
Abstract
Purpose
Thiamine-dependent enzymes (TDEs) linking glycolysis with the tricarboxylic acid cycle (TCA) pyruvate dehydrogenase (PDH), of the pentose phosphate pathway transketolases (TKTs), the TCA alpha-ketoglutarate deydrogenase (KGDH)/2-oxoglutarate dehydrogenase (OGDH) complex, and the amino acid catabolism branched-chain alpha-ketoacid dehydrogenase (BCKDH) complex are crucial factors for tumor metabolism. The expression of these enzymes has not been analyzed for carcinogenesis of oral squamous cell carcinoma (OSCC) with special focus on new targeted metabolic therapies as yet.
Methods
TDEs PDH, KGDH (OGDH), and BCKDH were analyzed in normal oral mucosa (n = 14), oral precursor lesions (simple hyperplasia, n = 21; squamous intraepithelial neoplasia, SIN I–III, n = 35), and OSCC specimen (n = 46) by immunohistochemistry and western blot (WB) analysis in OSCC tumor cell lines.
Results
Although the total numbers of PDH and KGDH (OGDH) positive samples decreased in OSCC, both enzymes were significantly overexpressed in the carcinogenesis of OSCC compared with normal tissue. BCKDH has been demonstrated to be significantly overexpressed in the carcinogenesis of OSCC. Specificity of the antibodies was confirmed by WB analysis.
Conclusions
This is the first study showing increased expression of TDEs in OSCC. Metabolic targeting of TDEs (including TKTs) by antagonistic compounds like oxythiamine or oxybenfothiamine may be a useful strategy to sensitize cancer cells to common OSCC cancer therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oral squamous cell carcinomas (OSCCs) and other tumor entities show different cancer metabolism properties compared with normal cells [1, 2]. It has been postulated that several pathways of carcinogenesis are associated with metabolic phases of transformation in tumor cells, which can be regarded as an integrated metabolic ecosystem [1, 3, 4]. Our recent studies in OSCCs demonstrated glycolysis, glutaminolysis, and mitochondrial oxidative phosphorylation (OXPHOS) to play major roles in tumor metabolism [1, 4–6]. Moreover, the pentose phosphate pathway (PPP) [7] is a crucial factor for survival of OSCC tumor cells [8, 9]. Intriguingly, the study conducted by Caneba et al. [10] demonstrated that actually a higher mitochondrial activity measured by higher pyruvate uptake and oxygen consumption is related to increased ovarian cancer invasiveness. Therefore, OXPHOS [5, 6] is clearly related to cancer cell proliferation and progression as currently suggested by our data [1].
Thiamine (vitamin B1)-dependent enzymes (TDEs) linking glycolysis with the tricarboxylic acid cycle (TCA) pyruvate dehydrogenase (PDH), of the PPP transketolase (TKT), the TCA alpha-ketoglutarate deydrogenase (KGDH)/2-oxoglutarate deydrogenase (OGDH) complex, and the amino acid catabolism branched-chain alpha-ketoacid dehydrogenase (BCKDH) complex are important enzymes for proliferative, anabolic, and survival purposes in tumor cells [11].
TKT, PDH, and KGDH (OGDH) are all important in carbohydrate metabolism. TKT is a cytosolic enzyme and is regarded as a key player in the PPP, a major route for the biosynthesis of nucleotides [8, 9]. Moreover, TKT-like 1 isoform TKTL1 has been shown to be responsible for generation of glutathione as an antioxidant of tumor cells (production of nicotinamide adenine dinucleotide phosphate, NADPH) linking the PPP with glutaminolysis and fatty acid synthesis [4, 12, 13].
PDH and KGDH (OGDH) are located in the mitochondria as essential parts of biochemical pathways that result in the generation of electron donors like nicotinamide adenine dinucleotide (NADH) and in the following to adenosine triphosphate (ATP). PDH links glycolysis to the TCA, while the reaction catalyzed by KGDH (OGDH) is a rate-limiting step in the TCA [11].
Valine, isoleucine, and leucine are essential branched chain amino acids (BCAAs). The metabolism of BCAAs involves transamination to succinyl-CoA (from valine and isoleucine) and acetyl-CoA (from leucine) that enter the TCA cycle [11, 14, 15]. BCKDH complex is located in the mitochondria [11, 14] and can serve as precursors for amino acid and protein synthesis as well as an energy source in cancer cells [11, 14, 16].
All in all, thiamine-dependent reactions of tumor cells link energy production of tumor cells with upregulation of their endogenous antioxidant capacity through accumulation of lactate (TKTL1, [12]), the redox couples glutathione/glutathione disulfide (TKTL1, [12]), NAD/NADH (PDH, KGDH), and NADP/NADPH (TKTL1, [12]), respectively [17–19]. These molecules constitute an intracellular redox buffer network that effectively scavenges reactive oxygen species (ROS) and free radicals preventing tumor cells from apoptosis.
