
Abstract
A liquid chromatography tandem-mass spectrometry (LC-MS/MS) method has been developed for the simultaneous determination of 22 veterinary drug residues, belonging to eight classes (coccidiostats, lincosamides, macrolides, tetracyclines, sulfonamides, benzimidazoles, diterpenes, and diaminopyrimidines), in eggs. Chromatographic separations were achieved on an XBridge BEH C18 column (150 × 2.1 mm, 3.5 μm, Waters, USA) maintained at 35 °C. The mobile phase was eluted at 400 μL min−1 in gradient mode between water and methanol/acetonitrile (20:80 v/v), both containing 0.1 % formic acid. The samples were prepared by protein precipitation with acetonitrile without additional cleanup steps. The method was successfully validated according to the Commission Decision 2002/657/EC and was demonstrated to be highly selective and free of matrix and residual effects. The method presented low limits of detection (0.37 to 7.5 μg kg−1) and quantification (1.25 to 20 μg kg−1). For banned substances, the decision limit values (CCα) and detection capability (CCβ) were 0.62–7.5 and 0.65–8.1 μg kg−1, respectively. For substances with a maximum residue limit, the CCα and CCβ values were 2.15–1061.5 and 2.3–1135.9 μg kg−1, respectively. All calibration curves showed excellent correlation (r ≥ 0.99). The recovery of the analytes and internal standards (49.0–103.7 %) was reached with high precision (RSD <8 %). At different concentration levels, the variations in precision and accuracy, in terms of repeatability and in-laboratory reproducibility, were <11 %. The new method is applicable to the routine analysis of commercial egg samples.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Many classes of veterinary drugs, such as coccidiostats, lincosamides, macrolides, tetracyclines, sulfonamides, benzimidazoles, diterpenes, and diaminopyrimidines, are widely administered to laying hens for the treatment and prevention of diseases and as growth promoters (Stolker and Brinkman 2005). The inappropriate use of these drugs, or not respecting withdrawal periods, can result in the presence of residues (parent compound and metabolites) in eggs, increasing the potential risk to consumers due to allergic reactions in hypersensitive individuals and the induction of resistant strains of bacteria (World Health Organization 2011).
To control the residue problem, and protect the human consumers, several governmental authorities such as the European Union (EU), the Food and Drug Administration (FDA), and Codex Alimentarius established maximum residue levels (MRLs) for a variety of veterinary drugs in different foodstuffs (Commission Regulation 2010; Codex Alimentarius Commission 2012; Food and Drug Administration 2015). In eggs, the European Medicines Agency (EMEA) has set tolerances of 2 μg kg−1 for narasin, 50 μg kg−1 for lincomycin, 150 μg kg−1 for lasalocid and erythromycin, 200 μg kg−1 for tetracyclines and tylosin, 400 μg kg−1 for flubendazole, and 1000 μg kg−1 for tiamulin (Commission Regulation 2009; Commission Regulation 2010). The FDA has established MRLs for tetracyclines, erythromycin, and tylosin of 400, 25, and 200 μg kg−1, respectively (Food and Drug Administration 2015). A Codex standard adopted an MRL of 400 μg kg−1 for tetracyclines, 50 μg kg−1 for erythromycin, and 300 μg kg−1 for tylosin at the 35th Session of the Codex Alimentarius Commission (Codex Alimentarius Commission 2012). According to EMEA, sulfonamides are prohibited for use in animals that produce eggs for human consumption and therefore no limit is set (Commission Regulation 2009).
A literature survey revealed several methods for the determination of veterinary drug residues in eggs, including liquid chromatography (LC) using ultraviolet (Christodoulou et al. 2007; de Paula et al. 2008), diode array (Furusawa 2003; Tolika 2011; Summa et al. 2015), and fluorescence detectors (Schneider and Donoghue 2003; Xie et al. 2011). However, these methods focused on one class of compounds, showing only moderate sensitivity and selectivity, and requiring long running times or complex extraction procedures. Recently, some published methods have used time-of-flight mass spectrometry (TOF-MS) to screen hundreds of compounds in eggs and other food matrices (Mol et al. 2008; Peters et al. 2009; Deng et al. 2011). However, methods based on TOF-MS are not considered in the Commission Decision 2002/657/EC regarding confirmatory methods (Commission Decision 2002).
LC coupled with tandem mass spectrometry (LC-MS/MS) provides higher selectivity and sensitivity than LC using conventional detectors. Furthermore, it fulfills the requirements demanded by the Commission Decision 2002/657/EC by combining analyte separation and structural information, providing unequivocal detection and confirmation of a substance and enabling the development of more reliable methods (Aerts et al. 1995; Kennedy et al. 1998).
Despite the advantages of LC-MS/MS, few analytical methods for the simultaneous analysis of multiclass drug residues in eggs have been described in the literature (Heller and Nochetto 2004; Heller et al. 2006; Jia et al. 2008; Mol et al. 2008; Frenich et al. 2010; Spisso et al. 2010; Jimenez et al. 2011; Capriotti et al. 2012; Kim et al. 2012; Nakajima et al. 2012; Robert et al. 2013; Dasenaki and Thomaidis 2015). This is probably due to differences in their chemical and physical properties, which make the extraction process and cleanup the most challenging steps. For these methods, sample preparation generally involves a solvent extraction step to promote the precipitation of proteins either alone (Mol et al. 2008; Spisso et al. 2010; Robert et al. 2013) or followed by an additional cleanup step using n-hexane (Dasenaki and Thomaidis 2015) or solid-phase extraction (SPE) (Jia et al. 2008; Frenich et al. 2010; Kim et al. 2012). The main advantage of the SPE procedure is the reduction in matrix effects and ion suppression. Nevertheless, additional cleanup steps involve time-consuming and laborious sample preparation protocols. Other extraction methodologies, including pressurized liquid extraction (PLE) (Jimenez et al. 2011) and quick, easy, cheap, effective, rugged, and safe (QuEChERS) (Capriotti et al. 2012; Nakajima et al. 2012), have been evaluated. In other cases, the LC-MS method was not fully validated (Robert et al. 2013) or did not cover some pharmacological classes of primary interest in the egg, such as tetracyclines, macrolides, and/or coccidiostats (Capriotti et al. 2012; Nakajima et al. 2012; Spisso et al. 2010). For laboratories involved in official controls, it is important to have analytical methods available that are suitable for the simultaneous determination of several classes of veterinary drugs while optimizing the time and cost of analysis. To our knowledge, no validated methods have been published that use an extraction with acetonitrile without additional cleanup steps and are able to identify and simultaneously quantify coccidiostats, lincosamides, macrolides, tetracyclines, sulfonamides, benzimidazoles, diterpenes, and diaminopyrimidines in eggs by LC-MS/MS.
