
Abstract
Aberrations in the PI3K signaling pathway are frequently observed in patients with breast cancer. Because of that, PI3K inhibitors are attractive options for the treatment of breast cancer because PI3K is the most proximal component of the pathway other than receptor tyrosine kinases. Buparlisib is a potent and highly specific oral pan-class I PI3K inhibitor, which is currently under investigation in patients with breast cancer. In this article, we describe the PI3K signaling pathway, the prognostic value of PI3K pathway mutations, as well as the mechanism of action of buparlisib. Lastly, we discuss preliminary results of preclinical and clinical studies showing the efficacy and safety profile of this agent in breast cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The alteration of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway is central to the growth and survival of many cancers, including that of colon, brain, stomach, liver, lung, and breast. Alterations include amplification of human epidermal growth factor receptor 2 (HER2), loss/mutation of the phosphatase and tensin homolog (PTEN), PI3K mutation/amplification, AKT overexpression/overactivation, and modulation of tuberous sclerosis protein 1 and 2 (TSC1/TSC2) tumor suppressors [1]. These aberrations in the PI3K signaling pathway are frequently observed in breast cancer, mainly in hormone-sensitive tumors. The use of PI3K pathway inhibitors is a valuable option in this setting.
PI3K inhibitors are attractive options for the treatment of cancer because PI3K is the most proximal component of the pathway other than receptor tyrosine kinases (RTKs). Therefore, targeting PI3K itself rather than AKT or mTOR could provide global inhibition of the downstream components within the pathway.
Buparlisib is a potent and highly specific oral pan-class I PI3K inhibitor, which is currently under investigation in patients with solid tumors. In this article, we describe the PI3K signaling pathway, as well as the main characteristics of buparlisib. Lastly, we discuss preliminary preclinical and clinical studies demonstrating the efficacy of this agent in breast cancer.
PI3K signaling pathway
The intracellular PI3K pathway regulates cellular functions such as cell proliferation, growth, survival, apoptosis, protein synthesis, and glucose metabolism. There are three classes of PI3Ks grouped according to structure and function (class I, II, and III). Class IA PI3K is the one most clearly implicated in human cancer and consists of a regulatory subunit and a catalytic subunit. Regulatory subunits are p85α (p85α, p55α, and p50α isoforms), p85β, and p55γ, which by convention are referred to collectively as p85. p110 are also regulatory subunits. They are encoded by the genes PIK3R1, PIK3R2, and PIK3R3, respectively. The catalytic subunits are p110α, p110β, and p110δ, which are encoded by genes PIK3CA, PIK3CB, and PIK3CD. Both PIK3CA and PIK3R1 are somatically mutated in cancers, and these mutations promote activation of the PI3K pathway [2].
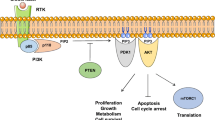
Class IA PI3Ks are activated by growth factor stimulation through RTKs. These receptors include HER2 and insulin-like growth factor-1 receptor (IGF-1R) among others [3–5]. The regulatory subunit, p85, directly binds to phosphotyrosine residues on RTKs and/or adaptors, such as the insulin receptor substrate 1 (IRS-1) [6]. This binding relieves the intermolecular inhibition of the p110 catalytic subunit by p85 and moves PI3K toward the plasma membrane where its substrate, phosphatidylinositol 4,5-bisphosphate (PIP2), resides. The catalytic subunit can also be triggered by activated RAS, which directly binds p110, and by G-protein coupled receptors [7, 8]. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3). In addition, the tumor suppressor PTEN dephosphorylates PIP3 to PIP2, thereby regulating PI3K-dependent signaling in a negative way [9] (Fig. 1).

PI3K/AKT/mTOR signaling pathway. 4E-BP1 eIF4E-binding protein, AKT protein kinase B, IRS-1 insulin-like growth factor-1, p70S6K 70 kDa ribosomal protein S6 kinase, PDK1 phosphoinositide-dependent kinase 1, PI3K phosphatidylinositol 3-kinase, PIP2 phosphatidylinositol 4,5-bisphosphate, PIP3 phosphatidylinositol 3,4,5-trisphosphate, PTEN phosphatase and tensin homolog, mTOR mammalian target of rapamycin, RAPTOR regulatory-associated protein of mTOR, RHEB RAS homolog enriched in brain, RICTOR rapamycin-insensitive companion of mTOR, RTK receptor tyrosine kinase, TSC1–TSC2 tuberous sclerosis protein 1 and 2
Following PIP3 formation, phosphoinositide-dependent kinase 1 (PDK1) and AKT bind to PIP3 through its pleckstrin homology (PH) domains in close proximity to the cell plasma membrane. PDK1 activates AKT by phosphorylating AKT at threonine 308 [10]. After this phosphorylation, AKT is fully activated by the subsequent phosphorylation at serine 473 by several protein kinases, such as PDK1, the complex mTORC2 formed by mTOR bound to rapamycin-insensitive companion of mTOR (RICTOR), or AKT itself [11–14] (Fig. 1).