Oral squamous cell carcinoma (OSCC) development is regarded as multistep carcinogenetic process mediated by several genetic alterations [20]. In this process, OSCC arises from a single transformed cell with a subsequent development through morphologically and clinically detectable precancerous stages [21]. Analysis of the mechanistic basis may harbor the availability of molecular tools to selectively and experimentally manipulate this multistep process. From these results, clinical oncologists expect subsequent implications for diagnosis and therapy of precursor lesions as well as OSCC.
Our recent studies [1, 4] demonstrated glycolysis, mitochondrial OXPHOS, and glutaminolysis as important cancer metabolism pathways for oral carcinogenesis. However, analysis of thiamine-dependent enzymes within the network of these pathways as indication for ‘metabolic therapies’ [22] in OSCC has not been reported yet. Therefore, the aim of this work was to investigate the relationship between TDEs PDH, KGDH (OGDH), and BCKDH with a multistep carcinogenesis of OSCC.
Materials and methods
Patients and tumor specimen
The records of healthy individuals (normal oral mucosal tissues, n = 14), patients with oral precursor lesions (simple hyperplasia, n = 21; squamous intraepithelial neoplasia SIN I, n = 5; SIN II, n = 9; SIN III, severe dysplasia, n = 10; SIN III, carcinoma in situ, n = 11), and patients with invasive OSCC (n = 46, Table 1) were retrospectively assessed from January 2009 to November 2014. The diagnosis of normal oral mucosal tissues, precursor lesions, and invasive squamous cell carcinoma was confirmed by the Department of Pathology, University Hospital Tuebingen. The material was archival formalin-fixed, paraffin-embedded tissue from routine histopathological work-ups. The material has been stored with permission of the local ethics committee of the University Hospital Tuebingen (approval number: 562-2013BO2), after informed consent obtained from the patients prior to surgical resection. Tumor blocks of paraffin-embedded tissue were selected by experienced pathologists, based on routine H&E stained sections. Sections from all available tissues underwent histopathological assessment, blinded to the prior histopathology report. Serial tissue sections (2 µm thickness) were cut from formalin-fixed paraffin-embedded (FFPE) blocks on a microtome and mounted from warm water onto adhesive microscope slides. First, we assessed H&E stained sections from each tissue section to differentiate between normal tissue, precursor lesions, tumor cell areas, stromal areas, and infiltrating immune cells. Oral precursor lesions were classified according to WHO criteria [21]. Tumor staging was performed according to the 7th edition of the TNM staging system by the UICC/AJCC of 2010 [23]. Grading of OSCC was defined according to WHO criteria [24].
Staining procedure and quantification of immunohistochemistry
We stained for PDH (Novus Biologicals, Cambridge, UK, rabbit pAb, NBP2-20024, E1 subunit, dilution 1:250), KGDH/OGDH (Thermo Fisher Scientific, IL, USA, rabbit pAb, PA5-28195, dilution 1:250), and BCKDH (antibodies-online Inc., Atlanta, USA, rabbit pAb, ABIN1496838, E1 subunit, dilution 1:250) in tissue sections. Staining was performed on serial sections of 2 µm thickness, which were deparaffinized in xylene and ethanol and rehydrated in water. Heat-induced epitope retrieval (HIER) was performed with either citrate buffer pH 6.0 (Dako, Hamburg, Germany) or EDTA buffer pH 9.0. Endogenous peroxidase activity was quenched with 0.3 % hydrogen peroxide. Endogenous biotin activity was blocked using the avidin/biotin blocking kit (Vector Laboratories, Burlingame, CA, USA). After incubation with the primary or rabbit control antibody (BD Pharmingen, Heidelberg, Germany [1]), the Dako LSAB2 peroxidase System (Dako, Hamburg) was used. Slides were subsequently incubated for 3–5 min in DAB (3,3′-diaminobenzidine, Biogenex) counterstained with hemalaun and mounted with Glycergel (Dako).
Five representative high power fields (1 HPF = 0.237 mm2, original magnification: ×200-fold) were analyzed for PDH, KGDH (OGDH), and BCKDH expression in normal tissue, oral precursor lesions, tumor tissue and averaged, respectively. The extent of the staining, defined as the percentage of positive staining areas of tumor cells in relation to the whole tissue area, was semi-quantitatively scored on a scale of 0–3 as the following: 0, <10 %; 1, 10–30 %; 2, 30–60 %; 3, >60 %. The intensities of the signals were scored as 1+, 2+, and 3+. Then, a combined score (0–9) for each specimen was calculated by multiplying the values of these two categories [25]. Cases were classified as negative, 0 points, positive, 1–9 points. Two observers blinded to the diagnosis performed scoring on identical sections marked by circling with a water-resistant pencil and finally with diamond-tipped pencil on the opposite side of the microscopic slide. Pictures were analyzed using a Canon camera (Krefeld, Germany). The photographed images were imported into the Microsoft Office Picture Manager.
Cell culture, Western blot and densitometric quantification
BICR3 and BICR56 OSCC cell lines [26, 27] were cultured in DMEM F-12 medium (Invitrogen, Belgium) containing 10 % fetal calf serum (Sigma-Aldrich, Germany), 1 % fungicide, and penicillin/streptomycin (Biochrom, Germany) at 37 °C and 5 % CO2.
