
Abstract
Background
Cancer stem cells (CSCs) have unique biological characteristics, including tumorigenicity, immortality, and chemoresistance. Colorectal CSCs have been identified and isolated from colorectal cancers by various methods. AKAP12, a scaffolding protein, is considered to act as a potential suppressor in colorectal cancer, but its role in CSCs remains unknown. In this study, we investigated the function of AKAP12 in Colorectal CSCs.
Methods
Herein, Colorectal CSCs were enriched by cell culture with a serum-free medium. CSC-associated characteristics were evaluated by Flow cytometry assay and qPCR. AKAP12 gene expression was regulated by lentiviral transfection assay. The tumorigenicity of AKAP12 in vivo by constructing a tumor xenograft model. The related pathways were explored by qPCR and Western blot.
Results
The depletion of AKAP12 reduced colony formation, sphere formation, and expression of stem cell markers in colorectal cancer cells, while its knockdown decreased the volume and weight of tumor xenografts in vivo. AKAP12 expression levels also affected the expression of stemness markers associated with STAT3, potentially via regulating the expression of protein kinase C.
Conclusion
This study suggests Colorectal CSCs overexpress AKAP12 and maintain stem cell characteristics through the AKAP12/PKC/STAT3 pathway. AKAP12 may be an important therapeutic target for blocking the development of colorectal cancer in the field of cancer stem cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the fourth most deadly cancer worldwide after lung, liver, and stomach cancer, killing almost 700,000 people each year [1]. Colorectal cancer has been the focus of numerous studies, including those involving cancer stem cells (CSCs).
CSCs utilize many signaling pathways found in normal stem cells. WNT, Notch, and Hedgehog are oncogenic cascades in CSCs [2, 3]. CSCs are a group of cells that exist in most types of liquid and solid cancers and contribute to tumor development, drug resistance, recurrence, and metastasis [4]. They have a strong self-renewal capacity and high plasticity. CSCs hiding in tumors are difficult to eradicate, and their initial identification is usually based on the expression of cell surface markers [5]. It is, therefore, essential to understand the role and possible mechanisms of CSCs in colorectal cancer.
A-kinase anchor protein 12 (AKAP12/AKAP250/gravin) is a member of the folding protein family (molecular weight 250 kD), which participates in multiple biological functions such as protein kinase C (PKC) and protein kinase A (PKA) complex formation [6], regulation of various signal transduction pathways [7], FAK-mediated cytoskeleton remodeling, and regulation of the β2 adrenergic receptor complex [8]. AKAP12 has also been proven to execute different functions in tumors, including invasion, metastasis, and angiogenic ability. AKAP12 was shown to be involved in regulating growth-related proteins such as p53, p21, p27, Bax, and Bcl [9], inhibiting cell migration through the PKC-Raf/MEK/ERK pathway [10], downregulating matrix metalloproteinase expression [11] to reduce degradation of the extracellular matrix [12] and inhibiting expression of vascular endothelial growth factor and epidermal growth factor receptors. Many classical studies have shown that AKAP12 expression was reduced in colorectal cancer, gastric cancer, esophageal cancer, acute myeloid leukemia [13], and other malignant tumors. These results suggest that AKAP12 acts as a tumor suppressor gene with a crucial role in inhibiting tumor growth, metastasis, and angiogenesis. However, the role of AKAP12 in CSCs remains unknown.
Signal transduction and transcription activator 3 (STAT3) is a STAT-family member that enters the nucleus to influence the transcription of genes in response to extracellular stimuli [14]. STAT3, which has been shown to be continually activated in a variety of solid tumors [15], can be activated by conversion to its phosphorylated form (p-STAT3) by a variety of protein kinases, such as JAK2 [16], PKA, and PKC [17]. Previous studies have shown that AKAP12 can positively regulate the phosphorylation of STAT3, thereby activating the key post-translational modifications required for tumor progression [18]. AKAP12 is a known scaffold ligand for PKC [19] and may, therefore, be directly involved in STAT3 phosphorylation. Recent studies show that STAT3 is essential for maintaining high levels of AKAP12 [20], and AKAP12 is a direct target for STAT3 transcription factors in lung epithelial cells [21].
The current study aimed to investigate the role of AKAP12 in colorectal CSCs (CCSCs) and to explore the potential mechanisms involved. A better understanding of the functions of AKAP12 in CCSCs may provide a new therapeutic approach for the early diagnosis, clinical treatment, and prognosis prediction in patients with colorectal cancer.
Materials and methods
Cell culture and enrich CSCs
LoVo and HCT116 colorectal cancer cells were purchased from the Shanghai Cell Bank at the Chinese Academy of Sciences and cultured in DME/F12 medium (Hyclone, USA) and McCoy’s 5A medium (Gibco, USA), respectively, supplemented with 10% fetal bovine serum (Gibco, Brazil) and 1% penicillin/streptomycin (Gibco, USA). Cells were cultivated at 37 °C in a humidified atmosphere containing 5% CO2. Serum-free medium preparation: B-27 (0.4%; Gibco, USA), epidermal growth factor (20 ng/ml; Peprotech, USA), and basic fibroblast growth factor (10 ng/ml; Peprotech, USA) were added to DME/F12 medium. HCT116 and LoVo cells were resuspended as a single-cell suspension using the prepared serum-free medium and uniformly inoculated in ultra-low attachment plates (plate diameter = 60 mm, Corning, USA). The cells were then cultured in a cell culture incubator containing 5%CO2 at 37 °C, and serum-free medium was changed weekly, and the non-stem cells that died due to lack of nutrition were discarded. Two weeks later, macroscopic 3D stem-like spheres formed and were collected by centrifugation.
