA/DHA supplementation increases hippocampal ACh levels, the principal neurotransmitter of parasympathetic nerves [31]. Hence, it is likely that AA/EPA/DHA supplementation increases brain ACh levels [32, 33], leading to an increase in the parasympathetic tone and thus an increase in HRV and protection from ventricular arrhythmias.
Vagus nerve stimulation also inhibits TNF synthesis in the liver, and ACh significantly attenuated the release of proinflammatory cytokines TNF-α, IL-16, IL-1β, and IL-18 [34–37]. Thus, one mechanism by which PUFAs suppress inflammation could be by augmenting the release of ACh and enhancing the parasympathetic tone.
PUFAs Modulate CETP Activity
High-density lipoprotein cholesterol (HDL-C) is an independent risk factor for CHD. Higher plasma HDL-C is associated with a decreased incidence of CHD. Cholesteryl ester transfer protein (CETP) inhibition produces an increase in HDL by an action that increases reverse cholesterol transport that could prevent atherosclerosis and CHD [38].
In HepG2 cells, AA, EPA, and DHA reduced the levels of CETP mRNA by more than 50% of the control levels, with a corresponding significant decrease in the CETP mass [39], and a significant negative correlation was noted between plasma CETP activity and n-3 fatty acids, suggesting that PUFAs suppress CETP activity [40].
Thus, PUFAs, especially n-3 EPA and DHA, inhibit CETP and HMG-CoA reductase enzyme, and lower plasma triglycerides, cholesterol, and low-density lipoprotein cholesterol with little or no change in HDL-C, but were reported to be effective in arresting atherosclerosis and preventing CHD [41]. These results suggest that EPA and DHA are of use in the prevention and treatment of CHD, and at least a part of these beneficial actions could be attributed to increased formation of LXs, protectins, resolvins, maresins, and nitrolipids.
EPA and DHA Function as Endogenous Anti-Arrhythmic Molecules
PUFAs increased the electrical threshold for the induction of ventricular fibrillation that reduces the risk of developing malignant cardiac arrhythmias [42••]. Mitochondrial dysfunction induced by EFA deficiency could be ameliorated by the presence of normal levels of the EFAs in the n-3—enriched mitochondrial membrane phospholipids [43] that may account for the ability of EPA/DHA to decrease cardiac arrhythmias during myocardial ischemia. The recovery of mitochondrial energy metabolism and myocardial pump function during reperfusion is significantly reduced in n-3 PUFA-enriched hearts, suggesting that EPA and DHA limit myocardial injury during ischemia and reperfusion [42••, 43]. Some of these beneficial actions could be attributed to the formation of LXs, resolvins, protectins, and maresins from various PUFAs [42••].
PUFAs Modulate Telomere and Telomerase Activity
The loss of telomere results in cellular senescence because the cell can no longer divide and replicate itself. Overexpression of telomerase reverse transcriptase (TERT) prevents telomere attrition and enables cells to proliferate indefinitely, a characteristic of cancer cells. Telomere and telomerase are central to aging and several diseases, such as cancer, atherosclerosis, CHD, type 2 diabetes, and hypertension, and to the biology of stem cells.
Telomere shortening has been reported in patients with type 2 diabetes mellitus, hypertension, and insulin resistance [44–48], which are low-grade systemic inflammatory conditions in which plasma levels of various PUFAs are low. NO activates telomerase and delays endothelial cell senescence [49]. Leukocyte telomere length is a predictor of future CHD in middle-aged, high-risk men, whereas pravastatin, an HMG-CoA reductase inhibitor, substantially abrogated shortening of the telomere length in high-risk subjects [50].
n-3 PUFAs are of benefit in type 2 diabetes, hypertension, and hypertriglyceridemia and prevent CHD, in part, by enhancing NO generation from endothelial cells and decreasing insulin resistance [51•]. Tumor cells undergo apoptosis on exposure to n-3 PUFAs (especially in response to EPA, DHA, and GLA) due to increase in intracellular free radical generation and formation of lipid peroxides [52, 53]. Because NO and lipid peroxides modify telomerase activity, it is likely that PUFAs enhance or decrease activity of TERT in endothelial cells and tumor cells, respectively. This is supported by the observation that EPA and DHA inhibit hTERT activity in human colorectal adenocarcinoma cells [54, 55]. Thus, PUFAs can prevent, reverse, or arrest atherosclerosis and CHD by their ability to enhance eNO synthesis and possibly that of LXs, resolvins, protectins, maresins, and nitrolipids that, in turn, augment hTERT activity and prevent endothelial senescence.