PDH, KGDH (OGDH), and BCKDH antibody specificity was confirmed by western blot analysis in BICR3, BICR56 cell lines. Protein extraction from OSCC cell lines BICR3 and BICR56 was performed as described previously [28]. Normal human oral keratinocyte protein lysate (HOK cell lysate) was purchased by ScienCell (Carlsbad, CA, USA) as control. The membranes were analyzed by immunoblotting using the same antibodies as used for IHC (above) PDH (dilution: 1:1000), KGDH/OGDH (dilution: 1:1000), BCKDH (dilution: 1:1000), or IgG control antibodies (BD Pharmingen, Heidelberg), and mAb GAPDH (Abcam, Cambridge, UK, dilution: 1:5000) overnight at 4 °C. Binding of the primary antibodies was detected with peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, CA, USA) and visualized by the enhanced chemiluminescence method (GE Healthcare, Freiburg, Germany).
Quantification of western blot bands was carried out using an automated densitometric quantification digitizing system (UN-SCAN-IT Gel software, version 6.1, Silk Scientific, Inc., Utah, USA) [29].
Statistical analysis
Statistical analysis was performed with MedCalc Software, Version 15.2.2 (Mariakerke, Belgium). Data were analyzed using the non-parametric Mann–Whitney U test or Kruskal–Wallis test when more than 2 groups were compared. All p values presented were 2-sided and p < 0.05 was considered statistically significant.
Results
Expression of PDH, KGDH (OGDH), and BCKDH in normal mucosa, oral precursor lesions and OSCC
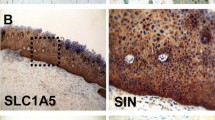
PDH (Figs. 1, 2) expression was found in all tissue types, normal oral mucosa (n = 14/14, 100 %), oral precursor lesions (simple hyperplasia, n = 21/21, 100 %; squamous intraepithelial neoplasia, SIN I–III, n = 25/35, 71 %), and OSCC specimen (n = 34/46, 74 %). In comparison with normal tissue/hyperplasia, a significantly increased expression of PDH was observed in SIN III lesions (CIS) and invasive OSCC. In comparison with SIN I, SIN II, and SIN III lesions (severe dysplasia), no significantly PDH expression was observed in OSCC. PDH expression was significantly increased in OSCC compared with SIN I–III lesions (p = 0.0008, Fig. 2).

Immunohistochemical staining of PDH in OSCC. Immunohistochemical staining shows representative images of PDH in N.T. (a) SIN (b), and OSCC (c). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, and the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: ×100-fold, left panel) which is also shown in larger magnification (×400-fold, right panel). PDH pyruvate dehydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue

Immunohistochemical analysis of PDH in normal oral mucosal tissue, oral precursor lesions—hyperplasia, SIN, and invasive OSCC. In comparison with normal tissue/hyperplasia, a significantly increased expression of PDH is observed in SIN III lesions (CIS) and invasive OSCC (p < 0.05, Kruskal–Wallis Test; a and b). In comparison with SIN I, SIN II, and SIN III lesions, no significantly difference of PDH expression is observed in OSCC. PDH expression is significantly increased in OSCC compared with SIN I-III (p = 0.0008, Mann–Whitney U test). Analysis refers to averaged scores. Red line indicates PDH expression results during carcinogenesis. Gray lines show 95 % confidence intervals. The triangles indicate negative samples. Analysis of significant statistically different single values is indicated in the table below (b). SIN III is subdivided into severe dysplasia and carcinoma in situ (CIS). PDH, pyruvate dehydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue
KGDH (OGDH) (Figs. 3, 4) expression was found in all tissue types, normal oral mucosa (n = 14/14, 100 %), oral precursor lesions (simple hyperplasia, n = 21/21, 100 %; squamous intraepithelial neoplasia, SIN I–III, n = 26/35, 74 %), and OSCC specimen (n = 33/46, 72 %). In comparison to normal tissue, hyperplasia, and SIN II lesions, a significantly increased expression of KGDH was observed in invasive OSCC. KGDH expression was significantly increased in OSCC compared with SIN I–III lesions (p = 0.0001, Fig. 4).

