
Abstract
The use of immune checkpoint inhibitors in solid organ malignancies has become widespread in the last decade. Accumulating evidence shows broad survival benefit as compared to traditional chemotherapies. At the same time, a need has emerged to stratify these drugs in various patient populations and histologies. Consequently, various immune biomarkers have been proposed to help in selecting patients for these therapies. Here, we review the evidence pertaining to biomarkers including programmed death-ligand 1, defective mismatch repair, tumor mutational burden, tumor-infiltrating lymphocytes, gene expression profiles, circulating blood cells, circulating DNA and the gut microbiome. The value of PD-L1 testing in certain malignancies, such as lung and urothelial cancer is highlighted as well as emerging data from trials such as GARNET and CheckMate142.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It is by now common knowledge among the medical community that immune checkpoint inhibitors (ICI) represent a sea change in the treatment of solid organ malignancies. Survival benefit from such therapies has been seen across a wide variety of histologies and patient populations [1]. However, only about twenty percent of tumors achieve a response [2]. Moreover, immune-related adverse events (irAE) can have ubiquitous organ involvement, prolonged courses and in some cases are even life threatening [3]. Thus, there remains a need for validated biomarkers. Doing so will allow for the identification of patients with the most potential for benefit. Conversely, biomarkers can be used to spare those who are unlikely to experience benefit or to better define subpopulations that are predisposed to toxicity. What’s more, biomarkers can lead to trial enrichment strategies and minimize cost burden to patients and payers. The goal of this article is, therefore, to review the available biomarkers pertinent to ICI and evaluate their clinical utility in the prediction of anti-tumoral response. For more information related to biomarker signals in other types of immuno-modulating agents, the SITC cancer immunotherapy resource document is recommended [4].
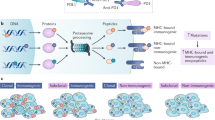
Immune checkpoint inhibitors work by augmenting the tumor microenvironment (TME). Under ideal circumstances, tumor-infiltrating lymphocytes (TIL) are recruited to the site of malignancy, recognize tumor cells (TC) and then destroy them through the release of cytolytic granules [5]. To summarize this recognition process, oncogenic mutations give rise to neoantigens, which are then captured by dendritic cells or other antigen presenting cells (APC), whereupon these proteins are displayed on Major Histocompatibility Complex (MHC). Cytotoxic T-cells, characterized by their expression of the CD8 receptor, then read these neoantigens using the T-cell Receptor (TCR), but this priming is heavily regulated by multiple costimulatory and inhibitory receptors. The receptors of most relevance to ICI being the Programmed Death Ligand (PD-L1) and Cytotoxic T Lymphocyte protein 4 (CTLA-4). PD-L1 is a transmembrane immunoglobulin expressed on TC, although not exclusively, and inhibits lymphocytes via its binding with PD-1 present on the surface of these T-cells. Antibody blockade of either the PD-1 or PD-L1 results in increased lymphocyte activation and proliferation [6, 7]. Similarly, T-cells can express CTLA-4, alongside the costimulatory molecule CD28, and both can bind CD80/86 expressed on APC. Dominance of CTLA-4-binding results in the suppression of cytotoxic T-cells. Vice versa, blockade by anti-CTLA-4 drugs allows these TILs to destroy the tumor. Currently, there are 7 FDA approved ICI. These include monoclonal antibodies targeting PD-1: nivolumab, pembrolizumab, cemiplimab and dostarlimab. Additionally, there are those ICI targeting PD-L1: atezolizumab, avelumab and durvalumab. The CTLA-4 inhibitor, ipilimumab, is also FDA approved.
Programmed death ligand
It was noted that in early clinical trials of ICI the duration of response (DOR) often exceeded overall survival (OS) thus suggesting that there was a subpopulation of individuals more likely to benefit from treatment [8]. PD-L1 expression, either by the tumor itself or the corresponding immune cell milieu, was seen as a potential biomarker. As such, companion immunohistochemical (IHC) tests were developed with many immune checkpoint agents, the details of which are summarized below (Table 1). The Blueprint Study was a project comparing these 22C3, 28–8, SP142 and SP263 assays [9]. This demonstrated that PD-L1 staining for TC was comparable between tests when the 22C3, 28–8 and SP263 assays were utilized, however, when SP142 was employed, fewer stained TC were observed. All the IHC assays demonstrated greater variability with IC staining than with TC. It is, therefore, recommended that pathologists be trained in the proper interpretations of these tests and that clinicians be aware of each IHC test as it relates to prognostic and predictive evidence in disease-specific clinical trials. Another industry-sponsored effort also found the SP142 test to be the outlier with significantly less PD-L1 detection in both TC and IC as compared with 22C3 or 28–8 [10]. Once more, pathology concordance was stronger for TC scoring than it was for IC.