The objective of this study was to develop a rapid and sensitive LC-MS/MS method for the detection and confirmation of residues of 22 veterinary drugs and 8 different classes in eggs by using a simple and cheap extraction procedure. The whole procedure was validated according to the quality criteria of the Commission Decision 2002/657/EC and applied to the determination of residues in commercial egg samples.
Materials and Methods
Chemicals and Reagents
Standards of chlortetracycline (92 %), deuterated sulfamerazine (internal standard, 98 %), doxycycline (98.7 %), erythromycin (95.3 %), oxytetracycline (97.5 %), sulfadiazine (99.5 %), sulfadimethoxine (98 %), sulfamerazine (99.2 %), and sulfamethoxazole (99.5 %) were obtained from Dr. Ehrenstorfer (Bavaria, Germany). Cambendazole (internal standard, 99.8 %), demeclocycline hydrochloride (internal standard, 92.3 %), flubendazole (99.2 %), lasalocid A sodium salt (98 %), lincomycin hydrochloride (100.3 %), nigericin sodium salt (internal standard, ≥98 %), sulfacetamide (99.7 %), sulfachloropyridazine (99.4 %), sulfametazine (99.7 %), sulfamethoxypyridazine (99.8 %), sulfaquinoxaline (96 %), sulfathiazole (99.9 %), tetracycline hydrochloride (97.5 %), tylosin tartrate (87.9 %), and trimethoprim (99.5 %) were purchased from Sigma-Aldrich (MO, USA). Certified reference standards of narasin (86.1 %) and tiamulin (≥96.5 %) were available from the US Pharmacopeial Convention (MD, USA).
Acetonitrile and methanol (HPLC grade) were obtained from Panreac Química (Catalonia, Spain). Ammonium formate (≥97 %) was obtained from Acros Organics (NJ, USA). EDTA (≥98.5 %) and anhydrous sodium acetate (≥99 %) were purchased from Sigma-Aldrich (MO, USA). Formic acid (98–100 %) was obtained from Merck (MA, USA). Ultrapure water was obtained using a Milli-Q purification system from Millipore (MA, USA).
Standard Solutions
Individual stock standard solutions at 500 μg mL−1 were prepared in methanol for macrolides, sulfonamides, tetracyclines, lincomycin, narasin, nigericin, and trimethoprim, at 1 mg mL−1 in methanol for tiamulin and at 100 μg mL−1 in acetonitrile/methanol (50:50 v/v) for benzimidazoles. Lasalocid stock solution was provided as an acetonitrile solution at 100 μg mL−1 and used directly for further dilutions. All stock solutions were stored in amber bottles at 4 °C. Intermediate working solutions were prepared at the required concentrations by dilution of the standard stock solutions with methanol.
Blank Egg Samples
Blank egg samples, produced organically on a farm in Paraná (Brazil), were used for extraction development and validation studies. The fresh eggs were broken and homogenized, then 1 g was transferred into a 15-mL polypropylene centrifuge tube (TPP Techno Plastic Products, Trasadingen, Switzerland) and stored at −40 °C.
Sample Preparation
An aliquot of 50 μL of the internal standard (IS) solution (to obtain a final concentration of 150 μg kg−1 of nigericin, 200 μg kg−1 of deuterated sulfamerazine, 200 μg kg 1 of demeclocycline, and 400 μg kg−1 of cambendazole) and 50 μL of methanol were added to 1 g of homogenized whole eggs. After mixing for 1 min and standing for 5 min, an aliquot of 400 μL of 100 mmol L−1 EDTA was added. The sample was vortexed for 1 min and allowed to rest for 5 min, then 5 mL of acetonitrile was added and the sample vortexed again for 1 min and sonicated for 10 min to homogenize the material with the solvent. After this, the sample was centrifuged (Eppendorf 5810-R, Hamburg, Germany) at 3220×g and 4 °C for 15 min. The supernatant was collected in a 15-mL polypropylene centrifuge tube and evaporated to 500 μL in a sample concentrator at 40 °C (CentriVap Labconco, MO, USA). The resultant extract was then diluted with 100 μL of methanol and 400 μL of water. The sample was vortexed for 1 min, sonicated for 10 min, and centrifuged at 3220×g and 4 °C for 15 min. Finally, 500 μL of supernatant was diluted with 500 μL of 5 mmol L−1 sodium acetate/acetonitrile/methanol (70:24:6 v/v/v). The sample was centrifuged at 20,817×g and 4 °C for 15 min and filtered through a PVDF syringe filter 0.22 μm pore size (Agilent HP, CA, USA) before injection.
