
Abstract
Inborn errors of metabolism have been considered as an infrequent cause of epilepsy. Improvement in diagnostics has improved the detection of a metabolic basis of recurrent seizures in neonates and children. The term 'metabolic epilepsy' is used to suggest inherited metabolic disorders with predominant epileptic manifestations as well as those where epilepsy is part of the overall neurological phenotype. Several of these disorders are treatable, and the physician should bear in mind the classical ages of presentation. As there are no specific clinical or electrographic features suggestive of metabolic epilepsies, an early suspicion is based on clinical and laboratory clues. Fortunately, with the advancement of gene sequencing technology, a diagnosis of these rare conditions is more straightforward and may not require invasive procedures such as biopsies, multiple metabolic stress-induced testing for abnormalities, and cerebrospinal fluid analysis. A gene panel may suffice in most cases and can be done from a blood sample. In many countries, many treatable metabolic disorders are now part of the neonatal screen. Early diagnosis and treatment of these disorders can result in the prevention of a full-scale metabolic crisis and improvement of neurological outcomes. Long-term neurological outcomes are variable and additional therapies may be required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although inborn errors of metabolism (IEMs) are a relatively infrequent cause of epilepsy, epileptic seizures are a common manifestation in several IEMs. The term 'metabolic epilepsy' has been used to connote epileptic seizures in two scenarios: (1) where epilepsy is a predominant manifestation as commonly seen in newborns and infants, and (2) where epileptic seizures occur in association with an IEM, especially during the periods of decompensation or acute deterioration [1]. A recent review of studies from 2012 to 2016 concluded that IEMs constitute a considerable proportion (42%) of all identified monogenic diseases associated with epilepsy or seizures [2]. Amongst these, disorders of energy metabolism (31%), amino-acidopathies (15%), congenital disorders of glycosylation (CDG) (14.5%), and lysosomal disorders (12%) were most common [2]. In the current review, metabolic epilepsy refers to both these scenarios; however, the discussion focusses on the treatable pediatric disorders relevant to the practicing pediatrician.
Pathophysiology of Epilepsy in IEMs
Interference with brain metabolism is the key pathophysiological mechanism in metabolic epilepsies. These metabolic derangements depend on the function of the gene involved. Some examples are an accumulation of toxic substrates or by-products (organic acidurias, urea cycle defects), deficiency of products (biotinidase deficiency), primary or secondary defects in the neurotransmitter pathways (phenylketonuria), defective transport and/or utilization of energy substrates (glucose transporter 1 deficiency), disturbances in neuronal membrane permeability (holocarboxylase synthetase deficiency), substrate deficiency (serine biosynthesis defect), reduced energy supply (glucose transporter 1 deficiency), impaired metal chelation/transport pathways (Menke's disease), or interference with embryonal cortical development resulting into malformations (peroxisomal disorders, congenital disorders of glycosylation) [3, 4]. The metabolic dysfunction may add to the already excitatory milieu of the neonatal brain owing to the immaturity of inhibitory pathways and predisposes to the neonatal-onset of epileptic seizures in the more severe forms of metabolic epilepsies [5, 6].
Classification of Metabolic Epilepsies
There are no specific clinical or electrographic features for metabolic epilepsies. However, an early suspicion is usually made based on some clues (Table 1), which represent the shared features of metabolic epilepsies [9]. For assigning a syndromic diagnosis, one may club the phenotypic presentations of metabolic epilepsies into one of the following categories [7]:
-
Neonatal seizures with acute/subacute encephalopathy or altered sensorium (e.g., pyridoxine dependent epilepsy, nonketotic hyperglycinemia, maple syrup urine disease)
-
Developmental delay or intellectual disability with seizures (e.g., phenylketonuria, creatine, serine or biotinidase deficiency)
-
Developmental delay and dysmorphism with seizures (e.g., Zellweger, pyruvate dehydrogenase deficiency)
-
Neurodegeneration with seizures, with/without neurovisceral storage (e.g., neuronal ceroid lipofuscinoses, gangliosidosis)
-
Movement disorders with epilepsy (e.g., glucose transporter 1 deficiency, neurotransmitter disorders)
-
Idiopathic epilepsy (in an otherwise healthy child) (e.g., glucose transporter 1 deficiency)
Different approaches may be used to classify these epilepsies in children further. These disorders may also be classified based on the typical age of presentation of IEMs in children. However, an overlap may occur with some IEMs such as mitochondrial disorders (Table 2). A brief description of some of the treatable metabolic epilepsies is provided below.
