
Abstract
Caffeine is the most widely studied psychoactive molecule in history due to its many pharmacological activities and a high number of biological and physiological effects. In literature, there is a great number of applications that describe extraction, identification, and quantification of caffeine in foods and beverages. For this purpose, an extraction step is followed by an analytical technique for the identification and quantification of caffeine. This work proposes an innovative method in which sample preparation, separation, and detection steps are unified in a single step. To the best of our knowledge, this is the first report on the determination of caffeine in coffee, tea, and cocoa by means of an online extraction coupled to a liquid chromatographic system equipped with a photodiode array detector. The developed methodology was validated in terms of sensitivity, detection limits, accuracy, and precision. The advantages of this technique are (i) a significant reduction of analysis time (more than 70%) and of solvents used (the extraction step is integrated in the chromatographic analysis), (ii) the whole procedure is thus completely automated drastically reducing possible operator errors to occur, and (iii) easily realized by using a conventional monodimensional liquid chromatography system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Caffeine is a heterocyclic organic compound built from coupled pyrimidinedione and imidazole rings (Talik et al. 2012). This molecule is a methylated derivative of xanthine, the 1,3,7-methylxanthine. It is naturally contained in coffee, tea, cocoa, mate, and guaranà in different concentration values. The caffeine content of raw Arabica coffee is 0.9–1.4%, while in the Robusta variety, it is 1.5–2.6% and constitutes 2.5–5.5% of the dry matter of tea leaves, 0.2% of cocoa, and 0.5–1.5% of dry mate (Belitz et al. 2009). These plants, tea, and coffee, in particular, are used for beverages and food production consumed daily by world population, this representing the major sources of caffeine intake through diet (Frary et al. 2005).
Many pharmacological activities and a high number of biological and physiological effects have been attributed to caffeine with a relatively low toxicity, making caffeine the most widely studied psychoactive molecule in history. In particular, these effects have been associated mainly to the stimulation of the central and sympathetic nervous system (Owens et al. 2014; Cappelletti et al. 2015), and in the increase of blood circulation reducing the risk of coronary heart disease and stroke (Okuno et al. 2002). Others effects are lower incidence of Alzheimer’s (Arendash and Cao 2010), Parkinson’s (Postuma et al. 2012), Machado-Joseph’s (Gonçalves et al. 2013), and Huntington’s diseases (Simonin et al. 2013); the anti-inflammatory (Lee et al. 2014) and anti-cancer (Yang et al. 2005; Lu et al. 2006) actions; inhibition of the elevation of body fat percentage (Inoue et al. 2006); and modulation of glucose metabolism (Sarriá et al. 2015). Moreover, the intake of caffeine increases in physical and endurance performances and capacity during short-term, high-intensity exercise, as recently reported in a positive opinion from the European Food Safety Authority (EFSA 2011). For this reason, its use was regulated by the International Olympic Committee (positive controls for more than 12 mg mL−1 of urine) (Chapman and Mickleborough 2009).
A great number of published literature data described the extraction, identification, and quantification of caffeine in foods and beverages. Several caffeine extraction methods have been employed such as solid-liquid extraction (Senol and Aydin 2006; Srdjenovic et al. 2008; Bermejo et al. 2013), solid phase extraction (Rodrigues et al. 2007; Hackett et al. 2008), supercritical fluid extraction (Tello et al. 2011; Gadkari and Balaraman 2015), and microwave-assisted extraction (Tzanavaras and Themelis 2007; Rostagno et al. 2011). Numerous analytical techniques (spectrophotometry, spectrofluorimetry, Fourier transform-infrared spectrophotometry, gas chromatography coupled to mass spectrometry, thin-layer chromatography, micellar electrokinetic chromatography, and capillary electrophoresis) were used for qualitative and quantitative analyses of caffeine in different extracts (Paradkar and Irudayaraj 2002; Armenta et al. 2005; Khanchi et al. 2007; Meinhart et al. 2010; Gałan et al. 2015; Pasias et al. 2017). Among them, reversed phase liquid chromatography (RP-LC) is the technique most commonly used for this purpose coupled to UV-Vis or mass spectrometer detectors (Smrke et al. 2015; Baeza et al. 2016; Magalhães et al. 2016; Regazzoni et al. 2016; Heeger et al. 2017).