LC-MS/MS Instrumentation and Conditions
LC-MS/MS analyses were performed using an Agilent 1200 HPLC System (Wilmington, USA) consisting of a G1312B binary pump, G1379B degasser, and G1316B column oven. The HPLC was connected to a CTC Sample Manager (Model 2777, Waters Corporation, Milford, USA) operated at 20 °C. The HPLC system was coupled to an Applied Biosystems/MDS Sciex API 3200 Triple Quadrupole Mass Spectrometer (Toronto, Canada) equipped with a syringe pump (Harvard Apparatus, South Natick, USA) and an electrospray ionization (ESI) ion source. The ESI source was operated in positive ion mode. Analyte separation was achieved on an XBridge BEH C18 2.1 mm × 100 mm, 3.5 μm particle size (Waters Corporation, Milford, USA) column coupled with an XBridge BEH C18 2.1 mm × 10 mm, 3.5 μm particle size (Waters Corporation, Milford, USA) sentry guard cartridge maintained at 35 °C. The mobile phase comprised solvent A (water) and solvent B (acetonitrile/methanol 80:20 v/v), both containing 0.1 % formic acid. The gradient elution program used the following: 0–0.25 min, 5 % B; 0.25–0.50 min, increase to 25 % B; 0.50–6 min, maintained 25 % B; 6–8 min, linear increase to 85 % B; 8–9 min, increase to 100 % B; 9–14 min, maintained 100 % B; 14–15 min, decrease to 5 % B; and finally, 15–20 min, 5 % B. The flow rate was 400 μL min−1 and the injection volume 10 μL. The needle was washed with acetonitrile/methanol (50:50 v/v) containing 0.1 % formic acid between injections. Data acquisition was performed with MS Workstation by Analyst software version 1.4.2 (AB Sciex, MA, USA). Quantitation was performed in multiple reaction monitoring (MRM) mode. The ion transitions and the individual compound parameters, including the declustering potential (DP), entrance potential (EP), collision cell entrance potential (CEP), collision energy (CE), and cell exit potential (CXP), are shown in Table 1. The ion-source parameters for ESI positive mode were as follows: curtain gas (CUR), 12 psi; collision gas (CAD), 10 psi; ion spray voltage (ISV), 5000 V; nebulizer gas (GS1), 45 psi; turbo gas (GS2), 50 psi; and temperature, 500 °C. The high-purity nitrogen and zero-grade air, used as the CUR, GS1, GS2, and CAD gases, were produced using a high-purity nitrogen generator from Peak Scientific Instruments (Chicago, USA).
Validation Procedure
The method was validated according to the requirements of the European Commission Decision 2002/657/EC (Commission Decision 2002) and Agência Nacional de Vigilância Sanitária do Brasil (Brasil 2012). The validation parameters included selectivity, limit of detection, limit of quantitation, decision limits, detection capability, linearity, carryover, matrix effect, recovery, precision, and stability.
Selectivity
Method selectivity was evaluated by analyzing 20 blank egg samples, obtained from two different sources, and checking the existence of interfering peaks in the analyte and IS retention time regions. The samples were prepared as described in the “Sample Preparation” section, but without the addition of the IS.
Limit of Detection, Limit of Quantitation, Decision Limits, and Detection Capabilities
Limit of detection (LOD) and limit of quantitation (LOQ) were estimated from signal-to-noise ratios. To conduct this study, an aliquot of 50 μL of work standard solution and 50 μL of methanol was added to 1 g of blank egg sample. The samples were vortexed for 1 min, and a 400-μL aliquot of 100 mmol L−1 EDTA was added. The samples were subjected to the sample cleanup procedure described in the “Sample Preparation” section. The extract obtained was diluted with 100 μL of methanol and 400 μL of water to obtain a final concentration of 250 μg kg−1 for each analyte. The samples were vortexed again for 1 min, sonicated for 10 min, and centrifuged at 3220×g and 4 °C for 15 min. Finally, the supernatant was diluted in series with 5 mmol L−1 sodium acetate/acetonitrile/methanol (70:24:6 v/v/v) and injected into the LC-MS/MS system until the smallest detectable peak was obtained. For each analyte, the LOD was calculated as the lowest concentration peak at a signal-to-noise ratio ≥3:1 and the LOQ was calculated as the lowest concentration peak at a signal-to-noise ratio ≥10:1.
Decision limits (CCα) were calculated using two different approaches, one for banned substances and one for MRL substances. For banned substances (doxycycline (DXC), sulfadiazine (SDZ), sulfadimethoxine (SDMX), sulfamerazine (SMR), sulfametazine (SMZ), sulfamethoxazole (SMX), sulfamethoxypyridazine (SMPZ), sulfacetamide (SCT), sulfachloropyridazine (SCP), sulfaquinoxaline (SQ), sulfathiazole (STZ), and trimethoprim (TMP)), the LOD was used as the CCα. For MRL substances (chlortetracycline (CTC), erythromycin (ERY), flubendazole (FLU), lasalocid (LAS), lincomycin (LIN), narasin (NAR), oxytetracycline (OTC), tetracycline (TC), tiamulin (TIA), and tylosin (TYL)), the CCα was calculated by analysis of 20 blank egg samples spiked with the analytes at MRL levels. The CCα was calculated as the concentration at the permitted limit plus 1.64 times the corresponding standard deviation.
For both substances, the CCβ was calculated by analysis of 20 blank egg samples spiked with the analytes at the CCα levels. The CCβ was calculated as the CCα plus 1.64 and multiplied by the corresponding standard deviation.
Linearity
The linearity of the method was demonstrated using blank matrix-matched calibration standards. An aliquot of 1 g of homogenized whole blank eggs was weighed in a polypropylene centrifuge tube (15 mL) and fortified with 50 μL of the appropriate work standard solution and 50 μL of the internal standard solution, to obtain final concentrations of 150 μg kg−1 of nigericin (NIG), 200 μg kg−1 of deuterated sulfamerazine (SMR-D4), 200 μg kg−1 of demeclocycline (DMC), and 400 μg kg−1 of cambendazole (CAM). The spiked samples were prepared as described in the “Sample Preparation” section.
The calibration curves were prepared at seven concentration levels plus a zero point (blank). For banned substances, the calibration curves were constructed at 0, 25, 37.5, 50, 62.5, 75, 87.5, and 100 μg kg−1 (equivalent to 0, 1, 1.5, 2, 2.5, 3, 3.5, and 4 lowest validation levels (LVL), respectively). For MRL substances, the calibration curves were constructed at 0, 1, 1.5, 2, 2.5, 3, 3.5, and 4 μg kg−1 for NAR; 0, 25, 37.5, 50, 62.5, 75, 87.5, and 100 μg kg−1 for LIN; 0, 75, 112.5, 150, 187.5, 225, 262.5, and 300 μg kg−1 for ERY and LAS; 0, 100, 150, 200, 250, 300, 350, and 400 μg kg−1 for CTC, OTC, TC, and TYL; 0, 200, 300, 400, 500, 600, 700, and 800 μg kg−1 for FLU; and 0, 500, 750, 1000, 1250, 1500, 1750, and 2000 μg kg−1 for TIA (all equivalent to 0, 0.5, 0.75, 1, 1.25, 1.5, 1.75, and 2 MRL, respectively). For each compound, the calibration curve was used to confirm the linear relationship between analyte peak areas/IS peak areas and the analyte concentration/IS concentration. The slope, intercept, and regression coefficient (r) were calculated as regression parameters by weighted (1/x) linear. The linear correlation coefficient (r) was also required to be equal to or greater than 0.99.