Reagents and antibodies
Enzastaurin [22] (LY317615, IC50 = 6 nM) (MedChem Express, USA) was dissolved in dimethyl sulfoxide (Sigma-Aldrich, USA) to a final concentration of 3 nM. Monoclonal antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), STAT3, p-STAT3, PKC, and PKA were purchased from Cell Signaling Technology (USA). Mouse polyclonal antibody against AKAP12 was purchased from Abcam (USA). Rabbit monoclonal antibody against p-STAT3 was purchased from Cell Signaling Technology (USA) and a monoclonal antibody against β-tubulin was purchased from Abways (China). Secondary antibodies were all purchased from Abcam (USA).
Lentivirus transfection assay
Lentivirus overexpressing human AKAP12 (Lv-hAKAP12) and empty vector (Lv-Null) were created by Shanghai Genechem (China). For lentiviral transfection of HCT116 and LoVo cells, Lv-hAKAP12 or Lv-Null were added to a well containing 5 × 104 cells (70%–80% confluent), 1 ml complete medium, and 5 μg/ml polybrene (Shanghai Genechem, China) for 24 h. The transfected cells were then selected with 2 μg/ml puromycin (Sigma-Aldrich, USA). Lv-Null was used as a control. Selected cells were maintained in growth medium with 0.5 μg/ml puromycin (Shanghai Genechem, China). Lentiviruses including specific small interfering RNA (siRNA) for AKAP12 (Lv-siAKAP12) or empty vector (Lv-Cont) were created by Shanghai Genepharma (China), as described above. In the process of cell infection of the lentiviral vector, Null acts as a negative control for Lv-hAKAP12 to rule out the effects of infection with the lentiviral vector itself. Correspondingly, Cont served as a negative control for Lv-siAKAP12. There is no essential difference between the sequence fragments.
Flow cytometry
Cells were harvested and resuspended in phosphate-buffered saline. The cells were then incubated at 4 °C for 15 min with anti-CD133-APC (130–098-829, MiltenyiBiotec, Germany), anti-Nanog-Alexa747 (561,300, BD Biosciences, USA), and anti-Sox2-Percp-CY5.5 (561,506, BD Biosciences, USA) or isotype controls. Flow cytometry analysis was conducted using a C6 FACS (BD Biosciences, USA).
Flow CD133 + cell sorting assay
HCT116 cells were harvested and resuspended in phosphate-buffered saline. The cells were then incubated at 4 °C for 15 min with anti-CD133-APC (130–098-829, MiltenyiBiotec, Germany). Flow CD133+ cell sorting assay was conducted using BD influx (BD Biosciences, Germany).
Cell cycle assay
After centrifugation, the supernatant was discarded, and the collected cells were washed three times with PBS. After adding pre-cooled 75% ethanol, it was placed at 4 °C overnight to fix. After washing the cells with PBS again, the cells were added to 500 uL of PBS containing 50 μg/mL ethidium bromide (PI, Multisciences, China), 100 μg/mL RNase A, 0.2% Triton X-100, and incubated at 37 °C. Flow cytometry was performed using standard procedures and cell cycle fitting analysis was performed using ModFit software.
Sphere-formation assay
HCT116 and LoVo cells were resuspended as a single-cell suspension using the prepared serum-free medium mentioned above and uniformly inoculated in ultra-low attachment plates (plate diameter = 60 mm, Corning, USA). The cells were then cultured in a cell culture incubator containing 5%CO2 at 37 °C, and serum-free medium was changed weekly, and the non-stem cells that died due to lack of nutrition were discarded. The number of stem-like cell spheres was counted after 14 days.
Limited dilution assay
Cells were seeded into ultra-low-attachment 96-well plates (Corning) at densities of 104, 103, 102, 101, and single cells per well, respectively, and cultured in 200 μl serum-free medium (DME/F12) as described above. Cell cultures were maintained in a humidified atmosphere at 37 °C and 5% CO2 for 14 days. The numbers of stem-like spheres with a diameter > 20 μm were then counted under an optical microscope.
Clonogenic assay
The cells were plated at a density of 1 × 103 cells per well in six-well plates with complete DME/F12 medium for 14 days. Due to the slow growth of cells in the siAKAP12 group, we have lengthened the culture time of this group of cells to 21 days, so Null cells and Cont cells appear to have a large difference in cell numbers. The used medium was removed, and the plate was fixed with 95% ethanol, and then stained with 0.1% crystal violet (w:v) for 15 min, and the excess dye solution was gently washed away. The number of cell colonies was counted under an ordinary optical microscope.
RNA extraction and quantitative polymerase chain reaction (qPCR) analysis
The total RNA of cells in six-well plates was extracted with TRIzol (Invitrogen, USA), and the process of converting total RNA into cDNA was performed by the PrimeScript RT kit (TaKaRa, China). Real-time fluorescent quantitative PCR (qPCR) amplification was performed using SYBR Green PCR Kit (TaKaRa, China) and a rapid real-time PCR system (Applied Biosystems, USA). mRNA levels were calculated using the 2-ΔΔct method. PCR primers for qPCR are shown in Supplemental Table I.
Protein isolation
Cells were harvested using a plastic scraper, washed three times with cold phosphate-buffered saline, and then lysed with ice-cold protein extract solution RIPA (Beyotime, China). The protein concentration was quantified using the BCA procedure according to the manufacturer’s protocol (Beyotime, China).