Immunohistochemical staining of KGDH (OGDH) in OSCC. Immunohistochemical staining shows representative images of KGDH in N.T. (a), SIN (b), and OSCC (c). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, and the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: ×100-fold, left panel), which is also shown in larger magnification (×400-fold, right panel). KGDH ketoglutarate dehydrogenase; OGDH 2-oxoglutarate dehydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue

Immunohistochemical analysis of KGDH (OGDH) in normal oral mucosal tissue, oral precursor lesions—hyperplasia, SIN, and invasive OSCC. In comparison to normal tissue/hyperplasia and SIN II lesions, a significantly (p < 0.05, Kruskal–Wallis Test; a and b) increased expression of KGDH is observed in invasive OSCC. KGDH expression is significantly increased in OSCC compared with SIN I-III (p = 0.0001, Mann–Whitney U test). Analysis refers to averaged scores. Red line indicates KGDH expression results during carcinogenesis. Gray lines show 95 % confidence intervals. The triangles indicate negative samples. Analysis of significant statistically different single values is indicated in the table below (b). KGDH is subdivided into severe dysplasia (sev. dysplasia) and carcinoma in situ (CIS). KGDH, ketoglutarate dehydrogenase; OGDH, 2-oxoglutarate deydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue
Branched-chain alpha-ketoacid dehydrogenase (BCKDH) (Figs. 5, 6) expression was found in all tissue types, normal oral mucosa (n = 14/14, 100 %), oral precursor lesions (simple hyperplasia, n = 20/21, 95 %; squamous intraepithelial neoplasia, SIN I–III, n = 34/35, 97 %), and OSCC specimen (n = 46/46, 100 %). In comparison with normal tissue/hyperplasia, a significantly increased expression of BCKDH was observed in SIN III lesions (severe dysplasia and CIS), and invasive OSCC. BCKDH expression was significantly increased in OSCC compared with SIN I–III lesions (p = 0.0393, Fig. 6).

Immunohistochemical staining of BCKDH in OSCC. Immunohistochemical staining shows representative images of BCKDH in N.T. (a), SIN (b), and OSCC (c). Brown chromogen color (3,3′-Diaminobenzidine) indicates positive staining, and the blue color shows the nuclear counterstaining by hematoxylin. The square box demonstrates the area of interest (original magnification: ×100-fold, left panel) which is also shown in larger magnification (×400-fold, right panel). BCKDH branched-chain alpha-ketoacid dehydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue

Immunohistochemical analysis of BCKDH in normal oral mucosal tissue, oral precursor lesions—hyperplasia, SIN, and invasive OSCC. In comparison with normal tissue/hyperplasia, a significantly increased expression of BCKDH is observed in SIN III lesions (severe dysplasia and CIS), and invasive OSCC (p < 0.05, Kruskal–Wallis Test; a and b). BCKDH expression is significantly increased in OSCC compared with SIN I-III (p = 0.0393, Mann–Whitney U test). Analysis refers to averaged scores. Red line indicates BCKDH expression results during carcinogenesis. Gray lines show 95 % confidence intervals. The triangles indicate negative samples. Analysis of significant statistically different single values is indicated in the table below (b). SIN III is subdivided into severe dysplasia and carcinoma in situ (CIS). BCKDH branched-chain alpha-ketoacid dehydrogenase; SIN squamous intraepithelial neoplasia; N.T., normal tissue
PDH, KGDH (OGDH), and BCKDH antibody specificity is confirmed by western blot analysis
Western Blot analysis of PDH, KGDH (OGDH), and BCKDH in BICR3 and BICR56 OSCC cell lines confirmed immunohistochemical staining specificity of antibodies used in immunohistochemistry (Fig. 7).

Western blot analysis in normal tissue, BICR3 and BICR56 OSCC cell lines. Western Blot of KGDH (OGDH), BCKDH, and PDH in BICR3 and BICR56 OSCC cell lines confirms immunohistochemical staining specificity of antibodies (left panel, a). Western blot analysis shows increased KGDH (OGDH) (~118 kDa), BCKDH (~46 kDa), and PDH (~38 kDa) expression compared to normal tissue. GAPDH (loading control) is shown as a band of approximately 35 kDa. Densitometric quantification (b) of western blot protein bands (pixel total) is given in the right panel (b). KGDH, ketoglutarate dehydrogenase; OGDH, 2-oxoglutarate deydrogenase; BCKDH, branched-chain alpha-ketoacid dehydrogenase; PDH, pyruvate dehydrogenase; GAPDH, glycerinaldehyd-3-phosphat-dehydrogenase; NHOK, normal human oral keratinocytes
Discussion
In vitro and in vivo studies regarding the function of TDEs TKT (including TKTL1 [12, 30]), KGDH (OGDH), and BCKDH [31, 32] have been associated with proliferation, anabolic, and prosurvival effects in tumor cells (reviewed in [11]). This is in concordance with our results showing increased expression of KGDH (OGDH) and BCKDH in the carcinogenesis of OSCC. However, the total numbers of KGDH positive samples decreased in OSCC, which could be indicative for a decrease of OXPHOS in some tumors leading to the more malignant aggressive metabolic Warburg phenotype [33]. The same phenomenon has been analyzed for PDH expression. Concerning head and neck squamous cell carcinomas (HNSCC) [34, 35] inhibition of PDH activity in cancer cells has been shown to contribute to the Warburg phenotype with subsequent poor outcome of HNSCC patients [34]. Nevertheless, as currently demonstrated in our study cohort [1], glycolysis and the PPP are concurrent upregulated with OXPHOS in OSCC samples. These metabolic pathways lead to the upregulation of the endogenous antioxidant capacity through production of lactate (e.g. by lactate dehydrogenase 5, LDH5 and TKTL1, [12]), the redox couples glutathione/glutathione disulfide [12], NAD/NADH (TCA) and NADP/NADPH (PPP), respectively, protecting tumor cells from apoptosis by increased reactive oxygen species (ROS) [17]. Therefore, elevated TCA activity (e.g. for energy generation by OXPHOS [6] or anaplerosis of fatty acids/amino acids [5, 10]) indicated by PDH and KGDH (OGDH) expression may not lead automatically to increased apoptosis in ‘natural occurring’ (not therapeutic manipulated) cancer cells due to several regulatory mechanisms counteracting ROS [1]. This in concordance with the results published by Koukourakis et al. [36], who demonstrated a high PDH expression with Hypoxia-inducible factor 1-alpha (HIF1-α)/LDH5 upregulation showing increased angiogenic activity of tumors. Concurrent PDH and HIF1/LDH5 upregulation could occur due to genetic events that allow a switch on the entire cellular metabolism. Such tumors with intensively upregulated aerobic and anaerobic metabolic potential were linked with poor postoperative outcome in lung cancer patients [36]. However, in our study cohort, we have no prognostic data demonstrating the outcome of KGDH (OGDH) or PDH positive/negative samples with a hypoxic phenotype (e.g. LDH5) but most of our studied tumors and precancerous lesions stained positive for both metabolic markers [1]. The prognostic impact of predominant hypoxic samples (PDH/KGDH (OGDH) negative, LDH5 positive) compared with tumors showing both metabolic phenotypes (PDH/KGDH (OGDH) positive, LDH5 positive) has to be evaluated.