AKT is the central mediator of the PI3K pathway and promotes cell growth and survival by several mechanisms. One of the most studied downstream effectors of AKT is mTOR, usually associated with the regulatory-associated protein of mTOR (RAPTOR), creating the complex mTORC1 [15]. AKT phosphorylates TSC2, thereby inhibiting the GTPase activity of the TSC1/TSC2 dimer, and the GTP-binding protein RAS homolog enriched in brain (RHEB) remain in its active GTP-bound state, causing a rise in mTORC1 [16, 17]. In the mTORC1 complex, mTOR phosphorylates 70 kDa ribosomal protein S6 kinase (p70S6K) and eIF4E-binding protein (4E-BP1), leading to an increased translation and synthesis of cell cycle regulating and ribosomal proteins [18, 19]. Activated p70S6K also forms a negative feedback reducing the activation of the PI3K pathway through phosphorylation and subsequent inhibition of ISR-1 [20] (Fig. 1).
PI3K pathway in cancer
The PI3K pathway is one of the most highly mutated in human cancer.
Disruption of this pathway can be due to a host of genetic aberrations, resulting in either amplification or mutation of PI3KCA, amplification or mutation of AKT, decreased expression of PTEN, and also amplification or overexpression of HER2 [21, 22].
PI3KCA encodes p110α, the catalytic subunit of PI3K, and is commonly mutated or amplified in cancers [23]. In breast cancer, the overall mutation rate of PI3KCA is around 40 % [24], and more than 85 % of the mutations occur in E542K and E545K at exon 9, which encodes the helical domain (HD), and H1047R at exon 20, which encodes the catalytic domain (KD). These mutations confer increased PI3K catalytic activity, leading to cellular transformation through growth factor and anchorage-independent cellular proliferation [25, 26].
Prognostic value of PI3K pathway mutations
The prognostic significance of PI3KCA mutations remains most frequently inconclusive in breast cancer. Kalinsly et al. analyzed the prognostic value of PI3KCA mutations in 590 patients with breast cancer [27]. They identified PI3KCA in 32.5 % of breast cancers. Compared with wild type (WT), PI3KCA mutations were significantly more likely to occur in elderly patients, lymph node negative, hormone receptor-positive, HER2-negative, and low-grade and early-stage breast cancer at diagnosis. Interestingly, patients harboring a PI3KCA mutated breast tumor show a marginally significant longer progression-free survival (PFS) compared with patients with WT tumors (p = 0.06) and a significant improvement in overall survival (OS) (p = 0.03). In particular, patients with a H1047R KD mutated tumor display a favorable prognosis when compared with WT. This improvement in OS is not observed in patients with a HD mutated tumors, showing that the OS benefit is a result of the H1047R KD mutation. These results demonstrate that the mechanism of PI3KCA mutation-associated protection may differ according to mutation type.
However, the fact that PI3KCA mutations are a marker of favorable outcome for breast cancer patients remains unexplained. One suggestion could be that cells harboring the mutation are more sensitive to systemic treatment. Another possibility is that there is a biphasic growth (i.e., an expansion of mutant cells followed by a senescence program of limited growth). Notably, the comparative group PI3KCA WT were mainly HER2-positive but were not treated with trastuzumab at this time [27].
A retrospective study conducted by Angulo et al. determined that there are differences in mutation status of components of the PI3K pathway and PTEN between primary tumors and metastases in breast cancer patients [28]. Also, Dupont et al. observed the discordance in PIK3CA mutations between primary and metastatic disease in breast cancer [29]. Overall, the rates of PI3KCA aberrations were similar between the primary tumors and matched metastases (around 41 %); however, the authors found marked discordances in PTEN levels (26 %), PIK3CA mutations (18 %), and receptor status (25 %) between the primary tumor and metastases. Unexpectedly, they also found that almost the same proportion of patients had activating PIK3CA mutations in their primary tumor and not in metastases as they had activating mutations in their metastases but not primary tumors. The gain and loss frequencies between primary and metastatic sites suggest that the aberrations in the PI3K pathway are not required for the metastatic process.
Antiestrogen treatment resistance
Boyault et al. showed that PI3K pathway mutations are associated with estrogen receptor-positive breast tumors irrespective of HER2 status [30]. Also, PIK3CA mutations frequently occur in estrogen receptor-positive or HER2-amplified breast tumors, which may be a major determinant of resistance to endocrine and HER2-targeted therapies [31].
Currently, one of the most widely accepted mechanisms linked with endocrine resistance is the amplification or overexpression of the HER2 proto-oncogene [32]. Moreover, a large body of experimental and clinical evidence suggests that activation of PI3K pathway promotes antiestrogen resistance. In fact, there is crosstalk between the PI3K and estrogen receptor pathway. PI3K activation was shown to induce estrogen receptor phosphorylation at Ser167 by AKT or p70S6K inducing estrogen-independent transcriptional activity [33, 34]. In addition, PI3K and RAS promote c-Jun phosphorylation. c-Jun complexes with c-Fos to form the AP-1 complex, which cooperates with estrogen receptor transcription [35]. These factors are the rationale for a combined targeting of the estrogen receptor and PI3K pathways, as shown in the section below [36].