Conclusions
PUFAs, especially EPA, DHA, and possibly GLA, DGLA, and AA, possess aspirin-like action; inhibit HMG-CoA and ACE enzymes; possess diuretic, anti-hypertensive, β-blocker—like actions; and modulate CETP and telomerase activities. PUFAs, especially n-3 fatty acids, enhance ACh levels and increase HRV due to their ability to augment parasympathetic tone. Furthermore, several studies suggested that PUFAs, especially n-3 fatty acids, are useful in the prevention and treatment of Alzheimer’s disease, schizophrenia, and depression [7••]. It is important to note that for their optimal action(s) PUFAs need many co-factors, such as folic acid, vitamin B12, vitamin B6, vitamin C, tetrahydrobiopterin (H4B), zinc, magnesium, calcium, L-arginine, and small amounts of selenium and vitamin E [4, 11]. Hence, it is important that these co-factors should also be provided in adequate amounts to bring about the beneficial actions of n-6 and n-3 PUFAs. Because statins, glitazones, and many antihypertensive and anti-arrhythmic drugs bring about their actions by modulating EFA/PUFA metabolism, it is likely that subclinical deficiency or altered metabolism of EFAs may subvert their actions/benefits. Some, if not all, of the beneficial actions of PUFAs could be attributed to their products’ LXs, resolvins, protectins, maresins, and nitrolipids, and their ability to enhance eNO formation that possess potent anti-inflammatory actions. CHD is a low-grade systemic inflammatory condition, whereas n-3 fatty acids and AA give rise to anti-inflammatory compounds such as LXs, resolvins, protectins, maresins, and nitrolipids. Hence, it is more than likely that the formation of LXs, resolvins, protectins, maresins, and nitrolipids from various PUFAs in the target tissues/cells is responsible for the beneficial actions observed (Fig. 2).

Schematic showing the relationship between polyunsaturated fatty acids (PUFAs) and cardiovascular diseases. COX-2—cyclooxygenase-2; HMGB1—high-mobility group box 1; iNOS—inducible nitric oxide synthase; LX—lipoxin; MIF—macrophage migration inhibitory factor; n—omega; NF—nuclear factor; ROS—reactive oxygen species; TNF—tumor necrosis factor
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Wald NJ, Law MR: A strategy to reduce cardiovascular disease by more than 80%. BMJ 2003, 326:1419. Erratum in: BMJ 2003, 327:586
Das UN: Essential fatty acid metabolism in patients with essential hypertension, diabetes mellitus and coronary heart disease. Prostaglandins Leukot Essent Fatty Acids 1995, 52:387–391
Das UN: Nutritional factors in the pathobiology of human essential hypertension. Nutrition 2001, 17:337–346
Das UN: A defect in the activity of delta6 and delta5 desaturases may be a factor predisposing to the development of insulin resistance syndrome. Prostaglandins Leukot Essent Fatty Acids 2005, 72:343–350
Mozaffarian D, Ascherio A, Hu FB, et al.: Interplay between different polyunsaturated fatty acids and risk of coronary heart disease in men. Circulation 2005, 111:157–164.
Das UN: Essential fatty acids—a review. Curr Pharm Biotechnol 2006, 7:467–482
•• Das UN: Can essential fatty acids reduce the burden of diseases? Lipids Health Dis 2008, 7:9. In this article, the author proposed that maintaining optimal levels of EFAs may reduce the occurrence of several diseases.
• Das UN: Cross talk among leukocytes, platelets, and endothelial cells and its relevance to atherosclerosis and coronary heart disease. Current Nutr Food Sci 2009, 5:75–93. This review provides a detailed discussion of the relationship between leukocytes, platelets, and endothelial cells in the pathogenesis of atherosclerosis and CHD, and the role of LXs, resolvins, and protectins in these diseases.
•• Das UN: Can endogenous lipid molecules serve as predictors and prognostic markers of coronary heart disease? Lipids Health Dis 2008, 7:19. For the first time, the author proposed that PUFAs, LXs, resolvins, and protectins could be used as predictors and prognostic indicators of CHD.