For all the above described analytical techniques, the most demanding step, in terms of time and resources, is the extraction method. Often, the sample pretreatment steps were carried out reacting a certain solvent (e.g., water, ethanol, methanol, and acetonitrile) volumes with the matrix, and subsequently required a prolonged heating to eliminate the extraction solvent. This leads to a loss of time energy and high solvent volume consumption. Moreover, a possible degradation of thermally sensitive compounds would be encountered during the extraction process. For these reasons, the development of an innovative technique that involves an online extraction coupled with a chromatographic system can help in saving energy and solvent consumption. An online extraction method can be more efficient and energy saving than a conventional pretreatment step, because all the extraction procedures (time for extraction and concentration) are eliminated reducing the risk of degradation of thermally sensitive compounds.
Previous literature reported applications of online extraction methods coupled to a liquid chromatographic system, such as microwave-assisted extraction (MAE-LC), dynamic sonication-assisted extraction (DSAE-LC), pressurized liquid extraction (PLE-LC), and pressurized hot water extraction (PHWE-LC) (Hyötyläinen 2007). Applications of the online supercritical fluid extraction (SFE) coupled with LC or supercritical fluid chromatography (SFC) (del Pilar Sanchez-Camardo et al. 2017; Zoccali et al. 2017) have been also reported. All these techniques are suitable for solid and semi-solid matrices, and for the analysis of both polar and non-polar analytes.
Recently, Ferreira and coworkers (Ferreira et al. 2016) described an innovative online extraction procedure from a solid matrix directly coupled to high-performance liquid chromatography (OLE-LC), in which sample preparation, separation, and detection steps are unified in a single step. The most important advantages of this approach are the greenest extraction (no extra solvents except for the mobile phases are consumed), sample pretreatment are not needed, risks of sample contamination reduction, and use of sample in milligram level. Moreover, unlike other methods above mentioned, no additional pumps and/or valves are necessary with respect to a conventional monodimensional LC system. This innovative strategy does not require any additional equipment to a classical LC instrumentation.
According to the previous considerations, this work presents an alternative strategy for the quantification of caffeine in foodstuffs using an OLE-LC method. The method was developed, optimized, and fully validated before its application to the analysis of different samples. To the best of our knowledge, this is the first report on the determination of caffeine in coffee, tea, and cocoa by means of an online extraction coupled to a liquid chromatographic system equipped with a photodiode array detector. Developed OLE-LC method was compared to conventional ultrasound-assisted solid-liquid extraction approach followed by LC analysis.
Experimental Conditions
Materials and Samples
This research was carried out on a 100% Arabica coffee, a decaffeinated 100% Arabica coffee, a black tea, and a cocoa powder. All the samples were purchased in a local market, and were stored at room temperature before the analysis. Coffees, tea, and cocoa samples were analyzed without any pretreatment for the OLE-LC analyses, while were subjected to ultrasound-assisted solid-liquid extraction (UASLE) before the RP-HPLC/PDA analysis in order to compare the two techniques.
Caffeine standard and C18 silica gel (particle size 200–400 mesh, 70-Å pore size) were purchased from Merck KGaA (Darmstadt, Germany). Solvents employed for the extraction procedure and for HPLC analyses were formic acid, methanol, water, and acetonitrile and were obtained from Merck KGaA (Darmstadt, Germany).
Instrumentation
Sample extractions were carried out in an Elma ultrasonic bath S 30H (Elma Schmidbauer GmbH, Germany), and the extracts were centrifuged in an Epperndorf 5804 R centrifuge (Eppendorf AG, Germany).
