
Abstract
Objective
To assess the feasibility, effectiveness, and safety of pulse methylprednisolone in comparison with intramuscular adrenocorticotropic hormone (ACTH) therapy in children with West syndrome (WS).
Methods
This open-label, pilot study with a parallel-group assignment included 44 recently diagnosed children with WS. Methylprednisolone therapy was given as intravenous infusion at a dose of 30 mg/kg/d for five days followed by oral steroids 1 mg/kg gradually tapered over 5–6 wk. The efficacy outcomes included a cessation of epileptic spasms (as per caregiver reporting) and resolution of hypsarrhythmia on electroencephalogram; safety outcome was the frequency of various adverse effects.
Results
By day 14 of therapy, 6/18 (33.3%) children in the methylprednisolone group and 10/26 (38.5%) children in the ACTH group achieved cessation of epileptic spasms [group difference − 5.2%; confidence interval (CI) -30.7 to 22.8; p = 0.73]. However, by six weeks of therapy, 4/18 (22.2%) children in the methylprednisolone group and 11/26 (42.3%) children in the ACTH group had cessation of epileptic spasms (group difference − 20.1%; CI -43.0 to 8.4; p = 0.17). Hypertension was more commonly observed in the ACTH group (10 children) than in the methylprednisolone group (2 children; p = 0.046). Pulse methylprednisolone therapy was relatively safe.
Conclusions
The study observed limited effectiveness of both ACTH and pulse methylprednisolone therapy, which may partially be due to preponderance of structural etiology and a long treatment lag. However, pulse methylprednisolone therapy appeared to be safe, tolerable, and feasible for management of WS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
West syndrome (WS) is an age-dependant epileptic encephalopathy characterized by epileptic spasms in clusters and presence of hypsarrhythmia on electroencephalogram (EEG) [1]. It is the commonest epileptic encephalopathy in infancy [2]. The underlying etiology may be structural, genetic, metabolic, etc. Stress-related excessive production of corticotrophin-releasing hormone (CRH) during early life has been proposed to underlie the pathogenesis of WS [3]. The presence of elevated CRH levels at a time of CRH-receptor abundance has been thought to result in epileptogenesis, thereby giving rise to epileptic spasms.
The main treatment strategy for WS consists of hormonal therapy [adrenocorticotropin hormone (ACTH) and oral prednisolone] and vigabatrin. The treatment goal is to achieve freedom from epileptic spasms and resolution of hypsarrhythmia in order to achieve better neurodevelopmental outcomes. Steroids, serendipitously discovered as efficacious, have been used for nearly six decades in WS [4]. They have been proposed to act via varied mechanisms such as anti-inflammatory action, correction of dysfunctional enzymes and intracellular hypoglycemia, maintenance of intracellular water and electrolyte balance, etc. [5]. Besides, ACTH has also been hypothesized to act through suppression of CRH, modulation of an intracellular neurosteroid and adenosine production, modulation of GABAA receptor due to second messenger effect, etc. [5]. Also, the role of steroids in other refractory epilepsies is affirmative [6].
ACTH has been recommended as first-line therapy option for WS [7]. Even long-term ACTH therapy (for months) has been tried in reports with positive results [8]. However, considering the huge expense involved in ACTH therapy, the search for relatively cheaper alternatives with a similar efficacy continues. Oral prednisolone, pulse methylprednisolone, and pulse dexamethasone are the options being used and evaluated, especially for low middle-income countries [9,10,11,12,13,14,15]. Although pulse methylprednisolone and dexamethasone have been found beneficial and of comparable efficacy, they lack sufficient evidence as a first-line therapy [11,12,13,14]. Further research is needed to document the efficacy, adverse effect profile, ease of administration, and cost of methylprednisolone as compared with ACTH therapy in WS. Hence, the current comparative study was done to assess the effectiveness and safety of pulse methylprednisolone in WS in comparison with the standard ACTH therapy.
Material and Methods
This open-label control trial was conducted at a tertiary-care referral hospital in Northern India over one year (July 2010–June 2011) after approval from the Institutional Ethics Committee.
All recently diagnosed children with WS were assessed for eligibility. Children (aged 3 mo-3 y) with a diagnosis of WS based on clinical assessment and signature EEG (based on Delphi consensus definition) were included [1]. Children with tuberous sclerosis (TSC) or where parents refused consent, were excluded from the study.