CAM was used as internal standard for ERY, FLU, LIN, TIA, and TYL, while DMC was used as internal standard for tetracyclines. SMR-D4 was used as internal standard for sulfonamides and TMP, while NIG was used as internal standard for LAS and NAR.
Carryover Test
A carryover test was performed by injecting a triplicate of samples prepared at higher concentration levels (4 LVL and 2 MRL) alternating with the blank egg sample. No interfering peaks should have been observed in the blank egg chromatogram, with areas greater than 20 % of the peak areas at the lower concentration level of each analyte and 5 % of the IS peak area (Brasil 2012).
Matrix Effect
Matrix effect was evaluated at two different levels, 1 and 4 LVL for banned substances and 0.5 and 2 MRL for allowed substances.
The matrix effect analysis was performed using two different blank egg samples in sextuplicate spiked with all analytes and IS post-extraction and standard solution samples at the same concentration. The normalized effect of matrix (NEM) was calculated for each concentration level (analyte response in matrix/IS response in matrix vs. the analyte response in solution/IS response in solution). The relative standard deviation (RSD) of NEM had to be less than 15 % to indicate that the matrix effect was not significant (Brasil 2012).
Recovery (Extraction Efficiency)
Recovery was evaluated at three levels (1, 2, and 3 LVL for banned substances and 0.5, 1, and 1.5 MRL for allowed substances) by comparing the peak areas obtained from blank egg samples spiked with all analytes and IS before extraction (A) with those obtained from blank egg samples spiked with all analytes and IS post-extraction (B) (Brasil 2003). The percentage ratio of B/A was defined as recovery.
Precision and Trueness (Recovery)
The precision and trueness (recovery) of the methods were evaluated using measurements of repeatability and in-laboratory reproducibility (Commission Decision 2002). To perform this assay, the blank egg samples spiked with all analytes and IS were prepared at three levels (1, 2, and 3 LVL for banned substances and 0.5, 1, and 1.5 MRL for allowed substances). Repeatability was determined in 18 replicates at each level for three consecutive days and by the same analyst. In-laboratory reproducibility was evaluated in 18 replicates at each level, by a second analyst for three consecutive days.
The precision of the method was evaluated at each concentration level by calculating the relative standard deviation (RSD%) in repeatability and in-laboratory reproducibility conditions. The trueness (recovery) was evaluated at each concentration level by calculating the relative error (RE%), expressed as the difference between the mean of the experimental concentrations and the theoretical spiked concentrations divided by the theoretical spiked concentration.
Stability
Stability in Solution
For stability studies of analytes and IS in solution, 50 mL of working solution was prepared with 5 mmol L−1 sodium acetate/acetonitrile/methanol (70:24:6 v/v/v) at a concentration of 50 ng mL−1 for banned substances and LIN; 2 ng mL−1 for NAR; 150 ng mL−1 for ERY and LAS; 200 ng mL−1 for CTC, OTC, TC, and TYL; 400 ng mL−1 for FLU; 1000 ng mL−1 for TIA; 150 ng mL−1 for NIG; 200 ng mL−1 for SMR–D4; 200 ng mL−1 for DMC; and 400 ng mL−1 for CAM. The working solution was fractionated into 40 aliquots (10 for each temperature). The aliquots were freshly evaluated and, after a period of 1, 2, and 3 weeks, stored at −40 and 4 °C (light-protected) and 20 °C (light-protected and light-exposed).
Stability was evaluated by comparing the peak areas obtained from freshly prepared working solution with those obtained after each period of storage. The results were expressed as the percentage of analyte remaining, considered stable for amounts ≥80 % (Croubels et al. 2003).
Stability in Matrix
The stability of analytes in matrix was assessed by spiking 1 g of the blank egg sample (n = 30) with 50 μL of analyte solution, obtaining a concentration level of 2 LVL for banned substances and 1 MRL for allowed substances. The samples were vortexed for 1 min. One aliquot (n = 6) was prepared as described in the “Sample Preparation” section and analyzed immediately. The remaining aliquots were evaluated after periods of 1, 2, 3, and 4 weeks stored at −40 °C.
Stability was evaluated by comparing the mean recovery of analytes obtained from the freshly prepared sample with those obtained after each period of storage. The results were expressed as a percentage of analyte remaining. It is considered stable when the remaining amount is within the range of 80–110 % (Croubels et al. 2003).
Stability in Sample Post-Processing
Stability of analytes in sample post-processing was assessed with a sample obtained as described in the “Stability in matrix” section.
The stability of the analytes and IS was evaluated by comparing the mean recovery obtained from fresh samples with those obtained after 24 and 48 h in the Sample Manager (20 °C, amber vials). The results were expressed as a percentage of analyte remaining, considered stable when the remaining amount is within the range of 80 to 110 % (Croubels et al. 2003).
Analysis of Real Samples
To evaluate the applicability of the validated method, 15 egg samples obtained from different local markets in Brazil were analyzed in triplicate. The samples were prepared as described in the “Sample Preparation” section.
Results and Discussion
LC-MS/MS Method Development
The mass spectrometer parameters were optimized by infusion experiments using individual working standard solutions of analytes and ISs prepared in acetonitrile/water (50:50 v/v) and containing different proportions of formic acid (0.05, 0.1, and 0.5 %) and ammonium formate (2, 3, and 5 mmol L−1) as additives. For all compounds, the best signal intensity was observed when 0.1 % formic acid was used. Protonated ([M+H]+) molecular ions were observed for the majority of analytes. However, for LAS, NAR, and NIG (IS), the sodium ([M+Na]+) adduct ions were predominant.