HPLC analyses were carried out using a Shimadzu Prominence LC-20A system (Shimadzu, Milan, Italy), including a CBM-20A controller, two LC-20 AD XR dual-plunger parallel-flow pumps, a DGU-20A3 online degasser, a 7725I Rheodyne® six-port two-position valve, and a CTO-20 AC column oven. As detector, an SPD-M10Avp diode array detector was employed, for determination of caffeine. Data acquisition was performed by the LCMSsolution ver. 3.7 software (Shimadzu).
Extraction of Caffeine by UASLE
The extraction procedure was carried out on 1 g of each sample, extracted with three aliquots of 10 mL of water (0.1% of formic acid), water/acetonitrile/formic acid (5:4.9:0.1 v/v/v), and acetonitrile (0.1% of formic acid) in an ultrasonic bath at room temperature (for 10 min at 50 Hz). The extracts were centrifuged at room temperature (for 5 min at 4000 rpm), combined, filtered on a filter paper, and brought to dryness in a rotary evaporator. The extracts thus obtained were dissolved in 1 mL of a mixture of water/acetonitrile/formic acid (5:4.9:0.1 v/v/v), filtered on Acrodisc filter 0.45 μm Sigma-Aldrich (Milan, Italy), and injected into HPLC. Each sample was analyzed in triplicate.
Extraction of Caffeine by OLE
Two milligrams of each sample was inserted in a SecurityGuard holder, and the chamber was filled with ca. 300 mg of C18 silica gel (Fig. 1a). The holder was equipped with two column frits (obtained from a disused HPLC column) in order to prevent the flow of particles through the separation system. As can be seen in Fig. 1, the SecurityGuard holder filled with the sample and C18 silica gel was connected to the chromatographic system and replaced instead of sample loop. Figure 1b shows the six-port two-position valve in “load” position. In this configuration, the mobile phase flow reached the LC column, bypassing the SecurityGuard holder. When the LC column was equilibrated, the six-port two-position valve was switched in “inject” position, and the mobile phase flow passed through the SecurityGuard holder filled with the sample and reached the LC column (Fig. 1c), where the separation was achieved. In this setup, the mobile phase represents both the extraction solvent for caffeine and the eluent for the chromatographic separation. The six-port two-position valve can be re-switched in load position when the chromatographic separation is over.

SecurityGuard holder (a) and setup of the six-port, two-position valve used for the online extraction (b “load” and c “inject” position)
HPLC Analytical Condition
Analyses were carried out on a LiChrosorb RP-18, 200 × 4.6 mm I.D. with particle size of 5 μm (Merck KGaA, Darmstadt, Germany). The injection volume was 2 μL, mobile phase consisted of water/formic acid (90:10, v/v) (solvent A) and water/acetonitrile/formic acid (40:50:10, v/v/v) (solvent B), and the step-wise gradient profile was as follows: 0 min, 0% B; 5 min, 0% B; 45 min, 30% B; 55 min, 100% B; and 60 min, 100% B. Flow rate was 1.0 mL min−1, data were acquired using a photodiode array detector in the range 200–400 nm, and the chromatograms were extracted at 280 nm. Time constant was 0.64 s and sample frequency 1.5625 Hz. Data acquisition was performed by Shimadzu LCMS solution software version 3.70.
Sample Extraction Method Validation
UASLE recovery of caffeine was determined by carrying out the extraction procedure used for the samples of our interest on a sample of decaffeinated coffee fortified of known amounts (0.5 and 5 mg L−1) of caffeine. Every extract thus obtained was analyzed in triplicate. Recovery was calculated using the following formula:
OLE-LC recovery of caffeine was determined by carrying out the online extraction directly coupled to HPLC analysis loading in the SecurityGuard holder both a decaffeinated coffee fortified of known amounts of caffeine and 2 μL of caffeine standard solution in a different concentration range (0.1–5 mg mL−1). Every extraction was carried out in triplicate.
RP-HPLC Method Validation and Statistical Analysis
To quantify caffeine content in the various samples tested, calibration curve has been constructed. Seven different concentrations of caffeine, in the range between 0.1 and 10 mg mL−1, prepared by diluting a stock solution of about 20 mg mL−1, using water as a solvent, were analyzed for five consecutive times by HPLC under the same chromatographic conditions optimized for the samples.