Mechanism of action of buparlisib
Buparlisib (Novartis Pharma AG, Basel, Switzerland) is an oral inhibitor of the pan-class I PI3K family of lipid kinases with antineoplastic activity. This agent specifically inhibits class I PIK3 in the PI3K/AKT kinase signaling pathway in an ATP-competitive approach, inhibiting both the production of the secondary messenger phosphatidylinositol-3,4,5-trisphosphate and the activation of the PI3K signaling pathway. This may result in inhibition of tumor cell growth and survival in susceptible tumor cell populations. Buparlisib does not significantly inhibit the related class III (Vps34) and class IV (mTOR) PI3K.
The mechanism of action of buparlisib consists of binding to the ATP-binding site of the KD of PI3K, preventing the phosphorylation of PIP2 to PIP3, which decreases the levels of phosphorylated AKT. This biological activity correlates with the inhibition of various AKT downstream signaling pathway components and with its antiproliferative activity [37, 38]. The vascularization of many tumors is in part due to VEGF-induced eNOS activation through a class IA PI3K-dependent mechanism involving AKT [39–41]. Buparlisib has shown in vivo antiangiogenic activity through the inhibition of PI3K [38]. Buparlisib has also demonstrated cell death irrespective of the level of PI3K when used in vitro at higher dose. This apparently PI3K-independent effect is due to the inhibition of microtubule dynamics upon direct binding to tubulin, causing a prometaphase to metaphase blockade [42].
Research with buparlisib
Several preclinical and clinical trials have evaluated or are currently evaluating the pharmacokinetic, pharmacodynamic and safety profile of buparlisib.
Preclinical studies
One of the preclinical investigations with buparlisib used 353 cell lines that varied with respect to key genetic determinants such as the status of the PIK3CA, PTEN, and KRAS genes [38]. Buparlisib exhibited preferential inhibition of tumor cells bearing PIK3CA mutations in contrast to either PTEN or KRAS mutant models. Also, buparlisib showed dose-dependent in vivo pharmacodynamic activity as measured by significant inhibition of phosphorylated AKT and tumor growth inhibition. Interestingly, it was observed that cotreatment of buparlisib with mitogen-activated protein kinase/extracellular-signal-regulated kinase (MEK) or HER2 inhibitors, or with cytotoxic agents such as docetaxel or temozolomide, could induce cell death [38].
In another preclinical study, the efficacy of several PI3K inhibitors in association with fulvestrant against estrogen receptor-positive breast cancer cell lines was evaluated [36]. Buparlisib induced high levels of apoptosis when combined with estrogen deprivation in sensitive cells. In addition, fulvestrant strongly potentiated apoptosis when combined with buparlisib treatment in MCF7 long-term estrogen-deprived (LTED) cells. This is the rationale behind the clinical studies combining buparlisib with fulvestrant in estrogen receptor-positive breast cancer patients, who progressed on an aromatase inhibitor.
Therapeutic options for triple-negative breast cancers are limited [43]. A small subset of these cancers has defects in homologous recombination (HR)-mediated DNA repair due to BRCA1/2 mutations. BCRA1/2 proteins are essential components of HR to repair double-strand breaks of DNA. Poly ADP-ribose polymerase (PARP) is needed for the repair of single-strand breaks of DNA, and the use of PARP inhibitors in tumors with BRCA1/2 mutations may be sufficient to cause lethal DNA damage. In cases without BRCA1/2 mutations, PARP inhibitors alone may be insufficient because of the functioning BRCA1/2 system. On the other hand, PI3K stabilizes and preserves double-strand break repairs by interacting with the HR complex. Triple-negative tumors also display aberrant activation of the PI3K pathway. Thus, direct inhibition of PI3K, together with PARP inhibition, could be an attractive strategy for this disease. Two preclinical trials studied the combination of buparlisib and the PARP inhibitor olaparib in triple-negative breast cancer cells [44, 45]. They observed that PI3K blockade results in HR impairment sensitization to PARP inhibition in triple-negative breast cancers without BRCA mutations. Also, an ongoing clinical trial is evaluating the combination of buparlisib and olaparib in patients with recurrent triple-negative breast cancer or recurrent high-grade serous ovarian cancer (NCT01623349).