Western blot
Equal amounts (60 μg) of protein samples were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) using 6% or 10% polyacrylamide gels and then transferred onto a nitrocellulose membrane (Millipore, USA). The membranes were blocked with 5% BSA Tris-buffered saline with 0.1% Tween-20 (TBST) buffer for 2 h at room temperature and then incubated with primary antibodies including anti-AKAP12 (1:1000), anti-STAT3 (1:1000), anti-p-STAT3 (1:1000), anti-PKC (1:1000), anti-PKA (1:1000), anti-β-tubulin (1:2000) and anti-GAPDH (1:3000) antibodies at 37 °C for 1 h or overnight at 4 °C. After washing with TBST, blots were incubated with the appropriate anti-rabbit or anti-mouse secondary antibodies (1:2000) and then detected using an Odyssey® CLx Infrared Imaging System (LI-COR Biosciences, USA). The intensities of the protein bands were analyzed using ImageJ software. GAPDH and β-tubulin were used as internal controls.
Evaluation of tumorigenicity
We determined the tumorigenicities of the different cancer cells by subcutaneous injection of 5 × 106 cells and negative control cells into either flank of 4-week-old female BALB/c-nu mice (Animal Center of Shanghai Tenth People's Hospital affiliated to Tongji University, China). All studies involving animals were approved by the Ethics Committee of Shanghai Tenth People's Hospital affiliated to Tongji University. Tumor size was measured 7 days after cell injection and every 3 days thereafter using calipers. We calculate its volume by measuring the longitudinal and transverse diameters of the tumor. The mice were euthanized on the 16th day after the cell injection. Due to the rapid tumor formation rate of the cells we inoculated, the nude mice did not show any obvious abnormal appearance during the whole process; the changes in body weight, feed, and drinking water intake were within the normal range, and the measurable clinical indicators were all normal; no significant change in induced behavior; no abnormality in response to external stimuli. There was no obvious macroscopic fluid accumulated internally as a result of tumor progression. The American Veterinary Association (AVMA) mentions that the tumor burden is no more than 10% of body weight; in an adult mouse, a tumor should not exceed 20 mm in any dimension, and we strictly abide by this requirement.
siRNA transient transfection
To achieve a cell density of 30%–50% during transfection, 50 × 104 cells were grown in a six-well plate 12 h before transfection. The medium was discarded overnight and 1 ml of OPTI-MEM (Invitrogen, Germany) serum-free and the antibody-free medium was added and the six-well plate was placed at 37 °C in a 5% CO2 cell incubator. Prepare transfection complexes: complex 1 is 125 μl of OPTI-MEM medium with 2.5 μl of lipo2000 (Invitrogen, Germany); complex 2 is 125 μl of OPTI-MEM medium with 50 nM siRNA. Mix complexes 1 and 2, incubate for 20 min at room temperature and then add dropwise to the culture dish. Discard the OPTI-MEM medium after 8 h and add 1.5 ml of fresh complete medium. Cells were collected at 24 h and 36 h post-transfection, and qPCR and immunoblotting assays were performed to verify the expression of genes and proteins, respectively.
Statistical analysis
All experiments were repeated at least three times independently. Data were statistically analyzed using Graph Pad Prism version 6.0 and SPSS version 21.0. Statistical comparisons were made using Student’s t-tests for differences between the two groups. Comparisons of results in three or more groups were made using the Bonferroni-Dunn test. A value of p < 0.05 was considered to indicate a significant difference.
Results
AKAP12 expression increased in stem-like cell spheres accompanied by increased expression of typical stemness markers
We investigated the relationship between AKAP12 and CCSCs in HCT116 and LoVo colorectal cancer cell lines. CSCs were enriched by cell culture in a serum-free medium to generate 3D stem-like spheres [23], the photos of 3D stem-like spheres are shown in Fig. 1A. AKAP12 expression levels were increased in stem-like spheres compared with the original cell lines (Fig. 1B, C). Furthermore, mRNA levels of some typical CCSC markers were significantly increased in sphere cells compared with the original cell lines, except for ABCG2 in LoVo stem-like spheres, which was reduced (Fig. 1D). HCT116-CD133+ cells were sorted and collected by flow cytometry. The expression level of AKAP12 in HCT116-CD133+ cells was significantly increased (Fig. 1E). These results suggest that AKAP12 may play a significant role in maintaining cancer stem-like phenotypes.

AKAP12 was upregulated in CCSCs. A 3D stem-like spheres were obtained from HCT116 cells. Bar = 100 μm. B–C AKAP12 protein and mRNA levels were detected in cell lines and their corresponding stem-like spheres by western blot and quantitative polymerase chain reaction (qPCR), respectively. **p < 0.01, *p < 0.05. D mRNA levels of the stemness markers CD133, EpCAM, Nanog, Oct-4, Sox-2, CD90, Bmi-1, and ABCG2 were confirmed by qPCR. **p < 0.01, *p < 0.05. E AKAP12 protein levels were detected in HCT116 CD133−/CD133+ cells by western blot
Induction of AKAP12 expression promoted maintenance of cancer stem-like phenotypes in colorectal cancer cells
AKAP12 was overexpressed in HCT116 and LoVo cells (HCT116-hAKAP12 and LoVo-hAKAP12) and confirmed its increased mRNA and protein levels (Fig. 2A, B) to appraise the role of AKAP12 in regulating CSC-associated characteristics. Overexpression of AKAP12 was associated with significantly upregulated mRNA levels of stem cell markers in HCT116-hAKAP12 and LoVo-hAKAP12 cells compared with negative control cells (HCT116-Null and LoVo-Null) (Fig. 2C). Furthermore, AKAP12 overexpression also increased the percentages of CD133+, Sox2+, and Nanog+ cells among HCT116-hAKAP12 and LoVo-hAKAP12 cells, as demonstrated by flow cytometry (Fig. 2D). AKAP12 overexpression also increased the number of clonogenic cells (Fig. 2E). According to limited dilution assays, hAKAP12 cells showed increased sphere formation at different cell-inoculum densities from 104 to 101 cells per well (Fig. 2F). At a dilution of 10 HCT116 cells/1 ml (1cell/well average), only 13/24 HCT116-Null wells and 11/24 HCT116-hAKAP12 received a single cell in the well, of which 0/13 HCT116-Null and 4/11 HCT116-hAKAP12 single-cell wells formed growing colonies. Last but not least, our cell cycle assay results showed that the proportion of cells staying in S-phase and G2M phase was increased in hAKAP12, especially staying in G2M phase. These results suggest that hAKAP12 cells have higher cleavage activity than Null cells (Sup. Figure 1, 2).