All children recently diagnosed with WS were screened for eligibility. Their parents or guardians were explained in detail about the study and about ACTH and pulse methylprednisolone therapy for WS, including the total cost for both treatments, in their own language and informed written consent was obtained. They were asked to make an informed choice based on the feasibility and expense of both the treatments. Those who consented for either ACTH or pulse methylprednisolone therapy were enrolled in the respective arms of the study. All the enrolled children were hospitalized for 5–7 d. A thorough clinical assessment was done for the enrolled children and recorded on a predesigned proforma. Pre-treatment weight, random blood sugar, and EEG were recorded. Sedated 30 min sleep with a brief period of wakefulness was recorded during the EEG recording. EEG recordings were analysed and scored using Jeavon’s scoring by a pediatric neurologist, who was unaware of the group allocation [16]. All scores between 13 to 30 were labelled as hypsarrhythmia and scores between 9 to 12 were taken as modified hypsarrhythmia.
The ACTH arm received synthetic corticotropin carboxymethylcellulose (Acton Prolongatum, Ferring Pharmaceuticals) intramuscularly once daily for six weeks. The regime followed was maximum-dose-at-initiation followed by a tapering regime. Therapy was initiated at a dose of 40 international unit (IU) per day for three weeks followed by tapering (30 IU/d, 20 IU/d and subsequently 10 IU/d, each given for a week with a total of six weeks therapy). The pulse methylprednisolone arm received an intravenous infusion of methylprednisolone at a dose of 30 mg/kg/d (in 200 ml saline) over 3 h for five days. This was followed by oral prednisolone at a dose of 1 mg/kg/d for three weeks followed by tapering (0.5 mg/kg/d for a week and 0.5 mg/kg every other day for a week). After six weeks of therapy, a repeat EEG was done and analysed. Failure of response was defined as no reduction or increase in the frequency of spasms after two weeks of starting intervention. After failure, these children were offered other antiepileptics drugs.
All the recruited children were monitored for blood pressure (BP) twice a day and for blood sugar, every alternate day during the hospital stay or at the nearest local health-care facility after discharge. Presence of at least two BP records above 95th centile on two successive occasions was considered as significant, and the child was initiated on oral propranolol at 1 mg/kg/d in 3 divided doses. Based on further recordings, the dose was titrated up to a ceiling of 2 mg/kg/d. Presence of random blood sugar record >180 mg/dl was taken as significant. Weight was recorded after six weeks to look for weight gain. Any infections occurring during this period were recorded.
During the hospital admission, parents were trained to recognize and count epileptic spasms. After discharge from the hospital, the parents of all children were followed-up via telephone to document the number of epileptic spasms, administration of intervention, and any adverse effects. A record of the daily frequency of epileptic spasms was maintained by parents. The primary outcome was a complete and persistent cessation of epileptic spasms at 6 wks after starting therapy. The secondary outcomes included a reduction in a number of epileptic spasms at two and six weeks of treatment initiation and improvement in EEG score by six weeks of therapy.
The measurable data were analyzed for its normality using Kolmogorov-Smirnov test. Student’s t test or Mann-Whitney U test were used for comparing continuous outcome variables wherever appropriate. The classified data were analyzed for two treatments using Chi-square test or Fisher’s exact test. A two-sided P value <0.05 was taken as statistically significant.
Results
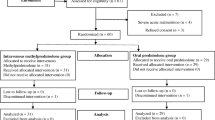
Forty-four children were included in the study to receive either ACTH (n = 26) or pulse methylprednisolone (n = 18). The mean age, gender, age at onset of spasms, and other baseline parameters were comparable among the two groups (Table 1). All patients were treated as per protocol. Two patients could not be followed up till the end of study (1 death in ACTH arm, 1 lost to follow-up in methylprednisolone arm). Three patients in the ACTH group did not get a repeat EEG done despite repeated requests but continued to come for follow-up (Fig. 1).
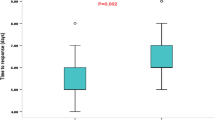
The two treatment arms did not differ significantly in terms of the cessation of epileptic spasms at two and six weeks respectively (Table 2). By day 14 of therapy, 6/18 (33.3%) children were spasms-free in the methylprednisolone group, whereas 10/26 (38.5%) children were spasms-free in the ACTH group (p = 0.73). However, by six weeks of therapy, 4/18 (22.2%) children in the methylprednisolone group and 11/26 (42.3%) children in the ACTH group had cessation of epileptic spasms (p = 0.17). Repeat EEG could be done for 39 children. There were no significant group differences on EEG scores (p = 0.57).

Study Flow. ACTH Adrenocorticotropic hormone; EEG Electroencephalogram
The most common adverse event observed during therapy was weight gain, (27 patients; 64.3%) without any significant difference between the two groups (Table 3). Infections were seen in 13 patients and included diarrhea (8), pneumonia (1), paronychia (2), impetigo (1), and scalp abscess (1). Hypertension was more commonly observed in the ACTH group (10 children) than in the methylprednisolone group (2 children; p = 0.046). Other than hypertension, the adverse event profile, and the frequency of adverse events was not significantly different between the two groups (Table 3).