Lapatinib-resistant cells were profiled for mutations in the PI3K pathway in a study by Rexer et al. [46]. The impact of PIK3CA mutations on the effect of HER2 and PI3K inhibitors combined was studied in HER2-amplified xenograft models with wild-type or mutant PIK3CA. Results suggest that the addition of a PI3K inhibitor further improved tumor regression and decreased tumor relapse after discontinuation of treatment. PIK3CA inhibition with buparlisib in combination with lapatinib and trastuzumab was required to achieve tumor regression in a PIK3CA-mutant HER2-positive xenograft. Hanker et al. observed that HER2-driven tumors in mice clustered with luminal breast cancers, whereas PIK3CA tumors were associated with claudin-low breast cancers [47]. The authors detected that HER2-positive/PIK3CA tumors were resistant to trastuzumab in monotherapy or in combination with lapatinib or pertuzumab. Drug resistance and enhanced mammosphere formation were reversed by treatment with a PI3K inhibitor.
Clinical studies
At this time, several clinical studies are evaluating the role of buparlisib in patients with breast cancer. One common issue in these trials is that there was no prior selection of patients according to PI3KCA status. However, these studies have a stratification of patients and are powered to answer whether PI3K activation is a predictor of response. The stratification ensures adequate power to assess the activity of buparlisib in patients presenting PI3K activated in comparison with all the patients in the context of the large phase III ongoing studies. So far, no data are conclusive on the activity of buparlisib in this specific patient population. Another important issue is that the PI3K signaling pathway seems relevant in all breast carcinoma subtypes, including also triple-negative breast carcinoma, i.e., PI3K signaling pathway seems relevant independently of molecular alterations in patients with breast cancer.
Dose-escalation studies of buparlisib
Table 1 shows the phase I trials conducted with buparlisib in different advanced solid tumors. In three of these trials [48–50], patients with solid tumors, mainly colon and breast cancer, received daily oral buparlisib from 12.5 to 150 mg. The maximum-tolerated-dose (MTD) of buparlisib was 100 mg/day, and dose-limiting toxicities observed were hyperglycemia, rash, epigastralgia, and mood alteration. In general, buparlisib was well tolerated. Most frequent treatment-related adverse events included rash, hyperglycemia, diarrhea, anorexia, and mood alteration. Skin rash was successfully managed with antihistamines and topical corticosteroids. Hyperglycemia was managed with metformin and insulin, but at 150 mg, discontinuation of buparlisib was required to control hyperglycemia. Mood alterations were reversed with a dose hold and subsequent dose reduction, as well as with the administration of selective serotonin reuptake inhibitors and anxiolytics. Regarding efficacy, between 52 and 58 % of patients achieved stable disease and 3 % of patients showed partial response.
However, these trials could not elucidate whether tumors bearing a PI3K mutation had a higher probability of response to buparlisib, although some of the patients treated for more than 8 months with buparlisib did have tumors with PI3K pathway abnormalities. Otherwise, preclinical data suggest that KRAS mutation may predict resistance to PI3K inhibitors [51]. This is particularly evident in colon cancer, although it could not be demonstrated in breast carcinoma. However, in one of these phase I trials, a breast cancer patient treated with buparlisib showed a partial response in spite of harboring a KRAS mutation [48].
In conclusion, these studies demonstrated the clinical safety and tolerability of buparlisib at the selected dose of 100 mg, as well as a favorable pharmacokinetic profile, which was consistent with its pharmacodynamic effects. Further clinical studies are needed to evaluate the predictive value of PI3K alterations.
A substudy of a previous phase I trial focused on metastatic breast cancer. MTD for buparlisib was 100 mg/day [48, 52], and drug-related adverse events were similar to those previously observed, which were manageable with treatment interruption and dose reduction. Regarding efficacy, 11 % of patients exhibited a partial response and 50 % had stable disease.
Subsequently, another phase I study evaluated the inhibitory effect of buparlisib in patients with solid tumors in the context of glucose metabolism regulation and tumor biology by surveying the phosphorylation of proteins downstream of the PI3K pathway [53]. Buparlisib appears to have the ability to induce an increase in C-peptide and inhibition of the immediate effector of PI3K, phosphorylated AKT, as well as to downregulate the phosphorylated proteins downstream of PI3K pathway, such as S6 and 4E-BP1 at MTD.
Finally, two dose-escalation phase I trials tested buparlisib in combination with other drugs. The first one combined buparlisib with paclitaxel in patients with solid tumors [54]. The MTD achieved was 100 mg/day for buparlisib and 80 mg/m2 for paclitaxel. Dose-limiting toxicities were asthenia, hyperglycemia, and depression. In the second trial, buparlisib was combined with the oral MEK1/2 inhibitor GSK1120212 in patients with mainly RAS/RAF mutations [55]. Grade 3 dose-limiting toxicities were stomatitis, dysphagia, LVEF decrease, creatine kinase increase, nausea, anorexia, and decreased oral intake. The MTD was 70 mg/day of buparlisib. The authors concluded that the combination can be safely administered to these patients and shows a promising clinical activity.