A summary of predictive biomarker signals for PD-L1 expression in solid tumor malignancies from clinical trials leading to FDA approval of ICI is provided below (Supplemental Table 1). Melanoma was the first malignancy whereby PD-(L)1 blockade was shown to improve clinical outcomes. Interestingly, across a broad spectrum of melanoma trials, PD-L1 expression has not proven to reliably differentiate ICI responders from non-responders. The lone exception to this is Keynote-006 where PD-L1 ≥ 1% did demonstrate superior OS for pembrolizumab treatment over ipilimumab. Non-small cell lung cancer (NSCLC) has the preponderance of both clinical trials and FDA approved indications for ICI. Initial studies (Keynote-024, Keynote-042) had seemed to suggest some predictive value for PD-L1, and the FDA approved indications adhere to these. However, the majority of subsequent studies have shown no benefit for PD-L1 testing. As for NSCLC histology, non-squamous may have more use for PD-L1 testing than squamous, if such a benefit does exist, as Keynote-189 suggested progression-free survival (PFS) benefit from PD-L1 ≥ 1% while Keynote-407 did not. Furthermore, in early-stage NSCLC, there may be disease-free survival (DFS) benefit but this is reserved to higher PD-L1 expression levels as seen with TC expression ≥ 50% in IMpower010 and ≥ 50% in CheckMate 189. To date, OS benefit has not been shown in early-stage NSCLC treated with immunotherapy. Conflicting results have been observed for Head and neck squamous cell carcinoma (HNSCC) with CheckMate 141 showing overall survival (OS) benefit from PD-L1 ≥ 1%, while Keynote-048 showed none. For urothelial carcinoma (UC), most studies have demonstrated predictive value for either TC or IC expression of PD-L1, although this benefit has ranged from only DFS all the way up to OS superiority. The trend appears to be that PD-L1 is less useful in the early muscle-invasive stage, as compared with unresectable and metastatic disease. Keynote-826 suggests that cervical cancer (CC) with PD-L1 ≥ 1% have improved survival with pembrolizumab. In renal cell cancer (RCC), variable results have been observed with some studies showing that PD-L1 is predictive, including in the post-nephrectomy setting (Keynote-564), whilst other studies have offered evidence to the contrary. No obvious differences are seen between those studies excluding non-clear-cell histology and those that do not. CheckMate 040 suggests that PD-L1 testing is not useful for hepatocellular carcinoma (HCC). Most studies in esophageal, gastroesophageal (GEJ) and gastric cancer have shown that high combined positive scores (CPS) predict improved OS from ICI therapy, but in Keynote-181 and Keynote-590 this finding was only statistically significant with expression greater than 10%. Results in triple-negative breast cancer (TNBC) have consistently identified high PD-L1 expressors as having benefit but this has ranged from pathological complete response (pCR) (Keynote-522) to OS advantage (IMpassion 130). Most recently, CheckMate 743 has shown that PD-L1 ≥ 1% predicts OS benefit from nivolumab plus ipilumumab in pleural mesothelioma.
A meta-analysis encompassing 6664 cancer patients across 41 ICI trials found that PD-L1 was predictive of favorable overall response rate (ORR) (OR 2.26, p < 0.001) [11]. These results include statistically significant benefit in NSCLC, UC, RCC, gastroesophageal, HNSCC and even melanoma, although the high proportion of PD-L1-negative patients with melanoma responding to treatment is duly noted. Other meta-analysis have largely affirmed these findings with the exception of breast cancer specific meta-analysis which have had conflicting results on the benefit of PD-L1 [12, 13].
These findings, while encouraging, indicate that detection of PD-L1 expression is not always associated with response and even patients with no PD-L1 detected on IHC have been found to achieve durable responses from ICI. Several reasons are postulated for the poor reliability of PD-L1 IHC as a biomarker for anti-PD-1 or anti-PD-L1 therapies. As an example, in breast cancer, the heterogeneity between hormone status, histological subtypes and metastasis make it difficult to assign a holistic PD-L1 status [14]. Discordant PD-L1 expression is also seen between primary and metastatic sites in melanoma and NSCLC [15, 16]. Finally, careful consideration must be made with regards to sampling time points and interventions as chemotherapy and radiation can increase both PD-L1 expression and TIL density [17].
Microsatellite instability and defective mismatch repair
Microsatellites are short nucleotide sequences with repeating motifs. These sequences usually range from 1 to 6 base pairs in unit length with anywhere from 10 to 60 repetitions per sequence. Together, they account for 3% of the entire genome [18]. Microsatellites by their very nature are prone to slippage and mispairing events [19]. Microsatellite instability (MSI) occurs when there are gains or losses to one or more of such repeats but researchers differ on the exact number of tandem repeats that constitute a microsatellite. Related to this concept is the mismatch repair (MMR) system, a conglomerate of DNA repair mechanisms that is responsible for correcting such errors. The key proteins involved in MMR are the gene products of MLH1, MSH2, PMS2, and MSH6 [20]. A defect in any one of these genes results in a defective mismatch repair (dMMR) mechanism, which in turn results in high microsatellite instability (MSI-H). The MSI-H phenotype has been associated with carcinogenicity, most notably that pertaining to Lynch Syndrome [21]. Evidence of MSI-H has been shown in several cancers including gastric, adrenocortical, CC, endometrial, CRC, mesothelioma, esophageal, breast, RCC and cholangiocarcinoma [22]. Furthermore, MSI-H status correlates with mutational burden, TIL presence and the expression of inhibitory immune checkpoint markers [23,24,25]. Because of this, it was theorized that these patients would be more likely to respond to PD-(L)1 blockade.
As previously stated, MSI status can be inferred by IHC staining for dMMR gene loss, providing a quick and relatively inexpensive means of assessing this. The sensitivity of IHC for MSI is more than 90% when all four dMMR proteins are tested together, but immunostaining is not without its own perils [26]. Missense mutations have been identified in these genes that can result in altered proteins that are non-functional but still recognizable by antibody staining [27]. This circumstance may then result in false negatives. Additionally, extensive loss of MSH6 immunoexpression is common in CRC following neoadjuvant chemoradiation [28]. MSI/dMMR status can also be detected by Polymerase Chain Reaction (PCR). The Bethesda panel which included 2 mononucleotide repeats and 3 dinucleotide repeats was initially proposed for the uniform analysis of MSI status [29]. Later, a Pentaplex panel consisting of 5 mononucleotide repeats was developed which did not require germline testing [30]. In a NCI workshop, this Pentaplex panel was recommended for the evaluation of MSI-H [31]. Alternatively, Next Generation Sequencing (NGS) affords a highly accurate and increasingly available way of evaluating for MSI/dMMR but false negatives can still occur due to tumor DNA dilution [32]. Currently, utilizing any of the methods described above to determine the MSI status of tumors is appropriate.