Full-scan data acquisition was performed, and the analyte concentrations varied, to obtain an adequate signal intensity to optimize the individual compound parameters (DP, EP, CEP, CE, and CXP) by automatic MRM. When the precursor ions of DXC and TC were set (m/z 445), the same product ions were observed (m/z 428 and 410). According to Nakazawa et al. (1999) and De Ruyck and De Ridder (2007), the product ions at m/z 428 corresponded to [M+H–NH3]+ and at m/z 410 to [M+H–H2O–NH3]+ for TC and [M+H–NH3–H2O]+ for DXC. However, the relative abundance of these product ions differed significantly; m/z 428 was the most intense for DXC and m/z 410 for TC. For sulfonamides, the common product ions were obtained (m/z 92, 108, and 156), indicating a well-defined fragmentation profile (Gehring et al. 1996). At the conclusion of MS/MS optimization, the two most intense fragment signals for each compound were obtained. The most intense was used for quantification and the second most intense for peak qualification (see Table 1).
Optimization of the source parameters (CUR, CAD, ISV, GS1, GS2, and temperature) was accomplished through flow injection analysis (FIA) by coupling the chromatograph with the mass spectrometer. The pump was operated at 200 μL min−1 using an isocratic system with a mobile phase of acetonitrile/water (50:50 v/v) containing 0.1 % formic acid.
Due to the distinct physicochemical characteristics of the substances, an XBridge BEH C18 column (100 mm × 2.1 mm, 3.5 μm particle size) coupled with an XBridge BEH C18 pre-column (10 mm × 2.1 mm, 3.5 μm particle size) was used during method development. For mobile phase composition, several combinations of methanol, water, and acetonitrile were tested, maintaining 0.1 % formic acid as the additive under all conditions. Flow rate (200–400 μL min−1) and column oven temperature (30–40 °C) were also varied to achieve optimum run times and peak shapes.
Various gradient programs were tested, taking the distinct physical–chemical characteristics of the compounds into consideration. In order to retain the most polar substances, an initial gradient with 95 % water was established. For all conditions tested, the acetonitrile/methanol mixture provided better ionization of analytes than acetonitrile when employed as an organic modifier, especially for the sodium adducts of NAR, LAS, and NIG (IS). Furthermore, complete separation of tetracycline and doxycycline isomers (with mass transitions in common; see Table 1) was achieved, indicating the excellent specificity of the developed method.
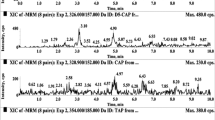
At the end of method development, the best overall sensitivity and peak shape were achieved by maintaining the column at 35 °C with a gradient mobile phase consisting of (A) water and (B) acetonitrile/methanol (80:20, v/v), both containing 0.1 % formic acid. The optimum run time was achieved by maintaining the flow rate at 400 μL min−1. Representative MRM chromatograms of the LC-MS/MS method are presented in Fig. 1.

MRM chromatograms of egg sample spiked with analytes and internal standards at concentrations of 4 LVL and 2 MRL
Sample Preparation Optimization
The simultaneous determination of 22 veterinary drug residues belonging to eight classes was a challenging task due to their very different chemical structures and properties. Therefore, appropriate sample treatment was essential for obtaining reliable results.
In order to remove lipids and proteins and achieve the best recovery of analytes and ISs, different extraction procedures were tested, including protein precipitation (PP) and SPE. All extraction procedures were performed by preparing a blank egg sample, spiked with analytes and ISs at 250 μg kg−1, in six replicates.
The most frequently organic solvents used as extracting agents are methanol and acetonitrile because they both allow protein precipitation as the analyte extraction. Many authors prefer acetonitrile (Mol et al. 2008; Dubreil-Cheneau et al. 2009; Peters et al. 2009; Frenich et al. 2010; Spisso et al. 2010; Robert et al. 2013) over methanol, because methanol extracts many matrix compounds, complicating further cleanup procedures and making difficult the sample analysis. Acetonitrile is commonly employed in sample preparation as it provides protein precipitation with high extraction efficiency and minimum co-extraction of lipids.
Different volumes of acetonitrile (2.5, 5, and 10 mL) were evaluated for 1 g of egg matrix. Using 5 mL of acetonitrile as a precipitating agent caused rapid protein precipitation, forming a dense precipitate that was easily removed by centrifugation. In addition, it demonstrated the highest efficiency for compound extraction (>50 %) with adequate reproducibility (RSD <8 %). During the experiments, it was observed that there was a slight improvement of the recoveries when increasing the volume of the acetonitrile, but not significant to justify the use of higher amount of solvent.
Tetracyclines can form complexes with metal ions (e.g., Ca2+ and Mg2+) present in the sample. Thus, an aliquot of 400 μL of 100 mmol L−1 EDTA was added to the sample as a competing chelating agent. This led to improved recoveries (>60 %) for CTC, OTC, and TC.
To improve extract cleanup, SPE purifications were performed with buffers at different pH values (pH 3–7) with Oasis HLB cartridges, recommended for polar and non-polar compounds. Briefly, the extract obtained from the precipitation of proteins with acetonitrile was evaporated until dryness and redissolved with the extraction buffer, which consisted of the following: pH 3 citric acid buffer, pH 4.5 acetic acid buffer, pH 6 acetic acid buffer, and pH 7 phosphate buffer. At pH 3 and 4.5, ERY recovery was <30 % as this analyte was unstable under acidic conditions. At pH 6 and 7, recoveries were higher than 60 % for most sulfonamides, but <40 % for DXC, TC, and DMC (IS). Among the pH evaluated, low recoveries and inadequate repeatability (RSD >20 %) were observed for the majority of analytes. According to Peters et al. (2009), egg proteins are smaller and water-soluble, interacting strongly with the cartridge sorbent and consequently interfering with analyte adsorption.