Buparlisib in HER2-positive disease
HER2 overexpression, which is found in 20–30 % of human breast cancers, has been linked to the activation of the PI3K pathway in patients with this disease. Trastuzumab is a monoclonal antibody that blocks HER2. Trastuzumab resistance has been associated with loss/downregulation of PTEN, which has been reported in 5–10 % of human breast cancers and also causes activation of the PI3K pathway, suggesting that PI3K-targeting therapies could overcome this resistance [56]. Also, in a study by Barbareschi et al. [57], the authors analyzed PI3KCA hot-spot mutations and PTEN immnunohistochemical expression in 129 HER2-positive infiltrating breast cancers treated with trastuzumab.
Out of the total 129 patients with HER2-positive infiltrating breast cancers which had been treated with trastuzumab, PI3KCA hot-spot mutations were detected in 19 % of them. No correlations were observed between mutations and pathological and biological parameters in these patients. In addition, in patients treated with neoadjuvant therapy and in metastatic breast cancer, no relationship was detected between response to trastuzumab-based therapy and this mutation. PTEN loss was found in 28 % of cases, 13 % of which presented also mutation for PI3KCA. PI3K pathway activation, defined as PI3KCA mutation and/or PTEN loss, was not related with response to treatment or clinical outcome in patients with metastatic breast cancer.
A trial by Saura et al. analyzed the safety and efficacy of buparlisib in combination with trastuzumab in patients with HER2-positive advanced breast carcinoma who have progressed on a trastuzumab-containing regimen. The rationale for this trial was the synergistic activity observed for both drugs in preclinical models. The primary objective of this phase I/II study was to determine the MTD of buparlisib in combination with weekly trastuzumab. Patients had received a median of 4 (1–10) antineoplastic regimens. Results from the phase Ib of this trial were recently reported [58]. In 18 patients evaluated, the MTD of buparlisib in combination with trastuzumab was set up at 100 mg/day. No grade 4 toxicity was reported, and grade 3 adverse events were asthenia, altered mood, rash, GGT increase, hypokalemia, and hypersensitivity in one patient. The preliminary pharmacokinetics data indicated that systemic drug exposure (C max and AUC) of buparlisib in combination with trastuzumab was similar to what is seen when used as single agent.
Also, preliminary results of the phase II of this trial have been recently presented at the European society for medical oncology (ESMO) [59]. A total of 53 patients previously treated with anti-HER2 regimens received 100 mg/day of buparlisib and the standard dose of weekly trastuzumab. Patients had been administered ≤4 prior anti-HER2 regimens, including trastuzumab (required), lapatinib, and/or trastuzumab and maytansine (T-DM1). Most patients discontinued the treatment due to disease progression (55 %), although 16 % of patients withdrew due to adverse events. Most common suspected study drug-related grade 3/4 adverse events were increased transaminases, rash, asthenia, nausea, anxiety, skin photosensitivity, and hyperglycemia. The disease control rate was 49 %. These results showed that buparlisib in combination with trastuzumab has an acceptable safety profile and an encouraging preliminary activity in heavily pretreated patients with HER2-positive metastatic breast cancer that are resistant to trastuzumab.
Buparlisib in hormone receptor-positive disease
Mutations in the PIK3CA gene present in 28–47 % of estrogen receptor-positive breast cancers have been associated with antiestrogen resistance [31]. However, antiestrogen-resistant cancers still retain estrogen receptors and estrogen sensitivity. This suggests that the administration of single agent PI3K-targeted therapy to patients with estrogen receptor-positive and PI3K mutant breast cancer may be insufficient to inhibit tumor growth [31].
A phase Ib trial has been recently conducted adding buparlisib 100 mg/daily or intermittently (5 days on/2 days off) to letrozol in postmenopausal patients with metastatic breast cancer [60]. Fifty-one patients were enrolled, of whom 49 had progressed on a previous aromatase inhibitor. The best responses were seen in the arm with daily buparlisib, where over 50 % of patients had >25 % tumor reduction evaluated by 2-(fluorine-18)fluoro-2-deoxy-d-glucose positron emission tomography (FDG-PET). Dose-limiting toxicities were transaminitis and depression. These results indicate that the combination of buparlisib with letrozole is safe and useful in patients with aromatase inhibitor-refractory estrogen receptor-positive metastatic breast cancer.
Ongoing research
Several trials are currently evaluating buparlisib alone or in combination for the treatment of patients with breast cancer.
A pharmacodynamic study of buparlisib is ongoing in patients with hormone receptor-positive, HER2-negative, and PI3KCA mutation breast cancer (NCT01513356). This is a phase 0 clinical trial, the main purpose of which is to determine the grade of inhibition of PI3K/AKT/mTOR signaling pathways in a surgical specimen after 4 weeks of treatment with buparlisib. Preliminary results were presented at SABCS 2013. To date, 47 patients have been included in the study and 17 out of 47 patients (36 %) had mutations in PI3KCA. Results from pRPS6 showed a marked inactivation in 8 out of 11 patients (72 %) that have completed 4 weeks of buparlisib treatment [61].There are several ongoing placebo-controlled, randomized phase III trials. In the first 2, the objective is to evaluate buparlisib plus fulvestrant in patients with hormone receptor-positive HER2-negative advanced or metastatic breast cancer that is refractory to aromatase inhibitor (BELLE-2 study, NCT01610284), or who progressed on or after treatment with mTOR inhibitor (BELLE-3 study, NCT01633060). Lastly, BELLE-4 study (NCT01572727) is a randomized seamless phase II/III trial evaluating the addition of buparlisib to paclitaxel in patients with HER2-negative, locally advanced or metastatic breast cancer.