Pyridoxine-Dependent Epilepsy
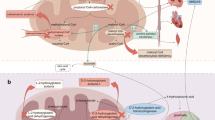
Pyridoxine-dependent epilepsy is a rare, autosomal-recessive, inborn error of lysine catabolism. It presents as recurrent, drug-refractory, neonatal seizures and responds dramatically to pyridoxine supplementation in therapeutic doses. Multifocal, erratic, myoclonic jerks, often mixed with tonic seizures and focal-onset motor seizures, are typically seen in the newborn, often associated with hypermotor features, tremors, hyperalertness, paroxysmal grimacing, and abnormal eye movements. Multiple seizure types may eventually follow, if untreated. Late-onset seizures may present until three years of age, and seizures may also occur in-utero (often perceived as recurrent hiccoughs) (Fig. 1). It is prudent to start pyridoxine supplementation at 100 mg intravenous or 30 mg/kg/d orally in 2 divided doses for 3–7 d in any neonate with drug-refractory seizures [10]. Seizures often respond within minutes, and the electrographic response can be seen during the bolus injection. Cardiorespiratory compromise may occur with the first dose and needs monitoring. Pathogenic variations in the ALDH7A1 gene confirm the diagnosis. The variations result in a deficiency of the α-aminoadipic semialdehyde dehydrogenase (antiquitin) enzyme, and accumulation of toxic intermediary substrates such as α-aminoadipic semialdehyde (which is used as a diagnostic biomarker), pipecolic acid, and piperidine-6-carboxylate (which inactivates the active form of pyridoxine and creates pyridoxine deficiency in the body). Therapy is life-long pyridoxine supplementation. Parents should be advised to double the dose during acute illnesses to prevent seizure recurrence. Despite early initiation of pyridoxine, the majority of cases are associated with mild to severe neurological problems, including secondary autistic features. Some children develop breakthrough seizures even on adequate doses and need additional anti-epileptic drugs; the cause for this is unknown. Oral folinic acid (3–5 mg/kg/d), dietary lysine restriction and L-arginine supplementation are often added to the drug regimen [11].

A 16 s sleep EEG epoch at 12 mo of age in a child diagnosed with pyridoxine-dependent epilepsy, showing asynchronous, asymmetric voltage with discontinuous background on the left side intermixed with a disorganized, chaotic activity and spike waves discharges (neonatal montage; sensitivity 15 mcV/mm; sweep speed 30 mm/s)
Pyridox(am)ine 5′-Phosphate Oxidase (PNPO) Deficiency
This distinct disorder is related to pyridoxine metabolism. It results from a deficiency of the rate-limiting enzyme PNPO that converts pyridoxine to its active metabolite pyridoxal 5′ phosphate [12]. Clinically, these neonates are often born prematurely, may demonstrate intra-uterine seizures, are often sicker in the postnatal period with recurrent drug-refractory seizures that are pyridoxine-resistant, and often have associated hypoglycemia, lactic acidosis, and encephalopathy [13]. The presentation may mimic hypoxic-ischemic encephalopathy. In the absence of a specific biomarker, biochemical suspicion is based on blood and urine profile reminiscent of L-aromatic acid decarboxylase deficiency (excessive vanillactic acid excretion in urine organic acid profile, elevated glycine and/or threonine, and reduced arginine) [12]. Diagnosis is confirmed by mutation analysis of the PNPO gene. Treatment is life-long supplementation of pyridoxal 5′ phosphate at 30 mg/kg/d in divided dosage. Mild gastrointestinal disturbance and liver enzyme derangement may be noted and may briefly need dose reduction. The outcome of early treatment is good. A sequential trial of pyridoxine and pyridoxal 5′ phosphate is recommended in all newborns and infants with drug-refractory seizures.