Levine L: Statins stimulate arachidonic acid release and prostaglandin I2 production in rat liver cells. Lipids Health Dis 2003, 2:1.
Das UN: Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot Essent Fatty Acids 2001, 65:37–40
Levy BD: Myocardial 15-epi-lipoxin A4 generation provides a new mechanism for the immunomodulatory effects of statins and thiazolidinediones. Circulation 2006, 114:873–875.
Birnbaum Y, Ye Y, Lin Y, et al.: Augmentation of myocardial production of 15-epi-lipoxin A4 by pioglitazone and atorvastatin in the rat. Circulation 2006, 114:929–935.
•• Ariel A, Serhan CN: Resolvins and protectins in the termination program of acute inflammation. Trends Immunol 2007, 28:176–183. This study outlines the ability of LXs, resolvins, and protectins to resolve inflammation.
• Das UN: A defect in the activity of Delta6 and Delta5 desaturases may be a factor in the initiation and progression of atherosclerosis. Prostaglandins Leukot Essent Fatty Acids 2007, 76:251–268. In this article, the author proposed that a defect in the activities of desaturases may initiate the process of atherosclerosis.
Nordoy A: Statins and omega-3 fatty acids in the treatment of dyslipidemia and coronary heart disease. Minerva Med 2002, 93:357–363.
O’Brien PM, Morrison R, Broughton Pipkin F: The effect of dietary supplementation with linoleic and gammalinolenic acids on the pressor response to angiotensin II—a possible role in pregnancy-induced hypertension? Br J Clin Pharmacol 1985, 19:335–342.
Kotchen TA, Welch WJ, Talwalkar RT: In vitro and in vivo inhibition of renin by fatty acids. Am J Physiol 1978, 234:E593–E599.
Kumar KV, Das UN: Effect of cis-unsaturated fatty acids, prostaglandins, and free radicals on angiotensin-converting enzyme activity in vitro. Proc Soc Exp Biol Med 1997, 214:374–379.
Okuda Y, Kawashima K, Sawada T, et al.: Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem Biophys Res Commun 1997, 232:487–491.
Jongstra-Bilen J, Haidari M, Zhu SN, et al.: Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med 2006, 203:2073–2083.
Wang W, Diamond SL: Does elevated nitric oxide production enhance the release of prostacyclin from shear stressed aortic endothelial cells? Biochem Biophys Res Commun 1997, 233:748–751.
•• Serhan CN, Yang R, Martinod K, et al.: Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J Exp Med 2009, 206:15–23. This study outlined the structure, formation, and properties of resolvins, protectins, and maresins.
•• Serhan CN: Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost 2009, 7(Suppl 1):44–48. This study also outlined the structure, formation, and properties of resolvins, protectins, and maresins.
Dusing R, Struck A, Gobel BO, et al.: Effects of n-3 fatty acids on renal function and renal prostaglandin E metabolism. Kidney Int 1990, 38:315–319.
Barcelli UO, Weiss M, Beach D, et al.: High linoleic acid diets ameliorate diabetic nephropathy in rats. Am J Kidney Dis 1990, 16:244–251.
Singer P, Berger I, Moritz V, et al.: N-6 and N-3 PUFA in liver lipids, thromboxane formation and blood pressure from SHR during diets supplemented with evening primrose, sunflower seed or fish oil. Prostaglandins Leukot Essent Fatty Acids 1990, 39:207–211.
Vaskonen T, Laakso J, Mervaala E, et al.: Interrelationships between salt and fish oil in stroke-prone spontaneously hypertensive rat. Blood Press 1996, 5:178–189.
Christensen JH, Gustenhoff P, Korup E, et al.: n-3 polyunsaturated fatty acids, heart rate variability and ventricular arrhythmias in post-AMI-patients. A clinical controlled trial. Ugeskr Laeger 1997, 159:5525–5529.
Christensen JH, Christensen MS, Dyerberg J, Schmidt EB: Heart rate variability and fatty acid content of blood cell membranes: a dose-response study with n-3 fatty acids. Am J Clin Nutr 1999, 70:331–337.
Minami M, Kimura S, Endo T, et al.: Dietary docosahexaenoic acid increases cerebral acetylcholine levels and improves passive avoidance performance in stroke-prone spontaneously hypertensive rats. Pharmacol Biochem Behav 1997, 58:1123–1129.