With regard to triple-negative breast cancer, a phase II clinical trial is now recruiting patients to evaluate the efficacy of buparlisib in patients with metastatic triple-negative breast cancer who have developed disease progression after standard chemotherapy in the adjuvant or the metastatic setting (NCT01629615). Another phase I trial is determining the highest possible dose of buparlisib that may be given safely, and also whether the combination of buparlisib with olaparib may be an effective therapy to treat patients with triple-negative breast cancer or with high-grade serous ovarian cancer (NCT01623349).
Finally, a phase IB/II study is evaluating the safety and efficacy profile of buparlisib in combination with lapatinib in HER2-positive, PI3K-activated, trastuzumab-resistant, locally advanced, recurrent, or metastatic breast cancer (NCT01589861). Other phase I trials in HER2-positive metastatic breast cancer are being designed to establish the safety, tolerability, and MTD of buparlisib in combination with capecitabine and/or trastuzumab or lapatinib in patients with metastatic breast cancer (NCT01300962).
Conclusions
Buparlisib is an oral pan-PI3K inhibitor that has allowed a full clinical development. It is a topic under discussion whether this drug possesses antiproliferative and antiangiogenic activity related to the inhibition of the PI3K pathway and its downstream effectors, as well as its capacity to cause cell death due to the inhibition of microtubule dynamics. Due to all its properties, alone or in combination with other drugs such as fulvestrant, buparlisib may be an effective approach especially for patients with HER2-negative, hormone receptor-positive breast cancer.
References
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62. doi:10.1038/nrc2664.
Cidado J, Park BH. Targeting the PI3K/Akt/mTOR pathway for breast cancer therapy. J Mammary Gland Biol Neoplasia. 2012;17(3–4):205–16. doi:10.1007/s10911-012-9264-2.
Escobedo JA, Navankasattusas S, Kavanaugh WM, Milfay D, Fried VA, Williams LT. cDNA cloning of a novel 85 kd protein that has SH2 domains and regulates binding of PI3-kinase to the PDGF beta-receptor. Cell. 1991;65(1):75–82. doi:10.1016/0092-8674(91)90409-R.
Otsu M, Hiles I, Gout I, Fry MJ, Ruiz-Larrea F, Panayotou G, et al. Characterization of two 85 kd proteins that associate with receptor tyrosine kinases, middle-T/pp60c-src complexes, and PI3-kinase. Cell. 1991;65(1):91–104. doi:10.1016/0092-8674(91)90411-Q.
Skolnik EY, Margolis B, Mohammadi M, Lowenstein E, Fischer R, Drepps A, et al. Cloning of PI3Kinase-associated p85 utilizing a novel method for expression/cloning of target proteins for receptor tyrosine kinases. Cell. 1991;65(1):83–90. doi:10.1016/0092-8674(91)90410-Z.
Myers MG Jr, Backer JM, Sun XJ, Shoelson S, Hu P, Schlessinger J, et al. IRS-1 activates phosphatidylinositol 3′-kinase by associating with src homology 2 domains of p85. Proc Natl Acad Sci USA. 1992;89(21):10350–4.
Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, et al. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci USA. 2008;105(24):8292–7. doi:10.1073/pnas.0707761105.
Stoyanov B, Volinia S, Hanck T, Rubio I, Loubtchenkov M, Malek D, et al. Cloning and characterization of a G protein-activated human phosphoinositide-3 kinase. Science. 1995;269(5224):690–3.
Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–8.
Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376(6541):599–602. doi:10.1038/376599a0.
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15(23):6541–51.
Balendran A, Casamayor A, Deak M, Paterson A, Gaffney P, Currie R, et al. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol. 1999;9(8):393–404. doi:10.1016/S0960-9822(99)80186-9.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi:10.1126/science.1106148.
Toker A, Newton AC. Akt/protein kinase B is regulated by autophosphorylation at the hypothetical PDK-2 site. J Biol Chem. 2000;275(12):8271–4.
Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868–80. doi:10.1038/nrd3531.
Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278(35):32493–6. doi:10.1074/jbc.C300226200C300226200.
Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4(9):648–57. doi:10.1038/ncb839ncb839.
Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J. 1996;15(3):658–64.
Chung J, Kuo CJ, Crabtree GR, Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell. 1992;69(7):1227–36. doi:10.1016/0092-8674(92)90643-Q.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166(2):213–23. doi:10.1083/jcb.200403069jcb.200403069.