Induction of AKAP12 expression promoted CSC-related properties in colon cancer cells. A–B AKAP12 expression levels in AKAP12-overexpressing (hAKAP12) and control cells (Null) were determined by qPCR and western blot. **p < 0.01, *p < 0.05. C Expression levels of stemness markers CD133, EpCAM, Nanog, Oct-4, Sox-2, CD90, Bmi-1, and ABCG2 in HCT116 and LoVo cells in response to overexpression of AKAP12 were assessed by qPCR. **p < 0.01, *p < 0.05. D Percentages of CD133+, Nanog+, and Sox-2+ cells in hAKAP12 and Null cells were determined by flow cytometry. **p < 0.01, *p < 0.05. E Null and hAKAP12 cells were subjected to clonogenic assays in six-well plates and the numbers of clonogenic cells per 1,000 cells were counted 10 days after seeding. **p < 0.01, *p < 0.05. F Null and hAKAP12 cells were subjected to limited dilution assays in ultra-low-attachment 96-well plates and the numbers of spheroids generated by 10,000, 1,000, 100, and 10 cells were counted 10 days after seeding. **p < 0.01, *p < 0.05
AKAP12 depletion reduced CSC-related properties in colorectal cancer cells
AKAP12 was further clarified in the modulation of CSC-related properties by depleting its expression with specific siRNA transfection to create the cell lines HCT116-siAKAP12 and LoVo-siAKAP12, respectively. AKAP12 mRNA and protein levels were reduced by transfecting siRNA (Fig. 3A, B). qPCR analysis revealed that mRNA expression levels of the stem cell markers were also reduced in HCT116-siAKAP12 and LoVo-siAKAP12 cells (Fig. 3C). Simultaneously, the numbers of stem-like sphere colonies were reduced by > 50% and > 60% in HCT116-siAKAP12 and LoVo-siAKAP12 cells, respectively, compared with the negative controls (HCT116-Cont and LoVo-Cont) (Fig. 3D). Furthermore, siAKAP12 cells showed reduced sphere-formation capabilities at each cell-inoculum density in a limited dilution assay (Fig. 3E). These findings demonstrate the important effects of AKAP12 depletion on the reduction of CSC-related properties.

AKAP12 knockdown reduced CSC-related properties in colon cancer cells. A–B AKAP12 expression levels in AKAP12-depleted (siAKAP12) and control (Cont) cells were determined by qPCR and western blot. **p < 0.01, *p < 0.05. C Expression levels of stemness markers CD133, EpCAM, Nanog, Oct-4, Sox-2, CD90, Bmi-1, and ABCG2 in HCT116 and LoVo cells in response to depletion of AKAP12 were determined by qPCR. **p < 0.01, *p < 0.05. D Control and siAKAP12 cells were subjected to clonogenic assays in six-well plates and the numbers of clonogenic cells per 1,000 cells were counted 14 days after seeding. **p < 0.01, *p < 0.05. F Control and siAKAP12 cells were subjected to limited dilution assays in ultra-low-attachment 96-well plates, and the numbers of spheroids generated by 10,000, 1,000, 100, and 10 cells were counted 14 days after seeding. **p < 0.01, *p < 0.05
AKAP12 promoted tumorigenicity in vivo
We established tumor xenograft models to assess the effects of AKAP12 on CSC-related properties in vivo by injecting six male BALB/c-nu mice with HCT116-siAKAP12 and control HCT116-Cont cells. The method by which we measure the volume of a tumor at a specific time is tumor volume = l*w2/2 (l, length: parallel to the head and tail of the mouse; w, width: perpendicular to the head and tail of the nude mouse). The data in Fig. 4B are the statistical data obtained by calculating the tumor volume in the body according to the above measurement method. Tumor volumes were significantly smaller on the experimental side compared with the control side in mice (Fig. 4A, B). These results indicate that AKAP12 contributes to the tumorigenicity of colorectal cancer stem-like cells.