Pyridoxine-Dependent Epilepsy Due to Pyridoxal 5′ Phosphate-Binding Protein
This recently described entity is associated with pathogenic variations in the PROSC gene, which encodes a pyridoxal 5′ phosphate (PLP)-binding protein [14]. The defect causes pyridoxine-dependent epilepsy responsive to pyridoxal 5′ phosphate supplementation.
Folinic Acid-Responsive Seizures
Folinic acid-responsive seizures are allelic to pyridoxine-dependent epilepsy due to antiquitin deficiency, have similar biomarkers, and are responsive to folinic acid (3–5 mg/kg/d) [15]. Although not practically done, an unknown peak 'X' has been identified in cerebrospinal fluid (CSF) by high-performance liquid chromatography, which may serve as a biomarker. As a pragmatic treatment, when genetic testing is not available, or results are pending, many clinicians begin PLP and folinic acid together in children suspected to have pyridoxine disorders. While prescribing, the physicians should note that this medication is distinct from folic acid (which can paradoxically worsen these patients) and is costlier, thus often affects compliance.
Cerebral Folate Deficiency
Cerebral folate deficiency is an example of a 'focal IEM' as it is characterized by low levels of 5-methyltetrahydrofolate (the active metabolite of folate) in the nervous system but normal folate metabolism in the rest of the body [16]. It may result from (a) defective folate transport across the blood-CSF barrier (associated with mutations in the FOLR1 gene encoding the folate receptor α), (b) an increased folate turnover in the nervous system (related to a deficiency of dihydrofolate reductase enzyme responsible for catalyzing the conversion of dihydrofolate to tetrahydrofolate) or, (c) the presence of autoantibodies against the folate receptor [9]. The intracerebral deficiency leads to severe developmental delay, movement disorder, white matter changes, bilateral basal ganglia calcification, and drug-refractory epilepsy. Low levels of 5-methyltetrahydrofolate in CSF are an important biomarker; further confirmation may be done by detection of pathogenic variations in the FOLR1 gene [16]. Treatment includes initiation of folinic acid (1–5 mg/kg/d to correct 5-methyltetrahydrofolate CSF levels).
Biotinidase and Holocarboxylase Synthetase Deficiency
Biotinidase enzyme deficiency is a rare, autosomal recessive IEM that results from defective endogenous recycling of biotin (vitamin B7 or vitamin H) in the body [17]. Enzyme activity below 10% is defined as profound, while 10–30% residual activity is defined as a partial deficiency. Classical clinical presentation includes recurrent seizures in the first few months of life (holocarboxylase deficiency presents soon after birth while biotinidase deficiency presents later at 2–3 mo of age), with alopecia, hypotonia, flexural rash often mimicking fungal infection or dermatitis, and scalp seborrhea (Fig. 2) [18]. Laryngeal stridor, lactic acidosis, and encephalopathy may be rarely seen. Presentations in older children include progressive optic atrophy, hearing deficit, myelopathy, and progressive spastic paresis [19].

A 10 s sleep EEG epoch at eight months of age in a child diagnosed with biotinidase deficiency, showing suppression-bursts background, with each burst of delta-theta activity lasting for 1–2 s followed by a period of 4–6 s suppression pattern seen throughout the record (neonatal montage; sensitivity 7 mcV/mm; sweep speed 30 mm/s)
Testing for biotinidase enzyme activity in the peripheral blood is readily available and is part of neonatal screening at birth in developed countries. Genetic testing is confirmatory. Treatment is life-long biotin supplementation at 5–20 mg/d regardless of weight or age for both these disorders [17]. Parents should be instructed to double the dose during infections or illnesses. Raw egg whites contain avidin, which binds to biotin and reduces its bioavailability, hence should be avoided. Concomitant valproate therapy also reduces biotin levels. Despite early and adequate biotin supplementation, optic atrophy and sensorineural hearing loss progress. Similar to pyridoxine, biotin supplementation should be considered in any infant or older child with unexplained seizures.