Finally, PP with acetonitrile was selected for sample preparation. When compared with SPE, this procedure presents more advantages as lower cost of analysis, smaller amount of residual chemicals generated, and less time consuming for sample preparation.
Considering the previous report (Dubreil-Cheneau et al. 2009; Spisso et al. 2010), a mixture of 5 mmol L−1 sodium acetate/acetonitrile/methanol (70:24:6 v/v/v) was used as the solvent for reconstitution of the extract to ensure the stability of sodium adducts.
Method Validation
Selectivity
The chromatograms obtained using different blank egg samples are presented in Fig. 2. The results showed no interfering peaks co-eluted with analytes and ISs. The developed method was, therefore, considered to be selective.

Chromatograms obtained by LC–MS/MS for the selectivity study: a blank egg sample A and b blank egg sample B
Limit of Detection, Limit of Quantitation, Decision Limits, and Detection Capabilities
As demonstrated in Table 2, the developed method demonstrated high sensitivity with low LOD (0.37–7.5 μg kg−1) and LOQ (1.25–20.0 μg kg−1). In general, these results are similar or better than previously published methods.
For banned substances, the CCα and CCβ values ranged from 0.62 to 7.5 and 0.65 to 8.1 μg kg−1, respectively. For MRL substances, the CCα and CCβ values ranged from 2.15 to 1061.5 and 2.3 to 1135.9 μg kg−1, respectively. When compared to the method proposed by Spisso et al. (2010), the CCα and CCβ values for ERY, LAS, LIN, NAR, and TYL were considerably lower (2.3 to 262.7 μg kg−1). For CTC, DXC, LAS, OTC, and SMX, the CCα and CCβ values were lower than those in Capriotti et al. (2012), which were in the range of 13.3 to 283 μg kg−1. The methods published by Heller and Nochetto (2004), Heller et al. (2006), Jia et al. (2008), Mol et al. (2008), Deng et al. (2011), Kim et al. (2012), Nakajima et al. (2012), and Dasenaki and Thomaidis (2015) did not report the values of CCα and CCβ. Considering the importance of performance characteristics required by the Commission Decision 2002/657/EC, the lack of evaluation of CCα and CCβ may compromise the confidence and suitability of the method to the intended use.
Linearity
The calibration curves obtained were linear with correlation coefficients (r) higher than 0.993 for all analytes in all concentration ranges. Additionally, at all concentrations, the results exhibited variations in precision and accuracy <15 %, guaranteeing a reliable response, regardless of the concentration.
Carryover
No significant interfering peaks (<1 % of the peak areas) were observed at the retention times of the analytes and ISs between injections. Thus, there was no risk of carryover contamination during sample analysis.
Matrix Effects
The variations of the NEM for each compound were <15 %, indicating that the effects of the biological matrix on analyte response was insignificant (Table 3).
Recovery (Extraction Efficiency)
For all compounds, satisfactory recoveries (49.0–103.7 %) were achieved, with high reproducibility (RSD <8 %). Recovery values were >80 % for more than 70 % of the compounds. Protein precipitation with acetonitrile was therefore considered effective for extracting veterinary drugs from hen eggs.
Precision and Trueness (Recovery)
The results of precision and trueness (recovery) are shown in Table 4. The new method was precise for all analytes, with RSD varying from 0.9 to 9.7 % for repeatability and from 0.8 to 10.6 % for in-laboratory reproducibility analysis. These results were in accordance with the “between one half and two thirds” value recommended in the Commission Decision 2002/657/EC for each concentration level.
The method also showed satisfactory trueness (recovery), with RE ranging from −6.8 to +6.4 % for repeatability and from −9.5 to +9.0 % for in-laboratory reproducibility analysis. These results were in accordance with the Commission Decision 2002/657/EC that recommended recoveries, in terms of bias, between −30 % and +10 % for analytes with concentrations of 1–10 μg kg−1 and between −20 % and +10 % for analytes with concentrations >10 μg kg−1.
Stability
Stability in Solution
A threshold of 80 % was applied for stability in solution, considering the high intrinsic variability of the MS detector (Croubels et al. 2003). After 1 week, degradation of DMC, DXC, LAS, NAR, SMX, SMZ, TC, TIA, and TMP was observed under all storage conditions. Fast photodegradation was observed in tetracyclines; similar observations have previously been reported (Oka et al. 1989; Soeborg et al. 2004). Only SDMX was stable under all storage conditions after 3 weeks. Thus, it was concluded that working solutions should be freshly prepared before analysis.
Stability in Matrix
For stability in matrix, a threshold of 80–110 % was applied, considering the minimum trueness of quantitative methods that establish an accuracy of −20 to +10 % for analytes with a concentration >10 μg kg−1.
It is known that the pH of the yolk and egg white are different, 6 and 7.6, respectively (Kan and Petz 2000). However, the egg white pH increases during the storage period and can reach up to 9.5 (Pereira et al. 2014). According to Soeborg et al. (2004), this alkaline pH contributes to the degradation of chlortetracycline (iso-chlortetracycline formation) and to its maximum complexation with Ca2+ ions. This could explain the low stability of tetracyclines, which was less than 2 weeks. Given the structural similarity of the tetracyclines, it is expected that both of them undergo the same type of degradation pattern.
Degradation of SMPZ and STZ was observed after 1 week of storage. After 2 weeks, degradation of SCT, SDZ, SMZ, SQ, LAS, TYL, TMP, and TIA was observed. SMX and ERY were stable for less than 3 weeks and SMR, SDMX, LIN, and FLU for less than 4 weeks. Only NAR and SCP remained stable after 4 weeks, keeping a threshold of 81 and 86 %, respectively. Therefore, egg samples fractionated should not be stored longer than 1 week before analysis.
Stability in Sample Post-Processing
No significant differences were demonstrated after 48 h in Sample Manager (20 °C). Thus, excellent stability was shown for all compounds.