AKAP12 impacted tumor growth in vivo. A Mice were implanted subcutaneously with Cont and siAKAP12 cells (n = 6). B Tumor volumes were measured and presented as the mean ± standard error. 0.01 < *p < 0.05
AKAP12 activation of STAT3 signaling pathway in colorectal cancer was PKC dependent
STAT3 and its relevant pathway play an important role in CSC self-renewal [24], epithelial–mesenchymal transition and metastasis [25]. In the current study, STAT3 was significantly increased in stem-like spheres (Fig. 5A, B; Sup. Figure 3A). The mRNA level of STAT3 reflects the expression level of total STAT3, while at the protein level, the expression level of total STAT3 = unactivated STAT3 expression level (STAT3 protein expression levels in Fig. 5C–F) + activated STAT3 expression level (p-STAT3 in Fig. 5C–F). AKAP12, STAT3, and p-STAT3 expression were verified in hAKAP12, siAKAP12 and negative control cells. Overexpression or knockdown of AKAP12 dramatically increased or reduced STAT3 mRNA levels and p-STAT3 protein expression, respectively, but STAT3 protein expression levels remained relatively stable (Fig. 5C–F; Sup. Figure 3B, C). These results indicated that AKAP12 modulated the phosphorylation of STAT3 via certain pathways. Interestingly, PKC expression levels varied in line with the upregulation/downregulation of AKAP12, while PKA levels remained relatively stable (Fig. 5C–F). Inhibition of PKC by the potent inhibitor enzastaurin significantly reduced STAT3 mRNA and p-STAT3 protein expression levels in hAKAP12 cells (Fig. 5G, H; Sup. Figure 3D). These results indicate that the elevated levels of AKAP12 sustain the phosphorylation of STAT3, probably via its ability to scaffold PKC.

AKAP12 activation of the STAT3 pathway in colon cancer cells depends on PKC. A–B STAT3 protein and mRNA levels in stem-like spheres were determined by qPCR and western blot, respectively. **p < 0.01, *p < 0.05. C–F AKAP12, STAT3, p-STAT3, PKA and PKC mRNA and protein levels were measured by qPCR and western blot, respectively, in Null, AKAP12, Cont, and siAKAP12 cells. **p < 0.01, *p < 0.05, #p = 0.568, ##p = 0.103. G–H PKC, STAT3, and AKAP12 mRNA and protein levels in HCT116-Null and HCT116-hAKAP12 cells treated with the PKC inhibitor enzastaurin were assessed by qPCR and western blot, respectively. **p < 0.01, *p < 0.05, #p = 0.092, ##p = 0.390
STAT3 positively affected AKAP12 expression in colorectal cancer cells
To clarify the relationship between STAT3 and AKAP12, we transfected HCT116 and LoVo cells with a STAT3-expressing plasmid and control vector, or with STAT3 or control siRNA to overexpress or deplete STAT3, respectively. The data in Figs. 6A–D show a positive correlation between AKAP12 and STAT3 expression, suggesting that there may be a positive feedback loop between STAT3 and AKAP12.

STAT3 positively affected AKAP12 expression levels in colon cancer cells. A–D HCT116 and LoVo colon cancer cells were transfected with STAT3-expressing plasmid or control vector or with STAT3 or control siRNA to overexpress or deplete STAT3, respectively. STAT3 and AKAP12 mRNA and protein levels were measured by qPCR and western blot. **p < 0.01, *p < 0.05
Discussion
The CSC (or cancer-initiating cell) theory suggests that only a very small number of cells in a tumor referred to as CSCs have the ability for cancer development [26, 27]. This doctrine believes that dysregulation of self-renewal may be the first step in carcinogenesis [28]. CSCs are products of self-renewal disorders and there are features specific for CSCs, including strong tumorigenic potential, the ability to grow into spheres under serum deprivation, high aldehyde dehydrogenase activity [29], and cell cycle disorders [30]. These cells generate clones and reshape tumors in vitro after transplantation into immunodeficient animals. Coincidentally, CSC also can xenograft and differentiate cancer into many heterogeneous cell populations.
Growing evidence has indicated the relevance of the CSC concept in colorectal cancer [31,32,33,34]. Previous studies have demonstrated that CSC in colorectal cancer are characterized by the expression of various markers such as BMI1, Sox2, Nanog, etc., which are involved in pathological processes such as colorectal carcinogenesis and development, and have an important impact on the tolerance of chemotherapy and radiotherapy in colorectal cancer [31,32,33,34]. Previous studies have shown the critical role of BMI1 as an epigenetic inhibitor in self-renewal, differentiation, and tumor initiation of CSC. Targeting BMI1 can successfully eliminate human colorectal cancer stem cells from mouse xenografts. Therefore, therapeutic strategies targeting cancer stem cells are important for the treatment of colorectal cancer [32].
Numerous CSC markers, including CD133, EpCAM, Nanog, and Oct-4, have been used in CSC research [35]. Cultivating cells as 3D spheres are considered a reliable means of enriching cultures for CSCs. We accordingly used this method to enrich CSCs from HCT116 and LoVo cancer cell lines and compared them with the original cells. However, the stem cell marker ABCG2 was not enriched in LoVo tumor-sphere cells, and further studies are needed to determine the reasons for this.
Although research on AKAP12 is relatively limited, AKAP12 has been shown to promote tumor cell apoptosis and inhibit invasion and angiogenesis [8, 18]. Our previous study showed that overexpression of AKAP12 inhibited the invasion and metastasis of colorectal cancer [8, 36]. Maki et al. confirmed that AKAP12 was required for oligodendrocyte differentiation in adult white matter in mice [37]. The latest research proves that HDAC6-dependent AKAP12 deacetylation led to its ubiquitination-mediated degradation and promoted colon cancer metastasis [38]. However, the role of AKAP12 in CSCs remains unclear. Herein, we showed that AKAP12 was upregulated by sphere formation in HCT116 and LoVo cells, while most stem cell markers were upregulated/downregulated by overexpression or reduction of AKAP12, respectively. Overexpression of AKAP12 also increased the percentages of CD133+, Sox2+, and Nanog+ cells, as demonstrated by flow cytometry. These results were supported by clonogenic and limited dilution assays. However, these results do not necessarily conflict with the idea of AKAP12 as a tumor suppressor. AKAP12 acts as an important scaffolding protein by binding certain key signaling media such as PKA, PKC, and other cell cycle-associated proteins to promote mitotic signaling and cytoskeletal remodeling [39, 40]. We, therefore, assumed that AKAP12 carried out its scaffolding functions in HCT116 and LoVo cells to control cytoskeleton remodeling and then promote stem cell characteristics by regulating a signal pathway or network.