In a phase 2 study evaluating pembrolizumab in 41 patients with metastatic carcinoma, superior PFS was observed in CRC tumors that were dMMR (78% at 20 weeks vs 11% with proficient MMR) [33]. In that same trial, no prognostic value for dMMR was observed with non-CRC tumors. A supplemental biologics licensing application was put forth containing data from 149 patients with MSI-H/dMMR cancers treated on 5 Keynote clinical trials (012, 028, 016, 158 and 164) [34]. While the majority of these patients were CRC, there were 59 patients that were not, these representing 14 other solid cancers. The responses from this heavily pretreated population was favorable (ORR 39%) with 78% having a DOR ≥ 6 months. Consequently, site-agnostic FDA approval was granted to pembrolizumab for the treatment of adult and pediatric patients with unresectable or metastatic solid tumors that are MSI-H or dMMR and who have progressed following prior treatment with no satisfactory alternative options. For colorectal malignancy specifically, the FDA granted further approval, this time in the frontline setting, after Keynote 177 showed that in 307 patients with metastatic CRC and MSI-H/dMMR status, treatment with pembrolizumab improved PFS [35]. Recent non-randomized data from cohort D of KEYNOTE-158 led to further approval of pembrolizumab in advanced endometrial carcinoma with MSI-H/dMMR. Likewise, Checkmate 142, a phase 2 study of nivolumab in patients with recurrent or metastatic CRC harboring MSI-H/dMMR, confirmed similar improvements in disease control and response [36]. This in turn led to the approval of nivolumab on a site agonist basis. An update from CheckMate 142 showed further benefit with the addition of ipilimumab, which resulted in expanded FDA approval in 2020 for MSI-H/dMMR metastatic CRC that had progressed on traditional chemotherapy [37]. More recently, results from the GARNET study, a phase 1 trial of dMMR or polymerase ὲ (POLE) mutant solid tumors, demonstrated a favorable ORR (38%) from treatment with dostarlimab [38]. From this, dostarlimab received FDA approval in August 2021 for the treatment of recurrent or advanced solid tumors harboring dMMR and having progressed on prior treatment with no satisfactory alternative options.
Tumor mutational burden
Tumor mutational burden (TMB) is the number of somatic mutations in a genomic region, but the precise calculation for this varies with both the sequence location, as well as the mutation’s characteristics, for instance if non-coding or silenced genes are included therein [39]. Preclinical studies have demonstrated that the strong immunogenicity of early tumors is a function of neoantigen expression [40]. More so, immunoediting gives rise to a population of tumors without an easily recognizable antigenic repertoire [41]. To that point, TMB by itself is of negative prognostic value [42]. As expected, higher TMB correlates with CD8-mediated cytotoxicity and these findings suggest that TC with an elevated TMB will be more likely to be impacted by lymphocyte-modulating therapy [43]. It is hypothesized that two factors determine a neoantigens ability to induce tumoral response from checkpoint inhibitors. First, the likelihood of presentation on MHC and second, recognition by the lymphocytes themselves [44]. Exome analysis from melanoma patients treated with CTLA-4 blockade has affirmed that TMB correlates with survival [45]. These missense mutations were found, using in silico translation, to bind MHC1 and, in vitro, to elicit a polyfunctional T-cell response. However, it is important to remember that not all tumors, nor epitopes, are created equally. For instance, in pancreatic cancer, it appears that neoantigen homology to peptides derived from certain infectious diseases may play a greater role than just the frequency of neoantigens alone [46].
TMB can be measured from tumor-derived tissue using NGS and involves testing of either specific gene panels or whole exome sequencing (WES). Originally considered the gold standard, WES has many limitations, including the specialized platforms, expertise, turnaround time and the need for germline comparisons [47]. Disparity between WES data from different vendors has been attributed to tumor heterogeneity, proprietary mutation cutoffs and sequencing artifacts from formalin fixation but pre-analytic microdissection can ameliorate this to some extent [48]. For these reasons and more, targeted gene panels were developed which differ from WES in terms of input requirements, covered region and bioinformatic algorithms. Because of these, variation in TMB can arise but tends to be low with one study showing only 9% of mutations being missed with comprehensive testing [49]. Unfortunately, panel TMB can overestimate, particularly at higher values and standardization between these two modalities is needed [50].
In addition to tissue sampling (tTMB), tumor mutational burden can also be established from peripheral blood (bTMB). This method of analyzing tumor DNA from serum is less invasive and expensive than tissue biopsy. Although not specific to TMB, cohort NGS studies have shown a 98% concordance between blood and tissue sampling in the identification of alterations [51]. A study evaluating data from two NSCLC studies, POPLAR and OAK, found clinically significant predictive value from bTMB [52]. For the phase 2 POPLAR study, OS benefit was observed for bTMB using a cutoff of \(\ge \) 16 mutations per megabase (mut/Mb), however, a \(\ge \) 20 mut/Mb cutoff for OS was statistically significant. Data from the subsequent phase 3, however, showed OS benefit with all cutoffs. Furthermore, bTMB and PD-L1 expression as biomarkers had little overlap, suggesting that these represent mostly divergent subgroups receiving benefit from immunotherapy.
Indeed, in a retrospective cohort analysis of 1662 cancer patients analyzed by NGS (MSK-IMPACT), the majority of whom had metastatic disease and all of whom had received prior ICI, higher TMB (TMB-H) was associated with improved OS (HR 0.52, p = 1.6 × 10–6) [53]. This was true across a broad distribution of histologies, the association being strongest in NSCLC, HNSCC, CRC, UC. However, it is also important to note that the predictive value of TMB was greatest when taken as a top percentage cutoff for each histology individually. As such, there does not appear to be a universal cutoff value for TMB but rather a disease-specific range at which this biomarker is most predictive. More recent cohort evidence has supported this hypothesis [54]. It is also worth noting that glioma failed to show even a trend toward significance with this approach. This result may portend a lack of efficacy on the part of immunotherapy, reflect the lower incidence of dMMR, or showcase the deleterious effects of hypermutation caused by alkylating therapies. While on the subject of tumor heterozygosity with respect to TMB, it is worth mentioning that in gastrointestinal cancers TMB and MSI appear to coincide, as opposed to melanoma, skin SCC and lung cancer where they do not [55].