Limit of detection (LOD) and limit of quantification (LOQ) values, following the EURACHEM guidelines (Magnusson and Örnemark 2014), were also calculated. The instrumental intraday and interday repeatability and recovery were calculated on six replicated injections at one concentration level of caffeine.
Results and Discussions
Many scientific publications have described both identification and quantification of caffeine in foods and beverages by means of reversed phase HPLC in combination with UV and MS detection (Tzanavaras and Themelis 2007; Rostagno et al. 2011; Baeza et al. 2016; Magalhães et al. 2016; Regazzoni et al. 2016; Heeger et al. 2017). All these studies started with a pretreatment sample, the extraction step, before the chromatographic analysis. The aim of this work was to quantify caffeine in coffee, tea, and cocoa with an innovative HPLC method in which preparation, separation, and detection steps are unified in a single step (OLE-LC). To the best of our knowledge, this is the first report for the quantification of an analyte using this online extraction coupled to liquid chromatography analysis. There is only one recent published article that describes, for the first time, this innovative technique, but only a chromatographic fingerprint of plant leaves was proposed (Ferreira et al. 2016).
The starting point was to validate the RP-HPLC/PDA method in terms of sensitivity, detection limits, accuracy, and precision. Calibration curve of caffeine was constructed under the same chromatographic conditions optimized for samples analysis in order to quantify its content (y = 14,146x + 34,646, R2 = 0.998). The validation process provided LOD value of 0.031 mg L−1 and LOQ 0.056 mg L−1. Concerning the intraday and interday repeatability, coefficient of variation (CV) values for both retention time and area of < 4% demonstrated good precision at the concentration level tested. Concerning recovery, a value of 90% was obtained for the caffeine at the concentration level tested, thus demonstrating good accuracy. The linear dynamic range of the PDA system was also evaluated. Solutions of caffeine were prepared at seven concentration levels (0.1–10 mg mL−1), with each level subjected to HPLC/PDA analysis. PDA response factors were calculated at each concentration level (n = 3), and plotted against mg mL−1 values in a graph (Fig. 2). As can be seen, detector linearity was maintained for concentration ranging from 1 to 10 mg mL−1, while for 0.1 and 0.5 mg mL−1, there was a slightly deviation from linearity. This could not be a problem for this application, because the caffeine content in coffee, tea, and cocoa is generally higher than 3 mg g−1 (Belitz et al. 2009).

Graph reporting the PDA dynamic range for caffeine
After LC method validation, it was necessary to optimize online extraction coupled to LC analysis, considering different parameters: concentration of caffeine loaded into the holder, mobile phase composition, and extraction time (the inject position time of the six-port, two-position valve; Fig. 1c), as reported in Table 1. As can be seen in Table 1, higher recovery values of caffeine ranging from 51 to 54% were obtained using water and acetonitrile acidified with 0.1% of formic acid as mobile phases, and maintaining the six-port, two-position valve in the inject position for all the analysis time. Recovery values were independent to the loaded caffeine concentration (2 μL of a standard solution ranging from 100 to 5000 mgL−1, or 2 mg of decaffeinated sample spiked with a 5000 mgL−1 solution of caffeine). Slightly lower recovery values (ca. 45%) were observed using water and methanol acidified with 0.1% of formic acid as mobile phase, when 2 μL of a 5000 mgL−1 caffeine standard solution, or 2 mg of decaffeinated sample spiked with a 5000 mgL−1 solution of caffeine, was loaded into the holder. It is interesting to note how the inject position time of the six-port, two-position valve affected recovery values: increasing the exposition to the mobile phase, recovery increased. In fact, the exposition of the sample to the mobile phase for 5 min or 10 min led to 10 and 20% recovery value increase, respectively. Based on the above considerations, the OLE-LC method was validated carrying out the online extraction with water and acetonitrile acidified with 0.1% of formic acid as mobile phase and maintaining the six-port, two-position valve in the inject position for all the analysis time.