To the best of our knowledge, no clinical data are available analysing the outcome of cancer patients with increased or decreased expression of KGDH (OGDH) and/or BCKDH in tumors. Though, in hypermetabolic states like cancer diseases, the release of BCAAs provides a pool of amino acids for oxidative energy and for the synthesis of proteins [11, 31]. Hence, analysis of BCKDH and KGDH (OGDH) expression may have prognostic impact of tumor patients and should be performed in further studies.
In the presence of thiamine pyrophosphate (TPP), the E1 subunits of PDH, KGDH (OGDH), and BCKDH perform decarboxylation steps [11, 37–39]. Therefore, we preferentially used antibodies detecting the E1 subunits in our analyses for the description of potential therapeutic targets of antagonistic compounds like oxythiamine [40–42] or oxybenfothiamine.
PDH inactivation is regulated by phosphorylation (p-PDH) whereas PDH is activated by PDH phosphatase [43]. Therefore, although different expression patterns in normal tissue and in the carcinogenesis of OSCC have been analyzed in this study detecting the E1 subunit (PDH, BCKDH), PDH activation status can be finally determined using p-PDH antibodies, which are difficult in the handling of immunohistochemical staining. Intriguingly, Hamabe et al. [43] recently demonstrated that patients in advanced stage colorectal cancer showing activated PDH (p-PDH negative) with concurrent upregulated hexokinase-2 (HK-2) in the tumor tissue have a worse prognosis making PDH inactivation attractive for cancer therapy. These data are in concordance with our observation and the data of Koukourakis et al. [36] that a glycolytic phenotype (HK-2, [1]) is upregulated in parallel to increased aerobic metabolic potential (PDH expression) within the same tumor but contradicts the results by Wigfield et al. [34] in HNSCC patients. Moreover, OXPHOS [5] measured by expression of PDH and KGDH (OGDH) in our study has been demonstrated to be upregulated in proliferating human breast cancer cells compared with non-proliferating (quiescent) tumor cells [32]. Altogether, these facts demonstrate that upregulated aerobic metabolic potential [5, 6, 10, 44] indicated by PDH and KGDH (OGDH) expression is even crucial for tumor development. This hypothesis is further supported that ROS generation by OXPHOS may promote epithelial–mesenchymal transition (EMT) and further metastasis as well as cancer invasion [10, 45].
TDEs (TKTL1 [46], PDH, and BCKDH) may increase the generation of acetyl-CoA, which is crucial for de-novo fatty-acid biosynthesis in proliferating tumor cells [13]. Therefore, targeting TDEs in cancer may decrease lipid synthesis, resistance to oxidative stress, and resistance to energy stress [13].
This is the first study showing TDEs in the carcinogenesis of OSCC. We analyzed increased/strong expression of PDH, KGDH (OGDH), and BCKDH in the multistep carcinogenesis of OSCC indicating thiamine as an important co-factor for cancer metabolism-related pathways in intermediary metabolism [11]. Western Blot analysis of PDH, KGDH (OGDH), and BCKDH confirmed staining specificity of antibodies used in immunohistochemistry. The clonal selection model and cancer stem cell hypothesis are commonly utilized to explain tumor heterogeneity and inherent differences of tumor-regenerating capacity [47]. The clonal selection model of multistep carcinogenesis implies that a random solitary cell undergoes malignant transformation, accumulates multiple mutations and subsequently acquires a survival advantage [48, 49]. Our results of significant upregulation of PDH, KGDH (OGDH), and BCKDH in precursor lesions suggest that a molecular `survival of the fittest´ scenario involving TDEs (including the TCA) plays out in the carcinogenesis of OSCC, making this model the better fit. Therefore, TDEs (TKTL1, PDH, KGDH, BCKDH) may contribute to the concept of competing the PPP [7, 41] and targeted anti-mitochondrial therapies leading consequently to apoptosis in tumor cells [5, 19, 38, 44, 50–52].