Sample Analyses
The newly developed and validated HPLC-MS/MS method was applied to 15 egg samples acquired from a local market. Figure 3 shows the MRM chromatogram of one sample containing residues of doxycycline (538.0 μg kg−1), sulfamethoxazole (635.2 μg kg−1), and trimethoprim (241.1 μg kg−1), the use of which is banned in animals that produce eggs for human consumption. This result shows the importance and potential application of the new method.

MRM chromatograms of an egg sample with residues of trimethoprim (TMP) at 241.1 μg kg−1, sulfamethoxazole (SMX) at 635.2 μg kg−1, and doxycycline (DXC) at 538.0 μg kg−1
Conclusion
A new, reproducible, sensitive, fast, and fully validated LC-MS/MS method was developed for the simultaneous determination of 22 veterinary drugs in eggs. Data from the validation study demonstrated that the method was selective, linear, precise, accurate, and free of residual and matrix effects. The newly developed method was successfully applied to real samples and found to be applicable to routine analysis, enabling sample compliance assessment according to regulatory limits established in the EU regulations.
References
Aerts MM, Hogenboom AC, Brinkman UA (1995) Analytical strategies for the screening of veterinary drugs and their residues in edible products. J Chromatogr B 667:1–40
Brasil, Agência Nacional de Vigilância Sanitária. Resolução n. 899, de 29 de maio de 2003. Determina a publicação do Guia para Validação de Métodos Analíticos e Bioanalíticos. Diário Oficial da República Federativa do Brasil, Brasília, DF, 02 fev. 2003.
Brasil, Agência Nacional de Vigilância Sanitária. Resolução RDC n. 27, de 17 de maio de 2012. Diário Oficial da União. Dispõe sobre os requisitos mínimos para a validação de métodos bioanalíticos empregados em estudos com fins de registro e pós-registro de medicamentos. Diário Oficial da República Federativa do Brasil, Brasília, DF, 22 maio 2012.
Capriotti AL, Cavaliere C, Piovesana S, Samperi R, Laganà A (2012) Multiclass screening method based on solvent extraction and liquid chromatography-tandem mass spectrometry for the determination of antimicrobials and mycotoxins in egg. J Chromatogr A 1268:84–90
Christodoulou EA, Samanidou VF, Papadoyannis LN (2007) Validation of an HPLC-UV method according to the European Union Decision 2002/657/EC for the simultaneous determination of 10 quinolones in chicken muscle and egg yolk. J Chromatogr B 859:246–255
Codex Alimentarius Commission (2012) Maximum residue limits for veterinary drugs in foods, updated as at the 35th Session of the Codex Alimentarius Commission, CAC/MRL 2-2012, July 2012, ftp://ftp.fao.org/codex/weblinks/MRL2_e_2012.pdf. Accessed 09 August 2015
Commission Decision 2002/657/EC implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results, 2002. Off J Eur Commun, No. L 221/8–36
Commission Regulation (EU) 124/2009 setting maximum levels for the presence of coccidiostats or histomonostats in food resulting from the unavoidable carry-over of these substances in non-target feed, 2009. Off J Eur Commun, No. L40/7–11
Commission Regulation (EU) 37/2010 of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin, 2010. Off J Eur Commun, No. L15/1–72
Croubels S, De Baere S, De Backer P (2003) Practical approach for the stability testing of veterinary drugs in solutions and in biological matrices during storage. Anal Chim Acta 483:419–427
Dasenaki ME, Thomaidis NS (2015) Multi-residue determination of 115 veterinary drugs and pharmaceutical residues in milk powder, butter, fish tissue and eggs using liquid chromatography-tandem mass spectrometry. Anal Chim Acta 880:103–121
de Paula FC, de Pietro AC, Cass QB (2008) Simultaneous quantification of sulfamethoxazole and trimethoprim in whole egg samples by column-switching high-performance liquid chromatography using restricted access media column for on-line sample clean-up. J Chromatogr A 1189:221–226
De Ruyck H, De Ridder H (2007) Determination of tetracycline antibiotics in cow’s milk by liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectr 21:1511–1520
Deng XJ, Yang HQ, Li JZ, Song Y, Guo DH, Luo Y, Du XN, Bo T (2011) Multiclass residues screening of 105 veterinary drugs in meat, milk, and egg using ultra high performance liquid chromatography tandem quadrupole time-of-flight mass spectrometry. J Liq Chromatogr R T 34:2286–2303
Dubreil-Cheneau E, Bessiral M, Roudaut B, Verdon E, Sanders P (2009) Validation of a multi-residue liquid chromatography-tandem mass spectrometry confirmatory method for 10 anticoccidials in eggs according to Commission Decision 2002/657/EC. J Chromatogr A 1216:8149–8157
Food and Drug Administration (2015) Code of Federal Regulations, Title 21, Part 556, Section 500, U.S. Government Printing Office, Washington DC, (Chapter 1), http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?cfrpart=556. Accessed 01 September 2015
Frenich AG, Aguilera-Luiz MD, Vidal JLM, Romero-Gonzalez R (2010) Comparison of several extraction techniques for multiclass analysis of veterinary drugs in eggs using ultra-high pressure liquid chromatography-tandem mass spectrometry. Anal Chim Acta 661:150–160
Furusawa N (2003) Rapid high-performance liquid chromatographic determining technique of sulfamonomethoxine, sulfadimethoxine, and sulfaquinoxaline in eggs without use of organic solvents. Anal Chim Acta 481:255–259
Gehring TA, Rushing LG, Churchwell MI, Doerge DR, McErlane KM, Thompson HC (1996) HPLC determination of sulfadiazine residues in coho salmon (Oncorhynchus kisutch) with confirmation by liquid chromatography with atmospheric pressure chemical ionization mass spectrometry. J Agric Food Chem 44:3164–3169
Heller DN, Nochetto CB (2004) Development of multiclass methods for drug residues in eggs: silica SPE cleanup and LC-MS/MS analysis of ionophore and macrolide residues. J Agric Food Chem 52:6848–6856