STAT3 is known to be activated in a variety of solid tumors, including colorectal cancer, suggesting that it may be essential for the survival and growth of tumor cells [41, 42]. Lin et al. also found that STAT3 played a crucial role in the proliferation and survival of tumor-initiating cells [43]. Peixoto et al. confirmed that AKAP12 positively regulated the phosphorylation of STAT3 in human glioblastoma [20], while PKA and PKC modulated the phosphorylation of STAT3 in human umbilical vein endothelial cells [18]. In the current study, STAT3 expression was increased in stem-like spheres and AKAP12 positively regulated p-STAT3, while PKC (but not PKA) was also upregulated/downregulated in accordance with AKAP12 overexpression or depletion, respectively. Enzastaurin (LY317615.HCl) is a selective protein kinase inhibitor that has been found to significantly inhibit the AKT pathway, thereby accelerating cell apoptosis and inhibiting the growth of glioblastoma and colorectal tumor grafts. Inhibition of PKC by enzastaurin significantly reduced p-STAT3 expression, whereas AKAP12 expression remained stable. These results suggested that elevated levels of AKAP12 sustained STAT3 phosphorylation, probably via its ability to scaffold PKC, thus revealing a possible AKAP12/PKC/STAT3 pathway in CCSCs. Regarding the mechanism whereby AKAP12 modulates PKC, Gelman et al. identified AKAP12 (SSeCKS in rats) in a screen for phosphatidylserine-dependent PKC-binding proteins and showed that PKC binding by AKAP12 was modulated by the level of PKC phosphorylation. However, the precise mechanism whereby AKAP12 modulates PKC in CCSCs remains unclear, and further studies are needed to clarify this. Notably, we detected a potential positive feedback loop between STAT3 and AKAP12 in HCT116 and LoVo cells by demonstrating that an increase/decrease in STAT3 could upregulate/downregulate AKAP12.
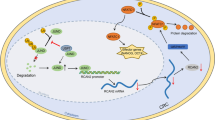
This study focused on the association between AKAP12 and CCSCs and the possible mechanisms involved in this relationship. We showed that AKAP12 acted as a promotor to maintain stem cell-like phenotypes in HCT116 and LoVo cell lines, thus improving our understanding of the role of AKAP12 in tumors from the perspective of CCSCs. Mechanistically, we identified the PKC/STAT3 signaling pathway as a possible link between AKAP12 and CSC-associated characteristics (Fig. 7). Overall, in this study, we demonstrated for the first time that in colorectal cancer cells, AKAP12 can activate STAT3 by regulating PKC, thereby promoting stem cell characteristics of colorectal cancer cells. The results of this study suggest that depletion of AKAP12 may help to suppress CSCs in patients with colorectal cancer. Nevertheless, there are some important caveats for limitations in this study. First, we focused on the colorectal cancer cell lines HCT116 and LoVo only, more experiments should be carried out in more colorectal cancer cell lines, primary colorectal cancer cells, and other solid tumor cells. Moreover, further studies in carcinomatous tissues are needed to elucidate the details of the mechanism. In addition, due to the time and funding constraints of the study, the majority of this research was done at the cellular level and remains to be explored in depth for specific mechanisms, and in vivo studies in animals need to be further refined. Our intended future work would focus on the depth mechanism and strive to improve the role of AKAP12 in colorectal cancer and related mechanisms. Regarding possible clinical applications, treatment options need to be considered in combination with the comprehensive effects of AKAP12 in other aspects of colorectal cancer.

AKAP12 helps maintain cancer stem cell-like phenotypes through the activation of STAT3 in colorectal cancer. AKAP12 promotes the precursors of PKC to synthesize PKCs. PKC modulates the phosphorylation of STAT3. This could be inhibited by PKC inhibitor enzastaurin. Upregulation of p-STAT3 increases the expression of stem cell markers like CD133, Nanog, Sox-2 and helps maintain cancer stem cell-like phenotypes
Conclusion
In conclusion, our current study demonstrates that AKAP12 promotes colorectal cancer cell stemness maintained through the activation of STAT3 in colorectal cancer. Moreover, CCSC overexpresses AKAP12, and AKAP12/PKC/STAT3 signaling pathway is an important pathway for AKAP12 to promote its stem cell characteristics, all of which indicate that AKAP12 may be an important therapeutic target for blocking the development of colorectal cancer from the perspective of cancer stem cells.
Data availability
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Abbreviations
- CSCs:
-
Cancer stem cells
- CCSCs:
-
Colorectal cancer stem cells
- AKAP12:
-
A-kinase anchor protein 12
- STAT3:
-
Signal transduction and transcription activator 3
- Lv-hAKAP12:
-
Lentiviruses overexpressing AKAP12
- Lv-Null:
-
Negative control
- Lv-siAKAP12:
-
Lentiviruses with AKAP12-siRNA
- Lv-siCont:
-
Negative control
- PKC:
-
Protein kinase C
- qPCR:
-
Quantitative polymerase chain reaction
- RT-PCR:
-
Reverse transcription PCR
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. Cancer J Clin. 2021;71(1):7–33.