Saura C, Bendell J, Jerusalem G, Grana-Suarez B, Su S, Ru Q et al. PD09-03: Phase I/II Study of BKM120 in Combination with Trastuzumab in Patients with HER2 Overexpressing Metastatic Breast Cancer Resistant to Trastuzumab-Containing Therapy. Cancer Res. 2011;71(suppl 24):Abstract PD09-03. doi:10.1158/0008-5472.sabcs11-pd09-03.
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–510. doi:10.1038/onc.2008.245.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi:10.1126/science.10965021096502.
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73. doi:10.1038/nature12113.
Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3(8):772–5.
Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65(23):10992–1000. doi:10.1158/0008-5472.CAN-05-2612.
Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009;15(16):5049–59. doi:10.1158/1078-0432.CCR-09-0632.
Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, et al. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011;10(6):1093–101. doi:10.1158/1535-7163.MCT-10-1089.
Dupont Jensen J, Laenkholm AV, Knoop A, Ewertz M, Bandaru R, Liu W, et al. PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res. 2011;17(4):667–77. doi:10.1158/1078-0432.CCR-10-1133.
Boyault S, Drouet Y, Navarro C, Bachelot T, Lasset C, Treilleux I, et al. Mutational characterization of individual breast tumors: tP53 and PI3K pathway genes are frequently and distinctively mutated in different subtypes. Breast Cancer Res Treat. 2012;132(1):29–39. doi:10.1007/s10549-011-1518-y.
Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–61. doi:10.1200/JCO.2010.34.4879.
De Laurentiis M, Arpino G, Massarelli E, Ruggiero A, Carlomagno C, Ciardiello F, et al. A meta-analysis on the interaction between HER-2 expression and response to endocrine treatment in advanced breast cancer. Clin Cancer Res. 2005;11(13):4741–8. doi:10.1158/1078-0432.CCR-04-2569.
Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276(13):9817–24. doi:10.1074/jbc.M010840200M010840200.
Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. S6 kinase 1 regulates estrogen receptor alpha in control of breast cancer cell proliferation. J Biol Chem. 2009;284(10):6361–9. doi:10.1074/jbc.M807532200.
DeNardo DG, Cuba VL, Kim H, Wu K, Lee AV, Brown PH. Estrogen receptor DNA binding is not required for estrogen-induced breast cell growth. Mol Cell Endocrinol. 2007;277(1–2):13–25. doi:10.1016/j.mce.2007.07.006.
Sanchez CG, Ma CX, Crowder RJ, Guintoli T, Phommaly C, Gao F, et al. Preclinical modeling of combined phosphatidylinositol-3-kinase inhibition with endocrine therapy for estrogen receptor-positive breast cancer. Breast Cancer Res. 2011;13(2):R21. doi:10.1186/bcr2833.
Voliva CF, Pecchi S, Burger M, Nagel T, Schnell C, Fritsch C et al. Biological characterization of NVP-BKM120, a novel inhibitor of phosphoinosotide 3-kinase in Phase I/II clinical trials. In: Proc AACR Annual Meeting, Washington, DC. Philadelphia (PA), EEUU, 2010.
Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther. 2012;11(2):317–28. doi:10.1158/1535-7163.MCT-11-0474.
Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68(16):6598–607. doi:10.1158/0008-5472.CAN-08-1044.
Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453(7195):662–6. doi:10.1038/nature06892.
Yuan TL, Choi HS, Matsui A, Benes C, Lifshits E, Luo J, et al. Class 1A PI3K regulates vessel integrity during development and tumorigenesis. Proc Natl Acad Sci USA. 2008;105(28):9739–44. doi:10.1073/pnas.0804123105.
Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11(8):1747–57. doi:10.1158/1535-7163.MCT-11-1021.
Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, et al. Triple-negative breast cancer: risk factors to potential targets. Clin Cancer Res. 2008;14(24):8010–8. doi:10.1158/1078-0432.CCR-08-1208.
Ibrahim YH, Garcia-Garcia C, Serra V, He L, Torres-Lockhart K, Prat A, et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012;2(11):1036–47. doi:10.1158/2159-8290.CD-11-0348.
Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, et al. Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2012;2(11):1048–63. doi:10.1158/2159-8290.CD-11-0336.
Rexer BN, Chanthaphaychith S, Dahlman KB, Arteaga CL. Direct inhibition of PI3K in combination with dual HER2 inhibitors is required for optimal antitumor activity in HER2+ breast cancer cells. Breast Cancer Res. 2014;16(1):R9. doi:10.1186/bcr3601.
Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sanchez V, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci USA. 2013;110(35):14372–7. doi:10.1073/pnas.1303204110.
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30(3):282–90. doi:10.1200/JCO.2011.36.1360.
Baselga J, De Jonge MJ, Rodon J, Burris HA, Birle DC, De Buck SS et al. A first-in-human phase I study of BKM120, an oral pan-class I PI3K inhibitor, in patients (pts) with advanced solid tumors. J Clin Oncol. 2010;28:15s (suppl; abstr 3003).