The online extraction method was then compared to a most commonly used UASLE, in order to highlight the differences in terms of recovery, time spent, and volume of solvents employed. Coffee, tea, and cocoa samples were extracted as reported in the “Materials and Samples” section. One gram of each sample was subjected to solvent extraction, with a total amount of solvent employed to carry out the extraction procedure of 30 mL. This volume has been completely reset in the OLE-LC. Total extraction time for the UASLE was more or less 3 h: ca. half of the time was employed for the extraction procedure and ca. 90 min was spent to brought to dryness the extract in a rotary evaporator. Also in this case, the extraction time has been completely reset in the OLE-LC. Moreover, in order to obtain the UASLE precision, the total analysis time was ca. 8 h (three parallel extraction procedures, time to brought to dryness the three extracts, and three HPLC analyses), with respect to the OLE-LC in which the total analysis time corresponded to the HPLC separation time. Another advantage of the online extraction method is that, being completely automated, drastically reduce possible operator errors to occur.
Concerning the recovery of the UASLE, known amount of caffeine was added to a decaffeinated coffee, and the sample was subjected to the same solvent extraction used for coffee, tea, and cocoa. Recovery value obtained with this method for caffeine was 77.2% (4.37 RSD %). This value was higher than that obtained with the OLE-LC method (recovery 51.3%, 2.04 RSD %). There are not significant differences in the solvent volume employed for the extraction and in the time of sample exposition to the mobile phase/extraction solvent for UASLE and OLE-LC methods. The principal differences between the two methods tested were the use of ultrasounds in the solid-liquid extraction and the percentage composition of the solvent employed for the extraction process. UASLE method started with a 100% of water acidified with the 0.1% of formic acid, and ended with a 100% of acetonitrile acidified with the 0.1% of formic acid. The OLE-LC method also being with a 100% of water acidified with the 0.1% of formic acid, but according to the LC step-wise gradient profile, caffeine eluted at 28.90 min, with the 16% of acetonitrile. Based on the considerations just made, dynamic caffeine extraction was carried out principally with acidified water with a low amount of acetonitrile, with respect to UASLE that used up to 100% of acetonitrile for the extraction procedure.
Perhaps it would be more analytically correct to compare an UASLE method in which acetonitrile percentage did not exceed 16%, but, in order to elucidate advantages and disadvantages of all the techniques tested in this work, a conventional ultrasound-assisted solid-liquid extraction (Rostagno et al. 2011) was chosen.
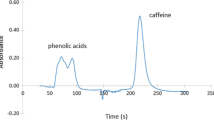
Figure 3a, b shows the UASLE-RP-HPLC/PDA and OLE-LC/PDA chromatograms of caffeine in cocoa. As can be seen in Fig. 3, retention time of all the molecules in OLE-LC/PDA chromatogram are delayed by 1 min compared to UASLE-RP-HPLC/PDA chromatogram. This delay is to be attributed to the two different instrumental setups. For the UASLE-RP-HPLC/PDA analyses, mobile phase flows through the sample loop (2 μL) in 0.12 s, while passed through the SecurityGuard holder (2-cm length × 0.8-cm diameter) in 1 min for OLE-LC/PDA analyses.

a UASLE-RP-HPLC/PDA and b OLE-LC/PDA chromatograms of caffeine in cocoa, extracted at 280 nm
Table 2 reports the concentration (mg kg−1 ± standard deviation) of caffeine in all analyzed samples. Among analyzed samples, tea is the richest in caffeine, while coffee and cocoa present lower amount of molecule of our interest. These results are in good accordance with those present in literature for caffeine in the samples of our interest (Brunetto et al. 2007; Belitz et al. 2009; Jeszka-Skowron et al. 2015). As reported in Table 2, quantitative data of caffeine for the same sample are not in accordance between the two methods used. These differences can be justified taking into consideration different recovery values obtained with the two methods used.