Marquardt S, Solanki M, Spitschak A, Vera J, Putzer BM. Emerging functional markers for cancer stem cell-based therapies: understanding signaling networks for targeting metastasis. Semin Cancer Biol. 2018;53:90–109.
Di Franco S, Bianca P, Sardina DS, Turdo A, Gaggianesi M, Veschi V, et al. Adipose stem cell niche reprograms the colorectal cancer stem cell metastatic machinery. Nat Commun. 2021;12(1):5006.
Najafi M, Farhood B, Mortezaee K. Cancer stem cells (CSCs) in cancer progression and therapy. J Cell Physiol. 2019;234(6):8381–95.
Dianat-Moghadam H, Heidarifard M, Jahanban-Esfahlan R, Panahi Y, Hamishehkar H, Pouremamali F, et al. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J Controll Releas : Off J Controll Releas Soc. 2018;288:62–83.
Jarrett SG, Wolf Horrell EM, D’Orazio JA. Retraction: AKAP12 mediates PKA-induced phosphorylation of ATR to enhance nucleotide excision repair. Nucleic Acids Res. 2020;48(20):11814.
Wu X, Luo Y, Wang S, Li Y, Bao M, Shang Y, et al. AKAP12 ameliorates liver injury via targeting PI3K/AKT/PCSK6 pathway. Redox Biol. 2022;53: 102328.
Liu W, Guan M, Hu T, Gu X, Lu Y. Re-expression of AKAP12 inhibits progression and metastasis potential of colorectal carcinoma in vivo and in vitro. PLoS ONE. 2011;6(8): e24015.
Suren D, Yildirim M, Alikanoglu AS, Kaya V, Yildiz M, Dilli UD, et al. Lack of relation of AKAP12 with p53 and Bcl-2 in colorectal carcinoma. Asian Pac J Cancer Prev : APJCP. 2014;15(8):3415–8.
Su B, Bu Y, Engelberg D, Gelman IH. SSeCKS/Gravin/AKAP12 inhibits cancer cell invasiveness and chemotaxis by suppressing a protein kinase C- Raf/MEK/ERK pathway. J Biol Chem. 2010;285(7):4578–86.
Lee SW, Jung KH, Jeong CH, Seo JH, Yoon DK, Suh JK, et al. Inhibition of endothelial cell migration through the downregulation of MMP-9 by A-kinase anchoring protein 12. Mol Med Rep. 2011;4(1):145–9.
Yang JM, Lee HS, Seo JH, Park JH, Gelman IH, Lo EH, et al. Structural environment built by AKAP12+ colon mesenchymal cells drives M2 macrophages during inflammation recovery. Sci Rep. 2017;7:42723.
Bots M, Verbrugge I, Martin BP, Salmon JM, Ghisi M, Baker A, et al. Differentiation therapy for the treatment of t (8; 21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. 2014;123(9):1341–52.
Rasanen K, Lehtinen E, Nokelainen K, Kuopio T, Hautala L, Itkonen O, et al. Interleukin-6 increases expression of serine protease inhibitor Kazal type 1 through STAT3 in colorectal adenocarcinoma. Mol Carcinog. 2016;55(12):2010–23.
Heichler C, Scheibe K, Schmied A, Geppert CI, Schmid B, Wirtz S, et al. STAT3 activation through IL-6/IL-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut. 2020;69(7):1269–82.
Venkatesan N, Wong JF, Tan KP, Chung HH, Yau YH, Cukuroglu E, et al. EZH2 promotes neoplastic transformation through VAV interaction-dependent extranuclear mechanisms. Oncogene. 2017. https://doi.org/10.1038/onc.2017.309.
Wang W, Guo C, Zhu P, Lu J, Li W, Liu C, et al. Phosphorylation of STAT3 mediates the induction of cyclooxygenase-2 by cortisol in the human amnion at parturition. Sci Signal. 2015;8(400):ra106.
Turtoi A, Mottet D, Matheus N, Dumont B, Peixoto P, Hennequiere V, et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis. 2012;15(4):543–54.
Mostafa MR, Yahia RS, Abd El Messih HM, El-Sisy E, El Ghannam DM. Gravin gene expression in acute myeloid leukemia. Med Oncol (Northwood, London, England). 2013;30(2):548.
Peixoto P, Blomme A, Costanza B, Ronca R, Rezzola S, Palacios AP, et al. HDAC7 inhibition resets STAT3 tumorigenic activity in human glioblastoma independently of EGFR and PTEN: new opportunities for selected targeted therapies. Oncogene. 2016;35(34):4481–94.
Jo U, Whang YM, Kim HK, Kim YH. AKAP12alpha is associated with promoter methylation in lung cancer. Cancer Res Treat : Off J Korean Cancer Assoc. 2006;38(3):144–51.
Cosenza M, Civallero M, Pozzi S, Marcheselli L, Bari A, Sacchi S. The combination of bortezomib with enzastaurin or lenalidomide enhances cytotoxicity in follicular and mantle cell lymphoma cell lines. Hematol Oncol. 2015;33(4):166–75.
Shaheen S, Ahmed M, Lorenzi F, Nateri AS. Spheroid-formation (Colonosphere) assay for in vitro assessment and expansion of stem cells in colon cancer. Stem Cell Rev. 2016;12(4):492–9.
Yang Z, He L, Lin K, Zhang Y, Deng A, Liang Y, et al. The KMT1A-GATA3-STAT3 Circuit is a novel self-renewal signaling of human bladder cancer stem cells. Clin Cancer Res : Off J Am Assoc Cancer Res. 2017. https://doi.org/10.1158/1078-0432.CCR-17-0882.