Grana B, Burris HA, Rodon Ahnert J, Abdul Razak AR, De Jonge MJ, Eskens F et al. Oral PI3Kinase inhibitor BKM120 monotherapy in patients (pts) with advanced solid tumors: an update on safety and efficacy. J Clin Oncol. 2011;29 (suppl; abstr 3043).
Ihle NT, Lemos R Jr, Wipf P, Yacoub A, Mitchell C, Siwak D, et al. Mutations in the phosphatidylinositol-3-kinase pathway predict for antitumor activity of the inhibitor PX-866 whereas oncogenic Ras is a dominant predictor for resistance. Cancer Res. 2009;69(1):143–50. doi:10.1158/0008-5472.CAN-07-6656.
Rodon J, Bendell JC, Abdul RAR, Homji N, Trandafir L, Quadt C et al. Safety profile and clinical activity of single-agent BKM120, a Pan-Class I PI3K inhibitor, for the treatment of patients with metastatic breast carcinoma. Cancer Res. 2011;71(suppl 24):Abstract P3-16-01. doi:10.1158/0008-5472.sabcs11-p3-16-01.
Rodon J, Bendell J, Razak ARA, De Jonge M, Eskens F, Di Tomaso E et al. A phase I dose escalation and expansion trial of BKM120, an oral pan-PI3K inhibitor, in patients with advanced solid tumors: analysis of pharmacodynamic biomarker data. In: ESMO Meeting Abstracts, Vienna, Austria, 2012. vol 9. p. ix158 (Abstract 457P). doi:10.1093/annonc/mds395.
Dirix L, Schuler M, Machiels J, Hess D, Awada A, Steeghs N et al. Phase IB dose-escalation study of BEZ235 or BKM120 in combination with paclitaxel (PTX) in patients with advanced solid tumors. In: ESMO Meeting Abstracts, Vienna, Austria, September 1 2012. vol 9. p. ix157 (Abstract 454P). doi:10.1093/annonc/mds395.
Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, et al. Ph lb dose escalation study of oral pan-PI3K inhibitor buparlisib (BKM120) with oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with advanced solid tumours. Clin Cancer Res. 2014. doi:10.1158/1078-0432.ccr-14-1814.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. doi:10.1016/j.ccr.2004.06.022S1535610804002107.
Barbareschi M, Cuorvo LV, Girlando S, Bragantini E, Eccher C, Leonardi E, et al. PI3KCA mutations and/or PTEN loss in Her2-positive breast carcinomas treated with trastuzumab are not related to resistance to anti-Her2 therapy. Virchows Arch. 2012;461(2):129–39. doi:10.1007/s00428-012-1267-2.
Saura C, Bendell J, Jerusalem G, Su S, Ru Q, De Buck S, et al. Phase Ib study of Buparlisib plus Trastuzumab in patients with HER2-positive advanced or metastatic breast cancer that has progressed on Trastuzumab-based therapy. Clin Cancer Res. 2014;20(7):1935–45. doi:10.1158/1078-0432.CCR-13-1070.
Pistilli B, Urruticoechea A, Chan S, Han HS, Jerusalem G, Kong A et al. Ph Ib/II study of BKM120 plus trastuzumab in patients with trastuzumab-resistant HER2+ advanced breast cancer. In: ESMO Meeting Abstracts, Vienna, Austria, 2012. vol 9. p. ix116 (Abstract 3180).
Mayer IA, Abramson VG, Balko JM, Isakoff SJ, Kuba MG, Sanders M et al. SU2C phase Ib study of pan-PI3K inhibitor BKM120 with letrozole in ER+/HER2− metastatic breast cancer (MBC). In: ASCO meeting abstracts, May 30 2012. vol suppl 15. p. Abstract 510.
Estevez L, Suarez A, Calvo I, Fernandez-Abad M, Perea S, Hidalgo M. Abstract P4-15-09: an exploratory analysis of inactivation of PI3K/AKT/mTOR signaling pathway using neoadjuvant BKM120 in PI3KCA mutated early breast cancer. Cancer Res. 2013;73(24 Supplement):P4-15-09. doi:10.1158/0008-5472.sabcs13-p4-15-09.
Acknowledgments
The authors wish to thank Dr. Fernando Sánchez-Barbero from HealthCo S.L. (Madrid, Spain) for his help in the preparation of the first draft of manuscript. The financial support of medical writing services was provided by Novartis. Novartis could comment the first draft of this manuscript, but all the decisions about its contents were made by the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
The study has been performed in accordance with the ethical standards of the Declaration of Helsinki and its later amendments. This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent statement
Additional informed consent was obtained from all individual participants for whom identifying information is included in this article.
Rights and permissions
About this article

Cite this article
Estévez, L.G., García, E. & Hidalgo, M. Inhibiting the PI3K signaling pathway: buparlisib as a new targeted option in breast carcinoma. Clin Transl Oncol 18, 541–549 (2016). https://doi.org/10.1007/s12094-015-1410-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-015-1410-z