Recovery values of 51.3% for an extraction technique are not excellent, but could be a good compromise considering all the advantages that this innovative technique leads. Certainly, it will be not the technique of choice for the quantification of trace molecules, but can represent a valid alternative for the analysis of the most abundant compounds in solid food samples, as the case of caffeine in cocoa, tea, and coffee.
Conclusions
This work provides for the first time a new method for the quantification of caffeine in coffee, tea, and cocoa by means of an online extraction coupled to liquid chromatography analysis. The main advantages of this technique are a significant reduction of analysis time (more than 70%) and of solvents used (the extraction step is removed) with respect to a classical solid-liquid extraction method. It is completely automated, drastically reduces possible operator errors to occur, and easily realizable by using a conventional monodimensional LC system. The developed methodology could be applied to the analysis of different analytes in solid food matrices, reducing analysis time, costs, and solvent consumption.
References
Arendash GW, Cao C (2010) Caffeine and coffee as therapeutics against Alzheimer’s disease. J Alzheimers Dis 20:S117–S126
Armenta S, Garrigues S, de la Guardia M (2005) Solid-phase FT-Raman determination of caffeine in energy drinks. Anal Chim Acta 547:197–203
Baeza G, Sarriá B, Bravo L, Mateos R (2016) Exhaustive qualitative LC-DAD-MSn analysis of Arabica green coffee beans: cinnamoyl-glycosides and cinnamoylshikimic acids as new polyphenols in green coffee. J Agric Food Chem 64:9663–9674
Belitz HD, Grosch W, Schieberle P (2009) Coffee, tea, cocoa. In: Food chemistry, 4th revised and extended edition. Springer-Verlag Berlin Heidelberg, Berlin, pp 938–970
Bermejo DV, Luna P, Manic MS, Najdanovic-Visak V, Reglero G, Fornari T (2013) Extraction of caffeine from natural matter using a bio-renewable agrochemical solvent. Food Bioprod Process 91:303–309
Brunetto MR, Gutiérrez L, Delgado Y, Gallignani M, Zambrano A, Gómez Á, Ramos G, Romero C (2007) Determination of theobromine, theophylline and caffeine in cocoa samples by a high-performance liquid chromatographic method with on-line sample cleanup in a switching-column system. Food Chem 100:459–467
Cappelletti S, Piacentino D, Sani G, Aromatario M (2015) Caffeine: cognitive and physical performance enhancer or psychoactive drug? Curr Neuropharmacol 13:71–88
Chapman RF, Mickleborough TD (2009) The effects of caffeine on ventilation and pulmonary function during exercise: an often-overlooked response. Phys Sportsmed 37:97–103
del Pilar Sanchez-Camardo A, Parada-Alfonso F, Ibanez E, Cifuentes A (2017) Online coupling of supercritical fluid extraction and chromatography techniques. J Sep Sci 40:213–227
EFSA Panel on Dietetic Products Nutrition and Allergies (EFSA NDA Panel) (2011) Scientific Opinion on the substantiation of health claims related to caffeine and increase in physical performance during short-term high-intensity exercise (ID 737, 1486, 1489), increase in endurance performance (ID 737, 1486), increase in endurance capacity (ID 1488) and reduction in the rated perceived exertion/effort during exercise (ID 1488, 1490) pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFSA J 9:2053
Ferreira VG, Leme GM, Cavalheiro AJ, Funari CS (2016) Online extraction coupled to liquid chromatography analysis (OLE-LC): eliminating traditional sample preparation steps in the investigation of solid complex matrice. Anal Chem 88:8421–8427
Frary CD, Johnson RK, Wang MQ (2005) Food sources and intakes of caffeine in the diets of persons in the United States. J Am Diet Assoc 105:110–113