In June 2020, the FDA granted accelerated to pembrolizumab for treatment-refractory cancers with a high TMB, defined as greater than 10 mut/Mb. Companion diagnostic approval for FoundationOne CDx was also granted for this purpose. This approval was based on Keynote-158, a phase 2 study of 1073 patients with advanced solid tumors and progression on prior therapy [56]. A prespecified cutoff of \(\ge \) 10 mut/Mb was used to define TMB-H. Amongst the 13% of patients with TMB-H, treatment with pembrolizumab resulted in an ORR of 29%. For comparison, the non-TMB-H group had an ORR of only 6%. There have been other signals of predictive value for TMB with most benefit observed in melanoma, NSCLC and UC [57]. CheckMate 227 was a phase 3 trial of 2876 patients with stage IV or recurrent NSCLC who were prospectively treated with nivolumab plus ipilimumab [58]. Of the 44% of patients with TMB-H tumors (\(\ge \) 10mut/Mb), ICI therapy resulted in a modest, but statistically significant, advantage in PFS and overall survival readouts from this study are much anticipated [59]. Similarly, in MYSTIC, a phase 3 of 1118 patients with advanced NSCLC treated with durvalumab plus tremelimumab, TMB with a cutoff of \(\ge \) 20 mut/Mb was found to be most predictive of clinical benefit, this finding made even more interesting by the fact that bTMB was superior to tTMB [60, 61]. However, the predictive value of TMB, and its recent FDA approval, is not without controversy. An analysis of 137 patients with advanced CRC treated with checkpoint inhibitors shows that any apparent benefit from TMB-H is abrogated once patients are stratified by MSI or POLE [62]. Many prospective trials, such as IM power 110, have shown no advantage from TMB and a TCGA analysis from over 10,000 patient tumors failed to associate this biomarker with immunotherapy outcomes [63]. Finally, it is important to consider if a high TMB reflects early or late branching events during the course of oncogenesis as clonal heterozygosity may contribute to resistance, the immuno-resistance of this event overcoming any positive effects from more robust neoantigen presentation [64].
Tumor infiltrating lymphocytes
As mentioned, TILS play a critical role in the recognition and suppression of tumors by the immune system. Their presence in the TME heralds a better prognosis across a wide variety of tumors such as melanoma, NSCLC and TNBC but negative associations have been observed in CRC [65,66,67,68]. Accordingly, the International Immuno-Oncology Biomarker Working Group (IIOBWG) has proposed standardized methods of TIL quantification by IHC, differentiating between stromal and tumor compartments as well as the 1 mm invasive margin that is frequently seen on histology, and excluding necrosis. These IIOBWG guidelines are specific to malignancy type, accounting for unique differences in each microenvironment [69].
In addition to their prognostic value, TILs have also been shown to predict benefit from immunotherapy. In a meta-analysis of 14,395 patients with NSCLC who were treated with immunotherapy including ICI, tumor vaccines and cellular therapy, CD8 TIL scores were found to improve the combined predictive utility of PD-L1 and TMB [70]. The optimal time for sampling of these TILS, however, remains an open question. Analysis from patients with metastatic melanoma has shown that those who respond to ICI are more likely to have preexisting CD8 TILs involving the invasive margin [71]. These TILS displayed proximity between PD-1 and PD-L1 expressing cells and had a more clonal, meaning less diverse, TCR repertoire. In contrast, other cohort studies have suggested that early on-treatment samples are more predictive of future immune response than preexisting TILS [72].
Circulating blood cells
White blood cell (WBC) differentials are routinely used in clinical practice and offer the potential of being a non-invasive and rapid predictor for benefit from immune checkpoint modulation. Indeed, it could be argued that circulating blood cells represent the first biomarker used in oncology [73]. Staging criteria, such as the Rai stage in chronic lymphocytic leukemia (CLL), rely on lymphocytosis as do prognostic indicators such as the International Prognostic Score for non-Hodgkin’s lymphoma. Circulating blood cells also have predictive value in hematological malignancies. For instance, high absolute lymphocyte counts (≥ 25 × 109/L) are at high risk for tumor lysis syndrome in CLL patients treated with venetoclax [74].
More recently, the utility of circulating blood cells in predicting response in solid tumors has been examined. In a study of 616 patients with melanoma treated with pembrolizumab, elevated baseline counts in eosinophils (≥ 1.5%) and lymphocytes (≥ 17.5%) were found to be independent predictors of favorable survival [75]. Similar findings have been observed with anti-CTLA-4 [76]. Immunoprofiling with flow cytometry further suggests that baseline peripheral CD8 counts play more of a role after treatment with CTLA-4 blockade than after PD-1 blockade [77]. Lending credence to this is a meta-analysis of 4647 patients with advanced stage cancer, where elevated pretreatment neutrophil to leukocyte ratio (NLR), as measured by peripheral blood, was associated with inferior OS after treatment with ICI (HR 2.16, p < 0.001) [78]. Subgroup analysis shows that NLR predicts worse survival in melanoma, NSCLC, RCC, sarcoma, UC, HNSCC, CRC, hepatobiliary, esophageal and mesothelioma [79]. Lastly, erythrocyte sedimentation rate (ESR) has also been associated with improved OS after immune checkpoint modulation [80].