Heller DN, Nochetto CB, Rummel NG, Thomas MH (2006) Development of multiclass methods for drug residues in eggs: hydrophilic solid-phase extraction cleanup and liquid chromatography/tandem mass spectrometry analysis of tetracycline, fluoroquinolone, sulfonamide, and beta-lactam residues. J Agric Food Chem 54:5267–5278
Jia X, Shao B, Wu Y, Yang Y, Zhang J (2008) Simultaneous determination of tetracylines and quinolones antibiotics in egg by ultra-performance liquid chromatography-electrospray tandem mass spectrometry. J AOAC Int 91:461–468
Jimenez V, Rubies A, Centrich F, Companyo R, Guiteras J (2011) Development and validation of a multiclass method for the analysis of antibiotic residues in eggs by liquid chromatography-tandem mass spectrometry. J Chromatogr A 1218:1443–1451
Kan CA, Petz M (2000) Residues of veterinary drugs in eggs and their distribution between yolk and white. J Agric Food Chem 48:6397–6403
Kennedy DG, McCracken RJ, Cannavan A, Hewitt SA (1998) Use of liquid chromatography mass spectrometry in the analysis of residues of antibiotics in meat and milk. J Chromatogr A 812:77–98
Kim E, Bahn K, Kang E, Kim M (2012) Quantitative analysis of lincomycin and narasin in poultry, milk and eggs by liquid chromatography-tandem mass spectrometry. Food Chem 132:1063–1070
Mol HGJ, Plaza-Bolanos P, Zomer P, de Rijk TC, Stolker AAM, Mulder PPJ (2008) Toward a generic extraction method for simultaneous determination of pesticides, mycotoxins, plant toxins, and veterinary drugs in feed and food matrixes. Anal Chem 80:9450–9459
Nakajima T, Nagano C, Sasamoto T, Hayashi H, Kanda M, Kanai S, Takeba K, Matsushima Y, Takano I (2012) Development and validation of rapid analysis method for multi-class veterinary drugs in livestock products by LC-MS/MS. Shokuhin Eiseigaku Zasshi 53:243–253
Nakazawa H, Ino S, Kato K, Watanabe T, Ito Y, Oka H (1999) Simultaneous determination of residual tetracyclines in foods by high-performance liquid chromatography with atmospheric pressure chemical ionization tandem mass spectrometry. J Chromatogr B 732:55–64
Oka H, Ikai Y, Kawamura N, Yamada M, Harada K, Ito S, Suzuki M (1989) Photodecomposition products of tetracycline in aqueous-solution. J Agric Food Chem 37:226–231
Pereira AS, Santos TTD, Coelho AFS (2014) Quality of eggs sold in different commercial establishments and the study of the conditions of storage. Food Sci Technol Int 34:82–87
Peters RJB, Bolck YJC, Rutgers P, Stolker AAM, Nielen MWF (2009) Multi-residue screening of veterinary drugs in egg, fish and meat using high-resolution liquid chromatography accurate mass time-of-flight mass spectrometry. J Chromatogr A 1216:8206–8216
Robert C, Gillard N, Brasseur PY, Pierret G, Ralet N, Dubois M, Delahaut P (2013) Rapid multi-residue and multi-class qualitative screening for veterinary drugs in foods of animal origin by UHPLC-MS/MS. Food Addit Contam A: Chem Anal Control Expo Risk Assess 30:443–457
Schneider MJ, Donoghue DJ (2003) Multiresidue determination of fluoroquinolone antibiotics in eggs using liquid chromatography-fluorescence-mass spectrometry. Anal Chim Acta 483:39–49
Soeborg T, Ingerslev F, Halling-Sorensen B (2004) Chemical stability of chlortetracycline and chlortetracycline degradation products and epimers in soil interstitial water. Chemosphere 57:1515–1524
Spisso BF, Ferreira RG, Pereira MU, Monteiro MA, Cruz TA, da Costa RP, Lima AMB, da Nobrega AW (2010) Simultaneous determination of polyether ionophores, macrolides and lincosamides in hen eggs by liquid chromatography-electrospray ionization tandem mass spectrometry using a simple solvent extraction. Anal Chim Acta 682:82–92
Stolker AA, Brinkman UA (2005) Analytical strategies for residue analysis of veterinary drugs and growth-promoting agents in food-producing animals—a review. J Chromatogr A 1067:15–53
Summa S, Lo Magro S, Armentano A, Muscarella M (2015) Development and validation of an HPLC/DAD method for the determination of 13 sulphonamides in eggs. Food Chem 187:477–484
Tolika EP, Samanidou VF, Papadoyannis IN (2011) Development and validation of an HPLC method for the simultaneous determination of ten sulfonamide residues in whole egg according to 2002/657/EC. J Liq Chromatogr R T 34:2396–2410
World Health Organization (2011) Tackling antibiotic resistance from a food safety perspective in Europe http://www.euro.who.int/__data/assets/pdf_file/0005/136454/e94889.pdf. Accessed 09 August 2015
Xie KZ, Jia LF, Yao YL, Xu D, Chen SQ, Xie X, Pei Y, Bao WB, Dai GJ, Wang JY, Liu ZP (2011) Simultaneous determination of thiamphenicol, florfenicol and florfenicol amine in eggs by reversed-phase high-performance liquid chromatography with fluorescence detection. J Chromatogr B 879:2351–2354
Acknowledgments
The authors would like to thank the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) for the scholarship, and Fundação Araucária for the financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Flávia Lada Degaut Pontes declares that she has no conflict of interest. João Cleverson Gasparetto declares that he has no conflict of interest. Thais Martins Guimarães de Francisco declares that she has no conflict of interest. Heloise Calzado Goetzke declares that she has no conflict of interest. Letícia Paula Leonart declares that she has no conflict of interest. Daniel Altino de Jesus declares that he has no conflict of interest. Roberto Pontarolo declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Rights and permissions
About this article

Cite this article
Pontes, F.L.D., Gasparetto, J.C., de Francisco, T.M.G. et al. Development and Validation of a Multiclass Method for the Analysis of Veterinary Drug Residues in Eggs Using Liquid Chromatography-Tandem Mass Spectrometry. Food Anal. Methods 10, 1063–1077 (2017). https://doi.org/10.1007/s12161-016-0670-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0670-8