Gadkari PV, Balaraman M (2015) Solubility of caffeine from green tea in supercritical CO2: a theoretical and empirical approach. J Food Sci Technol 52:8004–8013
Gałan A, Jesionek W, Majer-Dziedzic B, Lubicki L, Choma IM (2015) Investigation of different extraction methods on the content and biological activity of the main components in Coffea arabica L. extracts. J Planar Chromatogr - Mod TLC 28:178–183
Gonçalves N, Simões AT, Cunha RA, de Almeida LP (2013) Caffeine and adenosine A2A receptor inactivation decrease striatal neuropathology in a lentiviral-based model of Machado-Joseph disease. Ann Neurol 73:655–666
Hackett J, Telepchak MJ, Coyer MJ (2008) Analysis of total caffeine and other xanthines in specialty coffees using mixed mode solid-phase extraction and liquid chromatography-diode-array detection after microwave digestion. J Anal Toxicol 32:695–701
Heeger A, Kosińska-Cagnazzo A, Cantergiani E, Andlauer W (2017) Bioactives of coffee cherry pulp and its utilisation for production of cascara beverage. Food Chem 221:969–975
Hyötyläinen T (2007) Principles, developments and applications of on-line coupling of extraction with chromatography. J Chromatogr A 1153:14–28
Inoue H, Kobayashi-Hattori K, Horiuchi Y, Oishi Y, Arai S, Takita T (2006) Regulation of the body fat percentage in developmental-stage rats by methylxanthine derivatives in a high-fat diet. Biosci Biotechnol Biochem 70:1134–1139
Jeszka-Skowron M, Zgoła-Grzes’kowiak A, Grzes’kowiak T (2015) Analytical methods applied for the characterization and determination of bioactive compounds in coffee. Eur Food Res Technol 240:9–31
Khanchi AR, Mahani MK, Hajihosseini M, Maragheh MG, Chaloosi M, Bani F (2007) Simultaneous spectrophotometric determination of caffeine and theobromine in Iranian tea by artificial neural networks and its comparison with PLS. Food Chem 103:1062–1068
Lee I-A, Kamba A, Low D, Mizoguchi E (2014) Novel methylxanthine derivative-mediated anti-inflammatory effects in inflammatory bowel disease. World J Gastroenterol 20:1127–1138
Lu Y-P, Lou Y-R, Xie J-G, Peng Q-Y, Zhou S, Lin Y, Shih WJ, Conney AH (2006) Caffeine and caffeine sodium benzoate have a sunscreen effect, enhance UVB-induced apoptosis, and inhibit UVB-induced skin carcinogenesis in SKH-1 mice. Carcinogenesis 28:199–206
Magalhães LM, Machado S, Segundo MA, Lopes JA, Páscoa RNMJ (2016) Rapid assessment of bioactive phenolics and methylxanthines in spent coffee grounds by FT-NIR spectroscopy. Talanta 147:460–467
Magnusson B, Örnemark U (eds) (2014) Eurachem guide: the fitness for purpose of analytical methods – a laboratory guide to method validation and related topics, 2nd edn. Available from www.eurachem.org. Accessed 03 April 2018
Meinhart AD, Bizzotto CS, Ballus CA, Prado MA, Bruns RE, Filho JT, Godoy HT (2010) Optimisation of a CE method for caffeine analysis in decaffeinated coffee. Food Chem 120:1155–1161
Okuno T, Sugiyama T, Tominaga M, Kojima S, Ikeda T (2002) Effects of caffeine on microcirculation of the human ocular fundus. Jpn J Ophthalmol 46:170–176
Owens JA, Mindell J, Baylor A (2014) Effect of energy drink and caffeinated beverage consumption on sleep, mood, and performance in children and adolescents. Nutr Rev 72:65–71
Paradkar MM, Irudayaraj J (2002) Rapid determination of caffeine content in soft drinks using FTIR-ATR spectroscopy. Food Chem 78:261–266
Pasias IN, Kiriakou I, Proestos C (2017) Development of a rapid method for the determination of caffeine in coffee grains by GC-FID-a fully validated approach. Antioxidants 6:67
Postuma RB, Lang AE, Munhoz RP, Charland K, Pelletier A, Moscovich M, Filla L, Zanatta D, Rios Romenets S, Altman R, Chuang R, Shah B (2012) Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology 79:651–658