Lactate dehydrogenase
Lactate dehydrogenase (LDH) catalyzes the conversion of lactate to pyruvate and serves as a general marker for tissue damage. Regarding LDH as assessed by peripheral blood, some studies have shown that elevations are associated with favorable OS after ICI therapy whilst others have shown correlations in the opposite direction [75, 80, 81]. Worth mentioning is the fact that neither CheckMate 067 nor CheckMate 069 showed that baseline LDH exclusively predicted OS benefit from ICI in melanoma. Validation of this enzyme as a biomarker will require larger prospective studies in the future with attention to whether it is baseline values or trends during treatment that are most predictive [82].
TCR sequencing
In a retrospective pilot study of 12 patients with metastatic melanoma treated with anti-CTLA-4, TCR sequencing was performed on peripheral blood at baseline [83]. TCR diversity was graded based on ‘richness’, defined as the ratio between observed V-J rearrangements divided by theoretical, as well as ‘evenness’, reflecting the similarity between the frequencies of specific V-J rearrangements that were observed. Both high richness and high evenness were predictive of clinical benefit.
Gene expression profiles
Recently, a variety of gene expression profiles (GEP) relating to the TME have been identified as predictive for response to ICI. Early work focused on immune gene signatures inducible by interferon gamma (IFN- γ), including MHC-II. In two melanoma cohorts, MHC-II expression on TC was associated with improved response and OS after PD-1 blockade [84]. A T-cell inflamed GEP consisting of 18 genes was developed and validated with data from a 200-patient cohort accounting for 9 different malignancies to correlate with benefit from pembrolizumab [85]. Receiver operating characteristics (ROC) measure the ability for a test to differentiate between two groups, such as responders and non-responders, with area under curve (AUC) values of ≥ 0.8 being suggestive of most benefit. This T-cell inflamed GEP was comparable to PD-L1 IHC (AUC 0.75 vs 0.65, respectively). Another 8-gene panel defining effector T-cells (Teff) was developed for use with atezolizumab. Unfortunately, in the phase 3 IMpower150, Teff signatures were not found to be superior to PD-L1 expression at identifying NSCLC beneficiaries [86]. Along the same vein as this a companion GEP for durvalumab, consisting of IFN- γ, CD274, LAG3 and CXCL9 is currently under development [87]. Cross-modality meta-analysis shows that GEP is of greatest value when combined with other biomarkers. In comparison of 8135 patients representing 10 solid malignancies, multiplex IHC combined with immunofluorescence (IF) was found to have a superior AUC (0.79) to PD-L1 IHC alone (AUC 0.65) [88]. However, combining PD-L1 expression with GEP resulted in predictive value (AUC 0.74) which rivaled even that of multiplex IHC/IF.
Circulating tumor DNA
Circulating tumor DNA (ctDNA), not to be confused with cell-free DNA (cfDNA), has been proposed as a biomarker not only for initial response to ICI but also longitudinally for molecular relapse. An exploratory analysis from IMvigor010 found that ctDNA predicted OS benefit from atezolizumab versus observation in UC (HR 0.59, p = 0.0024) [89]. Transcriptomic analysis from ctDNA-positive patients benefiting from immunotherapy highlighted immune response signatures and basal-squamous genes. Analogous signals have been seen for OS in melanoma and pCR after neoadjuvant ICI in breast cancer [90] [91]. In prospective analysis of 94 patients with advanced solid tumors, ctDNA at baseline was correlated with OS benefit from pembrolizumab, and this association only became stronger once ctDNA kinetics were taken into account [92]. Among the 12 patients with ctDNA clearance, all were alive at 25 month follow-up.
Mature tertiary lymphoid
Much interest has also arisen in tertiary lymphoid structures (TLS) as predictive biomarkers for immune checkpoint blockade. These structures have a T-cell zones populated by mature APC and fibroblastic reticular cells. Interspersed within these are B-cell zones with germinal centers that house memory B cells and plasma cells [93]. In melanoma patients receiving neoadjuvant ICI, both the presence of TLS and its ratio to tumor area, as assessed by histology, correlates with response in early on-treatment specimens. However, these same findings were not significant for preexisting samples, this suggesting that biopsy after treatment initiation is of highest predictive value [94]. In line with this, gene signatures derived from TLS and taken from pretreatment samples are also associated with improved OS in patients treated with immune checkpoint blockade [95]. Another study has identified TLS gene signatures as predicative for response to PD-1 blockade in sarcoma populations [96].
Gut Microbiome
The microbiota has long been suspected to play a role in oncogenesis but recent evidence in the era of immunotherapy paints a much clearer picture of these interactions [97]. Preclinical studies in mice originally suggested an association between the gut microbiome and a host’s response to immunotherapy. In one such study using two syngeneic models of melanoma (JAX or TAC), differences in tumor groups were shown to be due to commensal microbiota and extinguishable both by cohousing as well as through unidirectional (JAX to TAC) fecal material transfer (FMT) [98]. Combination FMT with ICI was found to be synergistic with increased tumor control and IFNγ production. Using16s ribosomal RNA sequencing, Bifidobacterium was identified as the most likely causative species and these previous results could then be recapitulated with oral Bifidobacterim plus checkpoint blockade. Further study confirmed that antibiotic therapy impairs both anti-PD-1 and anti-CTLA-4 therapy in mouse models of sarcoma and melanoma [99].
Clinical studies have corroborated the role of the gut microbiome with respect to immunotherapy in humans. Decreased survival after immunotherapy has been observed with antibiotic use in patients with NSCLC, RCC and UC even after multivariate adjustment [99]. Akkermansia muciniphilia, a prodigious member of the ileum microbiota, was found to be most enriched in responders. In 26 patients with metastatic melanoma prospectively treated with ipilimumab, fecal microbiota composition was assessed using 16S rRNA sequencing at baseline and before administration. Patients with Bacteroides predominant microbiota had improved OS compared to those with Firmicutes such as Faecalibacterium [100]. In another study of 112 patients with metastatic melanoma undergoing PD-1 blockade, 16S sequencing revealed Clostridiales, most notably Ruminococcaceae, and Faecalibacterium as enriched in responders [101]. Contrarily, Bacteroides was preeminent in non-responders. Again, fecal microbiome transplantation improved ICI responses in mice with a statistically significant abundance of Faecalibacterium seen in responding animals.