Nonketotic Hyperglycinemia
Nonketotic hyperglycinemia or glycine encephalopathy is a rare, inborn error of glycine cleavage enzyme complex that leads to the accumulation of glycine in blood and CSF, as well as increased CSF: plasma glycine ratio (> 0.04 often used as a biomarker in simultaneously drawn samples) [20]. Neonatal and infantile presentations are common with progressive lethargy, hypotonia, myoclonic jerks, apnea, hiccups, burst-suppression on an electroencephalograph (EEG) and diffusion-restriction in myelinated tracts. Delayed presentations include global developmental delay with hypotonia, multifocal epilepsy, and progressive brain atrophy. The treatment consists of sodium benzoate to lower glycine, and dextromethorphan (often available as a constituent in cough syrups) to block glycinergic NMDA receptors [21]. Valproic acid is best avoided as it inhibits glycine metabolism.
Glucose Transporter 1 Deficiency
Glucose transporter 1 deficiency is characterized by impaired glucose transport into the brain due to the absence of its cerebral transporter 1 in the blood–brain barrier. The classical presentation includes infantile-onset refractory seizures, developmental delay, intellectual impairment, acquired microcephaly, hypotonia, and movement disorders [22]. An important clinical clue is the increased frequency of seizures before meals. The presence of a prominent movement disorder, especially paroxysmal exercise-induced dyskinesia in a child with intellectual impairment and epilepsy, should drive investigations for glucose transporter 1 deficiency [23]. Two specific epilepsy syndromes are early-onset absence epilepsy and myoclonic-astatic epilepsy. A CSF examination with low CSF glucose (absolute value < 2.22 mmoles/L), and/or CSF: blood glucose ratio < 0.4 (blood sample drawn immediately before CSF to prevent stress-associated hyperglycemia) in a fasting child is suggestive of the diagnosis. Pathogenic mutation in the SLC2A1 gene is diagnostic. Treatment consists of providing alternative sources of fuel to the brain such as ketone bodies via the ketogenic diet, and avoidance of drugs that impair glucose transporter 1 function such as caffeine, phenobarbital, diazepam, chloral hydrate, and tricyclic anti-depressants. Triheptanoin has recently been found useful in some cases.
Disorders of Creatine Biosynthesis and Transport
This rare IEM is characterized by reduced cerebral creatine, and accumulation of guanidinoacetate due to the deficiency of guanidinoacetate methyltransferase enzyme, arginine: glycine amidinotransferase enzyme and creatine transporter. Early-onset polymorphic seizures may present between 3 mo to 3 y of age. Older children have associated developmental delay/intellectual impairment, behavioral problems, hypotonia evolving into dystonia and movement disorders. Bilateral globus pallidum hyperintensities on T2-weighted magnetic resonance imaging and marked reduction/absence of a creatine peak on proton spectroscopy are unique radiological clues. They should be looked for in all children with unexplained intellectual disability and epilepsy [24]. Treatment for guanidinoacetate methyltransferase enzyme includes life-long creatine monohydrate (400 mg/d often available as body-building supplements), sodium benzoate, and ornithine aspartate supplementation (400–800 mg/kg/d), and dietary arginine restriction [25]. No effective treatment exists for creatine transporter defects.
Serine Biosynthesis Disorders
Serine biosynthesis disorders are another rare group of IEMs due to the deficient activity of one of the three key enzymes: 3-phosphoglycerate dehydrogenase, 3-phosphoserine aminotransferase, and phosphoserine phosphatase. Low CSF levels of serine characterize the disorder. Early-onset, refractory polymorphic seizures are associated with global developmental delay, microcephaly, cataract, spasticity, megaloblastic anemia, delayed myelination, and global cerebral atrophy [26]. Seizures may respond to serine supplementation (200–700 mg/kg/d) and glycine (200–300 mg/kg/d). Some cases may be associated with prominent dysmorphism and ichthyosis. Treatment is life-long, and outcomes are variable.