Discussion
This was an open-label parallel trial that compared ACTH and pulse methylprednisolone therapy for WS. Children with TSC were excluded, as vigabatrin is recommended as the first-line therapy for WS in TSC. The demographic and baseline characteristics (age at onset of spasms and presentation, gender) were comparable to the published literature from India and other developing countries [17,18,19,20,21]. However, these characteristics differed from the western literature, as children in this study had a higher age at presentation, and there was a male preponderance [8]. This could be probably due to relatively longer time to diagnosis and treatment, and lack of awareness which is not uncommon in developing countries [17, 18, 22, 23]. In the study population, the majority had a remote symptomatic cause of WS. This higher preponderance of structural etiology is similar to that reported in previous literature [17,18,19].
In this study, ACTH and pulse methylprednisolone therapy followed by oral prednisolone were comparable in terms of adverse events and clinical efficacy at two and six weeks, respectively. In the methylprednisolone group, cessation of epileptic spasms by two weeks was observed in one-third of patients while persistent cessation for six weeks was seen in around 22.2% of children. This is much lower than the response rates noted in the study by Mytinger et al. where 5/10 children were free of spasms by two weeks of methylprednisolone therapy and sustained remission and/ or brief, responsive relapse was seen in 3/10 children [11]. In the study by Mytinger et al., 7/10 children had resolution of spasms (after trying other drugs) at last follow-up [11].
In a similar study by Yeh et al., 9/14 children had electroclinical remission at three weeks of methylprednisolone therapy while sustained remission was observed in 5/14 of patients [12]. Similarly, the response rate for ACTH was much lower as compared with western literature [24]. However, this was comparable to the response rates in India [20, 21]. The relatively lower response rates for ACTH and methylprednisolone in Indian cohort may be due to longer treatment lag and predominance of structural etiology, which are known to be associated with poor outcomes [25]. Similar to previous studies, the tolerability of methylprednisolone therapy was good and comparable to ACTH therapy [11, 12].
Despite the recent advances in the understanding of WS, it continues to be a difficult-to-treat epilepsy with a dismal prognosis. The need of the hour is a treatment approach which should not only be effective in cessation of epileptic spasms with minimal adverse event, but also improve long-term outcome and should be cost-effective. ACTH therapy is a standard of care therapeutic option in many settings; however, it is quite expensive. The easy availability, ease of administration, better cost-benefit ratio, tolerability, and comparable effectiveness of pulse methylprednisolone therapy warrants its consideration for WS, especially in low middle-income countries [11, 12].
While this study does support the effectiveness of intravenous pulse methylprednisolone with prednisolone taper for treatment of WS, but it has some limitations. This was a small feasibility study with non-randomized study design. Another major limitation was a short follow-up period. Hence, a larger, randomized prospective study with long term follow-up is needed to make a strong case for this therapy. A longer follow-up period would help in better assessment of long-term neurodevelopmental outcome and remission of WS. The authors performed Jeavon’s EEG scoring for outcome assessment. The Burden of Amplitudes and Epileptiform Discharges (BASED) score is a recently developed simple and reliable tool with a good inter-rater agreement for EEG scoring in WS [26]. However, the authors could not apply BASED score as it was published after the completion of their study.
In conclusion, the current study shows that both ACTH and pulse methylprednisolone therapies had limited effectiveness in terms of a persistent cessation of epileptic spasms at six weeks in children with WS. This may partially be explained by a preponderance of structural etiology and a long treatment lag of the study participants. Overall, pulse methylprednisolone therapy appeared to be safe, tolerable, and feasible for management of WS. However, further evidence from a large multicentric trial looking at this aspect is the need of the hour. Future studies should also explore the effectiveness of intravenous pulse methylprednisolone with oral high-dose, steroid therapy, which is increasingly considered as a feasible, initial therapeutic option.
References
Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and west syndrome: consensus statement of the West Delphi group. Epilepsia. 2004;45:1416–28.
Sakakihara Y. Treatment of west syndrome. Brain Dev. 2011;33:202–6.
Frost DJ, Hrachovy RA. Pathogenesis of infantile spasms: a model based on developmental desynchronization. J Clin Neurophysiol. 2005;22:25–36.
Sorel L, Dusaucy BA. Findings in 21 cases of Gibbs’ hypsarrhythmia: spectacular effectiveness of ACTH. Acta Neurol Psychiatr Belg. 1958;58:130–41.
Aicardi J. Clinical approach to the management of intractable epilepsy. Dev Med Child Neurol. 1988;30:429–40.
Gupta R, Appleton R. Corticosteroids in the management of pediatric epilepsies. Arch Dis Child. 2005;90:379–84.