Conclusion
The age of immunotherapy comes with much promise but also several challenges, one of the greatest being identifying which patients are most likely to benefit from ICI and which can be spared potential toxicity. Accomplishing this will require biomarkers that are well validated, easily implemented and ideally capable of longitudinal monitoring to assess for recurrence. Several potential biomarkers predicting benefit from immune checkpoint blockade have been described. To date, no individual biomarker has been proven “best”. Yet, there is much that has been discovered. PD-L1, expression appears to be of much greater value in NSCLC, UC than in melanoma. MSI/dMMR appears to be gaining traction with trials such as GARNET and CheckMate 142 showcasing these alterations as a potential enrichment strategy. As for TMB, this remains a controversial biomarker despite recent accelerated approval by the FDA, in part because the perceived benefit may actually be due to confounding variables like MSI. TILS appear quintessential for NSCLC but several questions, such as optimal time for sampling, and challenges, specifically the standardization of IHC methods, remain. Circulating blood cells, peripheral TCR sequencing and ctDNA offer a cheap, non-invasive biomarkers whose predictive value may rival tissue-based assessment. Unfortunately, the data on LDH are too conflicted to offer much current value. GEP hold great potential, particularly when combined with other biomarkers such as PD-L1 expression. Finally, an improved understanding of the gut microbiome will likely yield insights into which patients are potential responders, but Faecalibacterium seems an ideal candidate at this time.
References
Yang F, Markovic SN, Molina JR, Halfdanarson TR, Pagliaro LC, Chintakuntlawar AV, et al. Association of Sex, Age, and Eastern Cooperative Oncology Group Performance Status With Survival Benefit of Cancer Immunotherapy in Randomized Clinical Trials: A Systematic Review and Meta-analysis. JAMA Network Open. 2020;3(8):e2012534-e.
Carretero-González A, Lora D, Ghanem I, Zugazagoitia J, Castellano D, Sepúlveda JM, et al. Analysis of response rate with ANTI PD1/PD-L1 monoclonal antibodies in advanced solid tumors: a meta-analysis of randomized clinical trials. Oncotarget. 2018;9(9):8706–15.
Wang Y, Zhou S, Yang F, Qi X, Wang X, Guan X, et al. Treatment-Related Adverse Events of PD-1 and PD-L1 Inhibitors in Clinical Trials: A Systematic Review and Meta-analysis. JAMA Oncol. 2019;5(7):1008–19.
Hu-Lieskovan S, Bhaumik S, Dhodapkar K, Grivel J-CJB, Gupta S, Hanks BA, et al. SITC cancer immunotherapy resource document: a compass in the land of biomarker discovery. Journal for immunotherapy of cancer. 2020;8(2):e000705.
Belli C, Trapani D, Viale G, D’Amico P, Duso BA, Della Vigna P, et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev. 2018;65:22–32.
Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7–H1 and PD-1 by Monoclonal Antibodies Potentiates Cancer Therapeutic Immunity. Can Res. 2005;65(3):1089.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7–H1 improves myeloid dendritic cell–mediated antitumor immunity. Nat Med. 2003;9(5):562–7.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–39.
Tsao MS, Kerr KM, Kockx M, Beasley M-B, Borczuk AC, Botling J, et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2018;13(9):1302–11.
Rimm DL, Han G, Taube JM, Yi ES, Bridge JA, Flieder DB, et al. A Prospective, Multi-institutional, Pathologist-Based Assessment of 4 Immunohistochemistry Assays for PD-L1 Expression in Non-Small Cell Lung Cancer. JAMA Oncol. 2017;3(8):1051–8.
Khunger M, Hernandez AV, Pasupuleti V, Rakshit S, Pennell NA, Stevenson J, et al. Programmed Cell Death 1 (PD-1) Ligand (PD-L1) Expression in Solid Tumors As a Predictive Biomarker of Benefit From PD-1/PD-L1 Axis Inhibitors: A Systematic Review and Meta-Analysis. JCO Precis Oncol. 2017;1:1–15.
Zhang M, Sun H, Zhao S, Wang Y, Pu H, Wang Y, et al. Expression of PD-L1 and prognosis in breast cancer: a meta-analysis. Oncotarget. 2017;8(19):31347–54.
Lotfinejad P, Asghari Jafarabadi M, Abdoli Shadbad M, Kazemi T, Pashazadeh F, Sandoghchian Shotorbani S, et al. Prognostic Role and Clinical Significance of Tumor-Infiltrating Lymphocyte (TIL) and Programmed Death Ligand 1 (PD-L1) Expression in Triple-Negative Breast Cancer (TNBC): A Systematic Review and Meta-Analysis Study. Diagnostics (Basel, Switzerland). 2020;10(9):704.
Dill EA, Gru AA, Atkins KA, Friedman LA, Moore ME, Bullock TN, et al. PD-L1 Expression and Intratumoral Heterogeneity Across Breast Cancer Subtypes and Stages: An Assessment of 245 Primary and 40 Metastatic Tumors. The American Journal of Surgical Pathology. 2017;41(3).
Madore J, Vilain RE, Menzies AM, Kakavand H, Wilmott JS, Hyman J, et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015;28(3):245–53.
Ilie M, Long-Mira E, Bence C, Butori C, Lassalle S, Bouhlel L, et al. Comparative study of the PD-L1 status between surgically resected specimens and matched biopsies of NSCLC patients reveal major discordances. A potential issue for anti-PD-L1 therapeutic strategies. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2015;27.