Approach to a Child with Suspected Metabolic Epilepsy
Although rare, metabolic epilepsies should be considered in the clinical scenarios described above (Table 1). Further clinical evaluation should include a detailed history and examination, including prenatal and perinatal period details, a three-generation pedigree analysis, and developmental milestones. The age of onset of epilepsy helps in narrowing the diagnostic possibilities (Table 2). Some of the specific clinical and investigational clues have been summarized again in Table 3. The investigations are generally tailored to each patient. They include a combination of the electroencephalogram, neuroimage with proton spectroscopy [27], basic metabolic screens (glucose, acidosis, ammonia, lactate, electrolytes including uric acid, ketones, and vitamin B12), blood and urine amino acids, urine organic acids, plasma carnitine profile, CSF assays for glycine, glucose, amino acids, neurotransmitters, pipecolic acid), muscle biopsies, specific biomarker testing, and genetic testing. Invasive tests such as biopsies are needed in selected cases where the genetic diagnosis is unrevealing, or a variant of unknown significance reported, or clinical suspicion is high.

A 16 s sleep EEG epoch at 14 mo of age in a child diagnosed with Menkes disease, showing asynchronous background with excessive slowing on the left side, intermixed with abundant multifocal epileptiform discharges (left temporal > right frontal-central) (neonatal montage; sensitivity 10 mcV/mm; sweep speed 30 mm/s)
Treatment
Specific treatments, as mentioned above, should be considered in the appropriate clinical setting. A therapeutic trial of pyridoxine, biotin, and folinic acid followed by pyridoxal 5′ phosphate is diagnostic and is recommended for drug-refractory seizures. Table 4 provides a summary of the available options for several treatable metabolic epilepsies. When no specific treatments are available, supportive care and prevention of catabolism should be initiated as per standard management guidelines for the individual groups of IEMs [1, 3, 7, 28]. Valproic acid (mitochondrial and hepatic toxin) and phenobarbitone (glucose transporter 1 inhibitor) should be avoided as mentioned above. Instead, broad-spectrum anti-seizures medications such as levetiracetam and benzodiazepines are a better initial choice. Targeted therapies for IEMs, are on the horizon but not commonly available in resource-limited settings. Hence, the focus of management in metabolic epilepsies should be to identify treatable causes and institute rescue therapy.
Conclusions
Although rare, metabolic epilepsies remain underdiagnosed. Several of these disorders are treatable, and the physician should remember the typical age of presentation of these disorders. Early diagnosis and treatment of these disorders can result in the prevention of a metabolic crisis and improvement of neurological outcomes. It is reasonable to start pyridoxine, folinic acid, and biotin as initial therapies for all children and neonates with refractory epilepsy. Long-term neurological outcomes are variable and additional therapies may be required.
References
Campistol J. Epilepsy in inborn errors of metabolism with therapeutic options. Semin Pediatr Neurol. 2016;23:321–31.
Tumiene B, Peterlin B, Maver A, Utkus A. Contemporary scope of inborn errors of metabolism involving epilepsy or seizures. Metab Brain Dis. 2018;33:1781–6.
van Karnebeek CDM, Sayson B, Lee JJY, et al. Metabolic evaluation of epilepsy: a diagnostic algorithm with focus on treatable conditions. Front Neurol. 2018;9:1016.
Lin Lin Lee V, KarMeng Choo B, Chung YS, P Kundap U, Kumari Y, Shaikh MF. Treatment, therapy and management of metabolic epilepsy: a systematic review. Int J MolSci. 2018;19:871.
Prasad AN, Hoffmann GF. Early onset epilepsy and inherited metabolic disorders: diagnosis and management. Can J Neurol Sci. 2010;37:350–8.
Pascual JM, Campistol J, Gil-Nagel A. Epilepsy in inherited metabolic disorders. Neurologist. 2008;14:S2-14.