OSCC is an aggressive tumor with low response to chemotherapy and basic resistance to most standard of care anticancer drugs [53, 54]. Therefore, the effectiveness of cytostatic chemotherapy (cisplatin, 5-FU) and radiation therapy with subsequent apoptosis in OSCC tumor cells might be enhanced by ROS generation [19, 52]. Accumulation of thiamine has been found to be increased in cancer cells when treated with chemotherapeutic drugs, such as 5-fluorouracil (5-FU) [55] but the mechanism is unknown. Patients who were treated with 5-FU were associated with a thiamine-deficient state [56]. Moreover, hypoxic stress (e.g. indicated by TKTL1 or LDH5 expression) may increase thiamine uptake by the thiamine transporter SLC19A3 in a tumor cells [57]. Independent of hypoxia, pyruvate accumulation [58] and TKTL1 expression [30] may lead to HIF1-α stabilization. Therefore, thiamine uptake and metabolism of tumor cells might also be enhanced in OSCC. In vitro and in vivo studies must show whether 5-FU or hypoxic conditions increase thiamine antagonist uptake like oxythiamine or oxybenfothiamine in OSCC cancer cells. However, during a clinical trial using thiamine antagonists, patients have to be observed for the development of Wernicke’s encephalopathy while therapy [11].
It has been shown that thiamine or thiamine derivates (e.g. benfothiamine, reviewed in [11]) may stimulate TKT and BCKDH activity. In the literature, conflicting results are available for the development of cancer by thiamine as a supplement in a western diet. Likewise, a western diet is characterized in part by excess thiamine supplementation and may be a factor for increased cancer incidence compared with other countries (e.g. Asia, Afrika) [59]. On the other hand, patients with severe malnutrition have exhibited osteosarcoma, and submandibular gland cysts that were cured without recurrence after thiamine supplementation, suggesting a role of thiamine deficiency in tumor development [60]. However, the study conducted by Kabat et al. [61] in 2008 did not reveal a relationship of vitamin B intake (including thiamine) with cancer incidence of the breast, endometrial, ovarian, colorectal, and lung cancer in women. At present, it is hypothetical whether thiamine supplementation impacts the metabolic phenotype of cancer cells [11].
Conclusions
This is the first study showing increased expression of TDEs PDH, KGDH (OGDH), and BCKDH in OSCC. Metabolic targeting of thiamin-depending enzymes (including TKTL1) by antagonistic compounds like oxythiamine or oxybenfothiamine may be a useful strategy to sensitize cancer cells to common OSCC cancer therapies.
Abbreviations
- SIN:
-
Squamous intraepithelial neoplasia
- OSCC:
-
Oral squamous cell carcinoma
- PDH:
-
Pyruvate dehydrogenase
- PPP:
-
Pentose phosphate pathway
- TKT:
-
Transketolase
- TCA:
-
Tricarboxylic acid cycle
- KGDH:
-
Ketoglutarate dehydrogenase
- OGDH:
-
2-oxoglutarate deydrogenase
- BCKDH:
-
Branched-chain alpha-ketoacid dehydrogenase
- TCA:
-
Tricarboxylic acid cycle
References
Grimm M, Cetindis M, Lehmann M, Biegner T, Munz A, Teriete P, et al. Association of cancer metabolism-related proteins with oral carcinogenesis—indications for chemoprevention and metabolic sensitizing of oral squamous cell carcinoma? J Transl Med. 2014;12:208. doi:10.1186/1479-5876-12-208.
Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Investig. 2013;123(9):3678–84. doi:10.1172/JCI69600.
Alfarouk KO, Shayoub ME, Muddathir AK, Elhassan GO, Bashir AH. Evolution of Tumor Metabolism might reflect carcinogenesis as a reverse evolution process (dismantling of multicellularity). Cancers Basel. 2011;3(3):3002–17. doi:10.3390/cancers3033002.
Cetindis M, Biegner T, Munz A, Teriete P, Reinert S, Grimm M. Glutaminolysis and carcinogenesis of oral squamous cell carcinoma. Eur Arch Oto Rhino Laryngol Off J Eur Fed Oto Rhino Laryngol Soc. 2015;. doi:10.1007/s00405-015-3543-7.
Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3(1):1. doi:10.1186/s40170-015-0128-2.
Zheng J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol Lett. 2012;4(6):1151–7. doi:10.3892/ol.2012.928.
Ramos-Montoya A, Lee WN, Bassilian S, Lim S, Trebukhina RV, Kazhyna MV, et al. Pentose phosphate cycle oxidative and nonoxidative balance: a new vulnerable target for overcoming drug resistance in cancer. Int J Cancer J Int Du Cancer. 2006;119(12):2733–41. doi:10.1002/ijc.22227.