Lim YJ, Koh J, Kim S, Jeon S-R, Chie EK, Kim K, et al. Chemoradiation-Induced Alteration of Programmed Death-Ligand 1 and CD8<sup>+</sup> Tumor-Infiltrating Lymphocytes Identified Patients With Poor Prognosis in Rectal Cancer: A Matched Comparison Analysis. Int J Radiat Oncol Biol Phys. 2017;99(5):1216–24.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Schlötterer C, Tautz D. Slippage synthesis of simple sequence DNA. Nucleic Acids Res. 1992;20(2):211–5.
Genschel J, Littman SJ, Drummond JT, Modrich P. Isolation of MutSβ from Human Cells and Comparison of the Mismatch Repair Specificities of MutSβ and MutSα *. J Biol Chem. 1998;273(31):19895–901.
Eshleman JR, Markowitz SD. Mismatch repair defects in human carcinogenesis. Human Molecular Genetics. 1996;5(Supplement_1):1489–94.
Bonneville R, Krook M, Kautto E, Miya J, Wing M, Chen H-Z, et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis Oncol. 2017;2017:1–15.
Lin EI, Tseng L-H, Gocke CD, Reil S, Le DT, Azad NS, et al. Mutational profiling of colorectal cancers with microsatellite instability. Oncotarget. 2015;6(39):42334–44.
Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001;91(12):2417–22.
Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015;5(1):43–51.
Engel C, Forberg J, Holinski-Feder E, Pagenstecher C, Plaschke J, Kloor M, et al. Novel strategy for optimal sequential application of clinical criteria, immunohistochemistry and microsatellite analysis in the diagnosis of hereditary nonpolyposis colorectal cancer. Int J Cancer. 2006;118(1):115–22.
Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition – Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20(4–5):269–76.
Bao F, Panarelli NC, Rennert H, Sherr DL, Yantiss RK. Neoadjuvant Therapy Induces Loss of MSH6 Expression in Colorectal Carcinoma. The American Journal of Surgical Pathology. 2010;34(12).
Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Can Res. 1998;58(22):5248.
Suraweera N, Duval A, Reperant M, Vaury C, Furlan D, Leroy K, et al. Evaluation of tumor microsatellite instability using five quasimonomorphic mononucleotide repeats and pentaplex PCR. Gastroenterology. 2002;123(6):1804–11.
Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96(4):261–8.
Stadler ZK, Battaglin F, Middha S, Hechtman JF, Tran C, Cercek A, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. 2016;34(18):2141–7.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res. 2019;25(13):3753.
André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability–high advanced colorectal cancer. N Engl J Med. 2020;383(23):2207–18.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
Lenz H-J, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: the phase II checkmate 142 study. J Clin Oncol. 2021. https://doi.org/10.1200/JCO.21.01015.
Andre T, Berton D, Curigliano G, Ellard S, Trigo Pérez JM, Arkenau H-T, et al. Safety and efficacy of anti–PD-1 antibody dostarlimab in patients (pts) with mismatch repair-deficient (dMMR) solid cancers: results from GARNET study. J Clin Oncol. 2021;39(3 suppl):9.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69.
Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Can Res. 2008;68(3):889.
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–4.
Valero C, Lee M, Hoen D, Wang J, Nadeem Z, Patel N, et al. The association between tumor mutational burden and prognosis is dependent on treatment context. Nat Genet. 2021;53(1):11–5.
McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science (New York, NY). 2016;351(6280):1463–9.
Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551(7681):517–20.
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189–99.
Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–6.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Qiu P, Pang L, Arreaza G, Maguire M, Chang KCN, Marton MJ, et al. Data interoperability of whole exome sequencing (WES) based mutational burden estimates from different laboratories. Int J Mol Sci. 2016;17(5):651.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–13.
Merino DM, McShane LM, Fabrizio D, Funari V, Chen S-J, White JR, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the friends of cancer research TMB harmonization project. J Immunother Cancer. 2020;8(1): e000147.
Müller JN, Falk M, Talwar J, Neemann N, Mariotti E, Bertrand M, et al. Concordance between comprehensive cancer genome profiling in plasma and tumor specimens. J Thorac Oncol. 2017;12(10):1503–11.
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. 2018;24(9):1441–8.
Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–6.
Valero C, Lee M, Hoen D, Zehir A, Berger MF, Seshan VE, et al. Response rates to anti-PD-1 immunotherapy in microsatellite-stable solid tumors with 10 or more mutations per megabase. JAMA Oncol. 2021;7(5):739–43.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–65.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44–56.
Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim S-W, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med. 2019;381(21):2020–31.
Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–104.
Rizvi NA, Cho BC, Reinmuth N, Lee KH, Luft A, Ahn M-J, et al. Durvalumab with or without tremelimumab vs standard chemotherapy in first-line treatment of metastatic non-small cell lung cancer: the MYSTIC phase 3 randomized clinical trial. JAMA Oncol. 2020;6(5):661–74.
Si H, Kuziora M, Quinn KJ, Helman E, Ye J, Liu F, et al. A blood-based assay for assessment of tumor mutational burden in first-line metastatic NSCLC treatment: results from the MYSTIC study. Clin Cancer Res. 2021;27(6):1631.
Rousseau B, Foote MB, Maron SB, Diplas BH, Lu S, Argilés G, et al. The spectrum of benefit from checkpoint blockade in hypermutated tumors. N Engl J Med. 2021;384(12):1168–70.
McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661–72.
Wolf Y, Bartok O, Patkar S, Eli GB, Cohen S, Litchfield K, et al. UVB-induced tumor heterogeneity diminishes immune response in melanoma. Cell. 2019;179(1):219-35.e21.
Huh JW, Lee JH, Kim HR. Prognostic significance of tumor-infiltrating lymphocytes for patients with colorectal cancer. Arch Surg. 2012;147(4):366–72.