Sharma S, Prasad AN. Inborn errors of metabolism and epilepsy: current understanding, diagnosis, and treatment approaches. Int J Mol Sci. 2017;18:1384.
Wolf NI, Garcia-Cazorla A, Hoffmann GF. Epilepsy and inborn errors of metabolism in children. J Inherit Metab Dis. 2009;32:609.
Rahman S, Footitt EJ, Varadkar S, Clayton PT. Inborn errors of metabolism causing epilepsy. Dev Med Child Neurol. 2013;55:23–36.
van Karnebeek CD, Jaggumantri S. Current treatment and management of pyridoxine-dependent epilepsy. Curr Treat Options Neurol. 2015;17:335.
van Karnebeek CD, Stockler-Ipsiroglu S, Jaggumantri S, et al. Lysine-restricted diet as adjunct therapy for pyridoxine-dependent epilepsy: the PDE Consortium Consensus Recommendations. JIMD Rep. 2014;15:1–11.
Mills PB, Camuzeaux SS, Footitt EJ, et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain. 2014;137:1350–60.
Mills PB, Surtees RA, Champion MP, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5’-phosphate oxidase. Hum Mol Genet. 2005;14:1077–86.
Tremino L, Forcada-Nadal A, Rubio V. Insight into vitamin B6 -dependent epilepsy due to PLPBP (previously PROSC) missense mutations. Hum Mutat. 2018;39:1002–13.
Gallagher RC, Van Hove JL, Scharer G, et al. Folinic acid-responsive seizures are identical to pyridoxine-dependent epilepsy. Ann Neurol. 2009;65:550–6.
Pope S, Artuch R, Heales S, Rahman S. Cerebral folate deficiency: analytical tests and differential diagnosis. J Inherit Metab Dis. 2019;42:655–72.
Wolf B. Biotinidase deficiency: “If you have to have an inherited metabolic disease, this is the one to have.” Genet Med. 2012;14:565–75.
Dhawan SR, Sharawat IK, Saini AG, Suthar R, Attri SV. Diaper rash in an infant with Seizures. J Pediatr. 2019;206:296.
Kasinathan A, Suthar R, Vyas S, Saini AG, Sankhyan N, Attri S. Subacute myelopathy: think beyond neuromyelitis optica spectrum disorder. Ann Indian Acad Neurol. 2019;22:541–2.
Applegarth DA, Toone JR. Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. J Inherit Metab Dis. 2004;27:417–22.
Van Hove JL, Vande Kerckhove K, Hennermann JB, et al. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis. 2005;28:651–63.
Gramer G, Wolf NI, Vater D, et al. Glucose transporter-1 (GLUT1) deficiency syndrome: diagnosis and treatment in late childhood. Neuropediatrics. 2012;43:168–71.
Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009;31:545–52.
Sharer JD, Bodamer O, Longo N, Tortorelli S, Wamelink MM, Young S. Laboratory diagnosis of creatine deficiency syndromes: a technical standard and guideline of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:256–63.
Clark JF, Cecil KM. Diagnostic methods and recommendations for the cerebral creatine deficiency syndromes. Pediatr Res. 2015;77:398–405.
van der Crabben SN, Verhoeven-Duif NM, Brilstra EH, et al. An update on serine deficiency disorders. J Inherit Metab Dis. 2013;36:613–9.
Dhawan SR, Saini AG, Vyas S, Attri SV. Teaching neuroimages: when MRI is a clue in episodic ataxia. Neurology. 2019;93:e2074–5.
Cosnahan AS, Campbell CT. Inborn errors of metabolism in pediatric epilepsy. J Pediatr Pharmacol Ther. 2019;24:398–405.
Author information
Authors and Affiliations
Contributions
CR: Drafting the manuscript; AGS: Drafting and revising the manuscript. AGS will act as guarantor for this paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Reddy, C., Saini, A.G. Metabolic Epilepsy. Indian J Pediatr 88, 1025–1032 (2021). https://doi.org/10.1007/s12098-020-03510-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-020-03510-w