Grimm M, Schmitt S, Teriete P, Biegner T, Stenzl A, Hennenlotter J, et al. A biomarker based detection and characterization of carcinomas exploiting two fundamental biophysical mechanisms in mammalian cells. BMC Cancer. 2013;13(1):569. doi:10.1186/1471-2407-13-569.
Grimm M, Munz A, Teriete P, Nadtotschi T, Reinert S. GLUT-1(+)/TKTL1(+) coexpression predicts poor outcome in oral squamous cell carcinoma. Oral Surg Oral Med Oral pathol Oral Radiol. 2014;117(6):743–53. doi:10.1016/j.oooo.2014.02.007.
Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab. 2012;303(8):E1036–52. doi:10.1152/ajpendo.00151.2012.
Zastre JA, Sweet RL, Hanberry BS, Ye S. Linking vitamin B1 with cancer cell metabolism. Cancer Metab. 2013;1(1):16. doi:10.1186/2049-3002-1-16.
Xu X, Zur Hausen A, Coy JF, Lochelt M. Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int J Cancer J Int Du Cancer. 2009;124(6):1330–7. doi:10.1002/ijc.24078.
Santos CR, Schulze A. Lipid metabolism in cancer. The FEBS journal. 2012;279(15):2610–23. doi:10.1111/j.1742-4658.2012.08644.x.
Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409–54. doi:10.1146/annurev.nu.04.070184.002205.
Harris RA, Joshi M, Jeoung NH, Obayashi M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J Nutr. 2005;135(6 Suppl):1527S–30S.
Platell C, Kong SE, McCauley R, Hall JC. Branched-chain amino acids. J Gastroenterol Hepatol. 2000;15(7):706–17.
Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18(20):5585–94. doi:10.1158/1078-0432.CCR-12-0858.
Sattler UG, Mueller-Klieser W. The anti-oxidant capacity of tumour glycolysis. Int J Radiat Biol. 2009;85(11):963–71. doi:10.3109/09553000903258889.
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–91. doi:10.1038/nrd2803.
Tanaka T, Tanaka M, Tanaka T. Oral carcinogenesis and oral cancer chemoprevention: a review. Patholog Res Int. 2011;2011:431246. doi:10.4061/2011/431246.
Driemel O, Hertel K, Reichert TE, Kosmehl H. Current classification of precursor lesions of oral squamous cell carcinoma principles of the WHO classification 2005. Mund Kiefer Gesichtschir. 2006;10(2):89–93. doi:10.1007/s10006-006-0675-3.
Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10(4):267–77. doi:10.1038/nrc2817.
Sobin LH, Ch W, editors. UICC TNM classification of malignant tumors Berlin. 7th ed. Berlin: Springer Verlag; 2010.
Hamilton SR, Aaltonen LA. Pathology and genetics. Tumours of the digestive system. 3rd ed. Lyon: IARC Press; 2000.
Walker RA. Quantification of immunohistochemistry–issues concerning methods, utility and semiquantitative assessment I. Histopathology. 2006;49(4):406–10. doi:10.1111/j.1365-2559.2006.02514.x.
Grimm M, Munz A, Teriete P, Nadtotschi T, Reinert S. GLUT-1 +/TKTL1 + co-expression predicts poor outcome in oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;117(6):743–53.
Edington KG, Loughran OP, Berry IJ, Parkinson EK. Cellular immortality: a late event in the progression of human squamous cell carcinoma of the head and neck associated with p53 alteration and a high frequency of allele loss. Mol Carcinog. 1995;13(4):254–65.
Alexander D, Schafer F, Olbrich M, Friedrich B, Buhring HJ, Hoffmann J, et al. MSCA-1/TNAP selection of human jaw periosteal cells improves their mineralization capacity. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2010;26(6):1073–80. doi:10.1159/000323985.
Grimm M, Alexander D, Munz A, Hoffmann J, Reinert S. Increased LDH5 expression is associated with lymph node metastasis and outcome in oral squamous cell carcinoma. Clin Exp Metastasis. 2013;30(4):529–40. doi:10.1007/s10585-012-9557-2.
Sun W, Liu Y, Glazer CA, Shao C, Bhan S, Demokan S, et al. TKTL1 is activated by promoter hypomethylation and contributes to head and neck squamous cell carcinoma carcinogenesis through increased aerobic glycolysis and HIF1alpha stabilization. Clin Cancer Res Off J Am Assoc Cancer Res. 2010;16(3):857–66. doi:10.1158/1078-0432.CCR-09-2604.
Liu S, Miriyala S, Keaton MA, Jordan CT, Wiedl C, Clair DK, et al. Metabolic effects of acute thiamine depletion are reversed by rapamycin in breast and leukemia cells. PLoS One. 2014;9(1):e85702. doi:10.1371/journal.pone.0085702.
Mandujano-Tinoco EA, Gallardo-Perez JC, Marin-Hernandez A, Moreno-Sanchez R. Rodriguez-Enriquez S (2013) Anti-mitochondrial therapy in human breast cancer multi-cellular spheroids. Biochim Biophys Acta. 1833;3:541–51. doi:10.1016/j.bbamcr.2012.11.013.
Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–90. doi:10.1101/gad.189365.112.
Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer. 2008;98(12):1975–84. doi:10.1038/sj.bjc.6604356.
McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim N, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–8.
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL, Tumor, et al. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 2005;7(1):1–6.
Stacpoole PW. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell. 2012;11(3):371–7. doi:10.1111/j.1474-9726.2012.00805.x.
Stuart SD, Schauble A, Gupta S, Kennedy AD, Keppler BR, Bingham PM, et al. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014;2(1):4. doi:10.1186/2049-3002-2-4.
Harris RA, Joshi M, Jeoung NH. Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem Biophys Res Commun. 2004;313(2):391–6.
Wang J, Zhang X, Ma D, Lee WN, Xiao J, Zhao Y, et al. Inhibition of transketolase by oxythiamine altered dynamics of protein signals in pancreatic cancer cells. Exp Hematol Oncol. 2013;2:18. doi:10.1186/2162-3619-2-18.
Rais B, Comin B, Puigjaner J, Brandes JL, Creppy E, Saboureau D, et al. Oxythiamine and dehydroepiandrosterone induce a G1 phase cycle arrest in Ehrlich’s tumor cells through inhibition of the pentose cycle. FEBS Lett. 1999;456(1):113–8.
Boros L, Puigjaner J, Cascante M, Lee W-N, Brandes J, Bassilian S, et al. Oxythiamine and dehydroepiandrosterone inhibit the nonoxidative synthesis of ribose and tumor cell proliferation. Cancer Res. 1997;57:4242–8.
Hamabe A, Yamamoto H, Konno M, Uemura M, Nishimura J, Hata T, et al. Combined evaluation of hexokinase 2 and phosphorylated pyruvate dehydrogenase-E1alpha in invasive front lesions of colorectal tumors predicts cancer metabolism and patient prognosis. Cancer Sci. 2014;105(9):1100–8. doi:10.1111/cas.12487.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi:10.1016/j.cmet.2007.10.002.
Hurd TR, DeGennaro M, Lehmann R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012;22(2):107–15. doi:10.1016/j.tcb.2011.11.002.
Coy JF, Dressler D, Wilde J, Schubert P. Mutations in the transketolase-like gene TKTL1: clinical implications for neurodegenerative diseases, diabetes and cancer. Clin Lab. 2005;51(5–6):257–73.
Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8(10):755–68. doi:10.1038/nrc2499.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8.
Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. 2007;6(19):2332–8. doi:10.4161/cc.6.19.4914.
Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–6. doi:10.1016/j.ccr.2006.08.015.
Frezza C, Gottlieb E. Mitochondria in cancer: not just innocent bystanders. Semin Cancer Biol. 2009;19(1):4–11. doi:10.1016/j.semcancer.2008.11.008.
Simon H, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–8.
Grimm M. Prognostic value of clinicopathological parameters and outcome in 484 patients with oral squamous cell carcinoma: microvascular invasion (V+) is an independent prognostic factor for OSCC. Clinical Transl Oncol Off Pub Fed Spanish Oncol Soc National Cancer Inst Mexico. 2012;14(11):870–80. doi:10.1007/s12094-012-0867-2.
Perez-Sayans M, Suarez-Penaranda JM, Pilar GD, Barros-Angueira F, Gandara-Rey JM, Garcia-Garcia A. Hypoxia-inducible factors in OSCC. Cancer Lett. 2011;313(1):1–8. doi:10.1016/j.canlet.2011.08.017.
Heier M, Dornish J. Effect of the fluoropyrimidines 5-fluorouracil and doxifluridine on cellular uptake of thiamin. Anticancer Res. 1989;9:1073–7.
Aksoy M, Basu T, Brient J, Dickerson J. Thiamin status of patients treated with drug combinations containing 5-fluorouracil. Eur J Cancer. 1980;16:1041–5.
Sweet R, Paul A, Zastre J. Hypoxia induced upregulation and function of the thiamine transporter, SLC19A3 in a breast cancer cell line. Cancer Biol Ther. 2010;10:1101–11.
Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277(26):23111–5. doi:10.1074/jbc.M202487200M202487200.
Boros L. Population thiamine status and varying cancer rates between western, Asian and African countries. Anticancer Res. 2000;20:2245–8.
Lee B, Yanamandra K, Bocchini J. Thiamin deficiency: a possible major cause of some tumors? Oncol Rep. 2005;14:1589–92.
Kabat G, Miller A, Jain M, Rohan T. Dietary intake of selected B vitamins in relation to risk of major cancers in women. Br J Cancer. 2008;99:816–21.
Acknowledgments
We thank Julia Grimm for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests.
Rights and permissions
About this article

Cite this article
Grimm, M., Calgéer, B., Teriete, P. et al. Targeting thiamine-dependent enzymes for metabolic therapies in oral squamous cell carcinoma?. Clin Transl Oncol 18, 196–205 (2016). https://doi.org/10.1007/s12094-015-1352-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-015-1352-5