Sahu JK. Infantile spasms--evidence based medical management. Indian J Pediatr. 2014;81:1052–5.
Madaan P, Negi S, Sharma R, Kaur A, Sahu JK. X-linked ALG13 gene variant as a cause of epileptic encephalopathy in girls. Indian J Pediatr. 2019;86:1072–3.
Raga SV, Wilmshurst J. Epileptic spasms: evidence for oral corticosteroids and implications for low and middle income countries. Seizure. 2018;59:90–8.
Wanigasinghe J, Arambepola C, Sri Ranganathan S, Sumanasena S, Attanapola G. Randomized, single-blind, parallel clinical trial on efficacy of oral prednisolone versus intramuscular corticotropin on immediate and continued spasm control in west syndrome. Pediatr Neurol. 2015;53:193–9.
Mytinger JR, Quigg M, Taft WC, Buck ML, Rust RS. Outcomes in treatment of infantile spasms with pulse methylprednisolone. J Child Neurol. 2010;25:948–53.
Yeh H-R, Kim M-J, Ko T-S, Yum M-S, You S-J. Short-term outcome of intravenous methylprednisolone pulse therapy in patients with infantile spasms. Pediatr Neurol. 2017;71:50–5.
Haberlandt E, Weger C, Sigl SB, et al. Adrenocorticotropic hormone versus pulsatile dexamethasone in the treatment of infantile epilepsy syndromes. Pediatr Neurol. 2010;42:21–7.
Yamamoto H, Asoh M, Murakami H, Kamiyama N, Ohta C. Liposteroid (dexamethasone palmitate) therapy for west syndrome: a comparative study with ACTH therapy. Pediatr Neurol. 1998;18:415–9.
Madaan P, Chand P, Linn K, et al. Management practices for West syndrome in South Asia: a survey study and meta-analysis. Epilepsia Open. 2020;5:461–74. https://doi.org/10.1002/epi4.12419.
Bower BD, Jeavons PM. Infantile spasms and hypsarrhythmia. Lancet. 1959;273:605–9.
Singhi P, Ray M. Profile of west syndrome in north Indian children. Brain Dev. 2005;27:135–40.
Gulati S, Jain P, Kannan L, Sehgal R, Chakrabarty B. The clinical characteristics and treatment response in children with west syndrome in a developing country: a retrospective case record analysis. J Child Neurol. 2015;30:1440–7.
Ibrahim S, Gulab S, Ishaque S, Saleem T. Clinical profile and treatment of infantile spasms using vigabatrin and ACTH--a developing country perspective. BMC Pediatr. 2010;10:1.
Angappan D, Sahu JK, Malhi P, Singhi P. Safety, tolerability, and effectiveness of oral zonisamide therapy in comparison with intramuscular adrenocorticotropic hormone therapy in infants with west syndrome. Eur J Paediatr Neurol. 2019;23:136–42.
Gowda VK, Narayanaswamy V, Shivappa SK, Benakappa N, Benakappa A. Corticotrophin-ACTH in comparison to prednisolone in West syndrome - a randomized study. Indian J Pediatr. 2019;86:165–70.
Hussain SA, Lay J, Cheng E, Weng J, Sankar R, Baca CB. Recognition of infantile spasms is often delayed: the ASSIST study. J Pediatr. 2017;190:215–21.e1.
Vaddi VK, Sahu JK, Dhawan SR, Suthar R, Sankhyan N. Knowledge, attitude and practice (KAP) study of pediatricians on infantile spasms. Indian J Pediatr. 2018;85:836–40.
Lux AL, Edwards SW, Hancock E, et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurol. 2005;4:712–7.
Widjaja E, Go C, McCoy B, Snead OC. Neurodevelopmental outcome of infantile spasms: a systematic review and meta-analysis. Epilepsy Res. 2015;109:155–62.
Mytinger JR, Hussain SA, Islam MP, et al. Improving the inter-rater agreement of hypsarrhythmia using a simplified EEG grading scale for children with infantile spasms. Epilepsy Res. 2015;116:93–8.
Author information
Authors and Affiliations
Contributions
MR, PS, AG planned the study design. MR collected data. MR, AG, JKS, PM and PS were involved in data interpretation. MR and PM wrote the draft. All authors approved the final draft. PS would be the guarantor of the study.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rajpurohit, M., Gupta, A., Madaan, P. et al. Safety, Feasibility and Effectiveness of Pulse Methylprednisolone Therapy in Comparison with Intramuscular Adrenocorticotropic Hormone in Children with West Syndrome. Indian J Pediatr 88, 663–667 (2021). https://doi.org/10.1007/s12098-020-03521-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-020-03521-7