Almeida T, Cunha RA, Ribeiro JA: Facilitation by arachidonic acid of acetylcholine release from the rat hippocampus. Brain Res 1999, 826:104–111.
Das UN: Alcohol consumption and risk of dementia. Lancet 2002, 360:490
Borovikova LV, Ivanova S, Zhang M, et al.: Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405:458–462.
Bernik TR, Friedman SG, Ochani M, et al.: Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 2002, 195:781–788.
Wang H, Yu M, Ochani M, et al.: Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421:384–388.
Das UN: Vagus nerve stimulation, depression and inflammation. Neuropsychopharmacology 2007, 32:2053–2054
Barter PJ, Brewer HB Jr, Chapman MJ, et al.: Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol 2003, 23:160–167.
Hirano R, Igarashi O, Kondo K, et al.: Regulation by long-chain fatty acids of the expression of cholesteryl ester transfer protein in HepG2 cells. Lipids 2001, 36:401–406.
Smaoui M, Hammami S, Attia N, et al.: Modulation of plasma cholesteryl ester transfer protein activity by unsaturated fatty acids in Tunisian type 2 diabetic women. Nutr Metab Cardiovasc Dis 2006, 16:44–53.
GISSI Prevenzione Investigators: Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet 1999, 354:447–455.
•• Das UN: Essential fatty acids and their metabolites could function as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules. Lipids Health Dis 2008, 7:37. This is the original proposal coined by the author that suggested that PUFAs may function as an endogenous “polypill.”
Demaison L, Sergiel JP, Moreau D, Grynberg A: Influence of the phospholipid n-6/n-3 polyunsaturated fatty acid ratio on the mitochondrial oxidative metabolism before and after myocardial ischemia. Biochim Biophys Acta 1994, 1227:53–59.
Obana N, Takagi S, Kinouchi Y, et al.: Telomere shortening of peripheral blood mononuclear cells in coronary disease patients with metabolic disorders. Intern Med 2003, 42:150–153.
Nakashima H, Ozono R, Suyama C, et al.: Telomere attrition in white blood cell correlating with cardiovascular damage. Hypertens Res 2004, 27:319–325.
Sampson MJ, Winterborne MS, Hughes JC, et al.: Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care 2006, 29:283–289.
Benetos A, Gardner JP, Zureik M, et al.: Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension 2004, 43:182–185.
Demissie S, Levy D, Benjamin EJ, et al.: Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell 2006, 5:325–330.
Vasa M, Breitschopf K, Zeiher AM, Dimmeler S: Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res 2000, 87:540–542.
Brouilette SW, Moore JS, McMahon AD, et al.; for the West of Scotland Coronary Prevention Study Group: Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet 2007, 369:107–114.
• Das UN: Beneficial actions of polyunsaturated fatty acids in cardiovascular diseases: but, how and why? Current Nutr Food Sci 2008, 4:2–31. This review described the mechanism(s) by which PUFAs are beneficial in CHD.
Das UN: Tumoricidal action of cis-unsaturated fatty acids and their relationship to free radicals and lipid peroxidation. Cancer Lett 1991, 56:235–243.
Das UN, Huang YS, Begin ME, et al.: Uptake and distribution of cis-unsaturated fatty acids and their effect on free radical generation in normal and tumor cells in vitro. Free Rad Biol Med 1987, 3:9–14.
Eitsuka T, Nakagawa K, Suzuki T, Miyazawa T: Polyunsaturated fatty acids inhibit telomerase activity in DLD-1 human colorectal adenocarcinoma cells: a dual mechanism approach. Biochim Biophys Acta 2005, 1737:1–10.
Eitsuka T, Nakagawa K, Miyazawa T: Dual mechanisms for telomerase inhibition in DLD-1 human colorectal adenocarcinoma cells by polyunsaturated fatty acids. Biofactors 2004, 21:19–21.
Acknowledgment
Dr. Das is in receipt of Ramalingaswami Fellowship of Department of Biotechnology, New Delhi, India during the tenure of this study.
Disclosure
No potential conflict of interest relevant to this article was reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Das, U.N. Lipoxins, Resolvins, Protectins, Maresins, and Nitrolipids: Connecting Lipids, Inflammation, and Cardiovascular Disease Risk. Curr Cardio Risk Rep 4, 24–31 (2010). https://doi.org/10.1007/s12170-009-0068-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12170-009-0068-x