Shien K, Papadimitrakopoulou VA, Ruder D, Behrens C, Shen L, Kalhor N, et al. JAK1/STAT3 Activation through a proinflammatory cytokine pathway leads to resistance to molecularly targeted therapy in non-small cell lung cancer. Mol Cancer Ther. 2017;16(10):2234–45.
Hatano Y, Fukuda S, Hisamatsu K, Hirata A, Hara A, Tomita H. Multifaceted interpretation of colon cancer stem cells. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18071446.
Wang R, Wei J, Zhang S, Wu X, Guo J, Liu M, et al. Peroxiredoxin 2 is essential for maintaining cancer stem cell-like phenotype through activation of Hedgehog signaling pathway in colon cancer. Oncotarget. 2016;7(52):86816–28.
Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci. 2019;234: 116781.
Srivastava AK, Banerjee A, Cui T, Han C, Cai S, Liu L, et al. Inhibition of miR-328-3p impairs cancer stem cell function and prevents metastasis in ovarian cancer. Can Res. 2019;79(9):2314–26.
Wang P, Wan WW, Xiong SL, Feng H, Wu N. Cancer stem-like cells can be induced through dedifferentiation under hypoxic conditions in glioma, hepatoma and lung cancer. Cell Death Discov. 2017;3:16105.
Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell. 2014;15(6):692–705.
Soheilifar MH, Moshtaghian A, Maadi H, Izadi F, Saidijam M. BMI1 Roles in cancer stem cells and its association with MicroRNAs dysregulation in cancer: emphasis on colorectal cancer. Int J Cancer Manag. 2018. https://doi.org/10.5812/ijcm.82926.
Zhu Y, Huang S, Chen S, Chen J, Wang Z, Wang Y, et al. SOX2 promotes chemoresistance, cancer stem cells properties, and epithelial-mesenchymal transition by β-catenin and Beclin1/autophagy signaling in colorectal cancer. Cell Death Dis. 2021;12(5):449.
Zhang M, Peng R, Wang H, Yang Z, Zhang H, Zhang Y, et al. Nanog mediated by FAO/ACLY signaling induces cellular dormancy in colorectal cancer cells. Cell Death Dis. 2022;13(2):159.
Yan X, Liu L, Li H, Qin H, Sun Z. Clinical significance of Fusobacterium nucleatum, epithelial-mesenchymal transition, and cancer stem cell markers in stage III/IV colorectal cancer patients. Onco Targets Ther. 2017;10:5031–46.
Liu W, Guan M, Su B, Ye C, Li J, Zhang X, et al. Quantitative assessment of AKAP12 promoter methylation in colorectal cancer using methylation-sensitive high resolution melting: correlation with Duke’s stage. Cancer Biol Ther. 2010;9(11):862–71.
Maki T, Choi YK, Miyamoto N, Shindo A, Liang AC, Ahn BJ, et al. A-kinase anchor protein 12 is required for oligodendrocyte differentiation in adult white matter. Stem Cells (Dayton, Ohio). 2018. https://doi.org/10.1002/stem.2771.
Deng Y, Gao J, Xu G, Yao Y, Sun Y, Shi Y, et al. HDAC6-dependent deacetylation of AKAP12 dictates its ubiquitination and promotes colon cancer metastasis. Cancer Lett. 2022;549: 215911.
Liu W, Kovacevic Z, Peng Z, Jin R, Wang P, Yue F, et al. The molecular effect of metastasis suppressors on Src signaling and tumorigenesis: new therapeutic targets. Oncotarget. 2015;6(34):35522–41.
Akakura S, Gelman IH. Pivotal role of AKAP12 in the regulation of cellular adhesion dynamics: control of cytoskeletal architecture, cell migration, and mitogenic signaling. J Sign Transduct. 2012;2012: 529179.
Yao C, Su L, Shan J, Zhu C, Liu L, Liu C, et al. IGF/STAT3/NANOG/Slug signaling axis simultaneously controls epithelial-mesenchymal transition and stemness maintenance in colorectal cancer. Stem Cells. 2016;34(4):820–31.
Jin W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells. 2020. https://doi.org/10.3390/cells9010217.
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, et al. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Can Res. 2011;71(23):7226–37.
Acknowledgements
All authors sincerely thank the Central Laboratory of Shanghai Tenth People's Hospital for providing experimental equipment and experimental platform for this research.
Funding
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
The research involving animals strictly abides by the ethical regulations of Longhua Hospital affiliated to Shanghai University of Traditional Chinese Medicine and has been approved by the ethics committee.
Consent for publication
Not applicable.
Informed consent statement
For this type of study, informed consent statement is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
12094_2023_3230_MOESM1_ESM.tif
Sup. Fig. 1:The proportion of cells staying in S-phase and G2M phase was increased in HCT116-hAKAP12 cells, Supplementary file1 (TIF 1007 KB)
12094_2023_3230_MOESM2_ESM.tif
Sup. Fig. 2:The proportion of cells staying in S-phase and G2M phase was increased in LoVo-hAKAP12 cells, Supplementary file2 (TIF 124 KB)
12094_2023_3230_MOESM3_ESM.tif
Sup. Fig. 3:Statistical graph of the corresponding WB results in Figure 5. A-D correspond to Figure 5B, D, F and H respectively, Supplementary file3 (TIF 201 KB)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, K., Wu, X., Li, Y. et al. AKAP12 promotes cancer stem cell-like phenotypes and activates STAT3 in colorectal cancer. Clin Transl Oncol 25, 3263–3276 (2023). https://doi.org/10.1007/s12094-023-03230-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-023-03230-5