Regazzoni L, Saligari F, Marinello C, Rossoni G, Aldini G, Carini M, Orioli M (2016) Coffee silver skin as a source of polyphenols: high resolution mass spectrometric profiling of components and antioxidant activity. J Funct Foods 20:472–485
Rodrigues CI, Marta L, Maia R, Miranda M, Ribeirinho M, Máguas C (2007) Application of solid-phase extraction to brewed coffee caffeine and organic acid determination by UV/HPLC. J Food Compos Anal 20:440–448
Rostagno MA, Manchón N, D’Arrigo M, Guillamón E, Villares A, García-Lafuente A, Ramos A, Martínez JA (2011) Fast and simultaneous determination of phenolic compounds and caffeine in teas, mate, instant coffee, soft drink and energetic drink by high-performance liquid chromatography using a fused-core column. Anal Chim Acta 685:204–221
Sarriá B, Martínez-López S, Sierra-Cinos JL, Garcia-Diz L, Goya L, Mateos R, Bravo L (2015) Effects of bioactive constituents in functional cocoa products on cardiovascular health in humans. Food Chem 174:214–218
Senol A, Aydin A (2006) Solid–liquid extraction of caffeine from tea waste using battery type extractor: process optimization. J Food Eng 75:565–573
Simonin C, Duru C, Salleron J, Hincker P, Charles P, Delval A, Youssov K, Burnouf S, Azulay J-P, Verny C, Scherer C, Tranchant C, Goizet C, Debruxelles S, Defebvre L, Sablonniere B, Buee L, Romon-Rousseaux M, Krystkowiak P (2013) Association between caffeine intake and age at onset in Huntington’s disease. Neurobiol Dis 58:179–182
Smrke S, Kroslakova I, Gloess AN, Yeretzian C (2015) Differentiation of degrees of ripeness of Catuai and Tipica green coffee by chromatographical and statistical techniques. Food Chem 174:637–642
Srdjenovic B, Djordjevic-Milic V, Grujic N, Injac R, Lepojevic Z (2008) Simultaneous HPLC determination of caffeine, theobromine, and theophylline in food, drinks, and herbal products. J Chromatogr Sci 46:144–149
Talik P, Krzek J, Ekiert RJ (2012) Analytical techniques used for determination of methylxanthines and their analogues—recent advances. Sep Purif Rev 41:1–61
Tello J, Viguera M, Calvo L (2011) Extraction of caffeine from Robusta coffee (Coffea canephora var. Robusta) husks using supercritical carbon dioxide. J Supercrit Fluids 59:53–60
Tzanavaras PD, Themelis DG (2007) Development and validation of a high-throughput high-performance liquid chromatographic assay for the determination of caffeine in food samples using a monolithic column. Anal Chim Acta 581:89–94
Yang H, Rouse J, Lukes L, Lancaster M, Veenstra T, Zhou M, Shi Y, Park Y-G, Hunter K (2005) Caffeine suppresses metastasis in a transgenic mouse model: a prototype molecule for prophylaxis of metastasis. Clin Exp Metastasis 21:719–735
Zoccali M, Giuffrida D, Dugo P, Mondello L (2017) Direct online extraction and determination by supercritical fluid extraction with chromatography and mass spectrometry of targeted carotenoids from red habanero peppers (Capsicum chinense Jacq.). J Sep Sci 40:3905–3913
Acknowledgements
Authors wish to thank Shimadzu Corporation for constantly supporting their research work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Marina Russo declares that she has no conflict of interest. Paola Dugo declares that she has no conflict of interest. Chiara Fanali declares that she has no conflict of interest. Laura Dugo declares that she has no conflict of interest. Mariosimone Zoccali declares that he has no conflict of interest. Luigi Mondello declares that he has no conflict of interest. Laura De Gara declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Russo, M., Dugo, P., Fanali, C. et al. Use of an Online Extraction Technique Coupled to Liquid Chromatography for Determination of Caffeine in Coffee, Tea, and Cocoa. Food Anal. Methods 11, 2637–2644 (2018). https://doi.org/10.1007/s12161-018-1247-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1247-5