Thomas NE, Busam KJ, From L, Kricker A, Armstrong BK, Anton-Culver H, et al. Tumor-infiltrating lymphocyte grade in primary melanomas is independently associated with melanoma-specific survival in the population-based genes, environment and melanoma study. J Clin Oncol. 2013;31(33):4252–9.
Zeng D-Q, Yu Y-F, Ou Q-Y, Li X-Y, Zhong R-Z, Xie C-M, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes for clinical therapeutic research in patients with non-small cell lung cancer. Oncotarget. 2016;7(12):13765.
Adams S, Gray RJ, Demaria S, Goldstein L, Perez EA, Shulman LN, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32(27):2959–66.
Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immuno-oncology biomarkers working group: part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv Anat Pathol. 2017;24(6):311–35.
Yu Y, Zeng D, Ou Q, Liu S, Li A, Chen Y, et al. Association of survival and immune-related biomarkers with immunotherapy in patients with non-small cell lung cancer: a meta-analysis and individual patient-level analysis. JAMA Netw Open. 2019;2(7):e196879.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Chen P-L, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6(8):827.
Hallek M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol. 2019;94(11):1266–87.
Venetoclax Package Insert. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/208573s000lbl.pdf. AbbVie Inc. Issued 04/2016. Accessed 10 Sept 22.
Weide B, Martens A, Hassel JC, Berking C, Postow MA, Bisschop K, et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin Cancer Res. 2016;22(22):5487–96.
Delyon J, Mateus C, Lefeuvre D, Lanoy E, Zitvogel L, Chaput N, et al. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Ann Oncol. 2013;24(6):1697–703.
Tietze JK, Angelova D, Heppt MV, Reinholz M, Murphy WJ, Spannagl M, et al. The proportion of circulating CD45RO<sup>+</sup>CD8<sup>+</sup> memory T cells is correlated with clinical response in melanoma patients treated with ipilimumab. Eur J Cancer. 2017;75:268–79.
Jiang T, Qiao M, Zhao C, Li X, Gao G, Su C, et al. Pretreatment neutrophil-to-lymphocyte ratio is associated with outcome of advanced-stage cancer patients treated with immunotherapy: a meta-analysis. Cancer Immunol Immunother. 2018;67(5):713–27.
Valero C, Lee M, Hoen D, Weiss K, Kelly DW, Adusumilli PS, et al. Pretreatment neutrophil-to-lymphocyte ratio and mutational burden as biomarkers of tumor response to immune checkpoint inhibitors. Nat Commun. 2021;12(1):729.
Kelderman S, Heemskerk B, van Tinteren H, van den Brom RRH, Hospers GAP, van den Eertwegh AJM, et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol Immunother. 2014;63(5):449–58.
Martens A, Wistuba-Hamprecht K, Geukes Foppen M, Yuan J, Postow MA, Wong P, et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin Cancer Res. 2016;22(12):2908–18.
Simeone E, Gentilcore G, Giannarelli D, Grimaldi AM, Caracò C, Curvietto M, et al. Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunol Immunother. 2014;63(7):675–83.
Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3:23.
Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7(1):10582.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Investig. 2017;127(8):2930–40.
Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med. 2018;378(24):2288–301.
Higgs BW, Morehouse CA, Streicher K, Brohawn PZ, Pilataxi F, Gupta A, et al. Interferon gamma messenger RNA signature in tumor biopsies predicts outcomes in patients with non-small cell lung carcinoma or urothelial cancer treated with durvalumab. Clin Cancer Res. 2018;24(16):3857–66.
Lu S, Stein JE, Rimm DL, Wang DW, Bell JM, Johnson DB, et al. Comparison of biomarker modalities for predicting response to PD-1/PD-L1 checkpoint blockade: a systematic review and meta-analysis. JAMA Oncol. 2019;5(8):1195–204.
Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595(7867):432–7.
Seremet T, Jansen Y, Planken S, Njimi H, Delaunoy M, El Housni H, et al. Undetectable circulating tumor DNA (ctDNA) levels correlate with favorable outcome in metastatic melanoma patients treated with anti-PD1 therapy. J Transl Med. 2019;17(1):303.
McDonald BR, Contente-Cuomo T, Sammut S-J, Odenheimer-Bergman A, Ernst B, Perdigones N, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med. 2019;11(504):7392.
Bratman SV, Yang SYC, Iafolla MAJ, Liu Z, Hansen AR, Bedard PL, et al. Personalized circulating tumor DNA analysis as a predictive biomarker in solid tumor patients treated with pembrolizumab. Nat Cancer. 2020;1(9):873–81.
Domblides C, Rochefort J, Riffard C, Panouillot M, Lescaille G, Teillaud J-L, et al. Tumor-associated tertiary lymphoid structures: from basic and clinical knowledge to therapeutic manipulation. Front Immunol. 2021;12:698604.
Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–55.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5.
Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577(7791):556–60.
Wastyk HC, Fragiadakis GK, Perelman D, Dahan D, Merrill BD, Yu FB, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. 2021;184(16):4137-53.e14.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (New York, NY). 2015;350(6264):1084–9.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science. 2018;359(6371):91.
Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K, Monot C, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol. 2017;28(6):1368–79.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (New York, NY). 2018;359(6371):97–103.
Funding
No external funding or material support was received for this work. Likewise, no funded writing assistance was provided.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the conception, data acquisition and analysis and drafting of the work. They are in agreement with the interpretations, have approved the final version for publication, and are accountable for its integrity.
Corresponding author
Ethics declarations
Conflict of interest
WJK and VK have no personal, professional or financial relationships that would constitute a conflict of interest.
Ethical approval
No ethical approval was needed as this was a review article.
Informed consent
None.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kapoor, V., Kelly, W.J. Biomarkers for immune checkpoint inhibitors in solid tumors. Clin Transl Oncol 25, 126–136 (2023). https://doi.org/10.1007/s12094-022-02942-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-022-02942-4