
Abstract
Congenital sucrase–isomaltase deficiency (CSID) is a rare autosomal carbohydrate malabsorption disorder caused by mutations in the sucrase–isomaltase gene. While the prevalence of CSID is high in the indigenous populations of Alaska and Greenland, it is imprecise and ambiguous in the Turkish pediatric population. In this cross-sectional case–control study, which is retrospective in nature, next-generation sequencing (NGS) results obtained from records of 94 pediatric patients with chronic nonspecific diarrhea were reviewed. Demographic characteristics, clinical symptoms and treatment responses of those diagnosed with CSID were evaluated. We identified one new, homozygous frame-shift mutation and 10 other heterozygous mutations. Two cases were from the same family and nine were from different families. While the median age at onset of symptoms was 6 months (0–12), median age at diagnosis was 60 months (18–192) with a median delay of 5 years and 5 months (10 months -15 years and 5 months) in diagnosis. Clinical symptoms included diarrhea (100%), abdominal pain (54.5%), vomiting after consuming sucrose (27.2%), diaper dermatitis (36.3%) and growth retardation (81%). Our clinical study revealed that sucrase-isomaltase deficiency may have been underdiagnosed in patients with chronic diarrhea in Turkey. In addition, the frequency of heterozygous mutation carriers was significantly higher than that of homozygous mutation carriers and those with a heterozygous mutations responded well to the treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
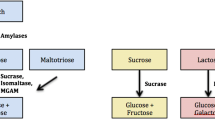
Congenital sucrase–isomaltase deficiency (CSID) is a rare autosomal recessive intestinal disease characterized by decreased sucrase and isomaltase enzyme levels and/or decreased or absent enzyme activities in the small intestine (Reinshagen et al. 2008). It occurs as a result of a homozygous or compound heterozygous mutation in the SI gene on the 3rd chromosome, which encodes the sucrase–isomaltase enzyme protein. The disease has five sub-phenotypes upon differences in genetic mutations. Not all phenotypes have sucrase activity, however, isomaltase activity can range from zero to normal. As lactase activity is also insufficient in most of these phenotypes, lactose intolerance may be often seen along with an increased risk of colon cancer and other gastrointestinal diseases. Osmotic diarrhea, meteorism, abdominal pain, nausea-vomiting and growth retardation are commonly noted in the course of disease due to the absence of enzyme activities in the intestinal lumen. Clinical symptoms usually emerge with increased intake of sucrose-containing foods after initiating supplementary feeding. It is well known that the prevalence of CSID is between 1/500 and 1/2000 in Europe, 10% in Greenland Eskimos, ~3% in Alaska Natives, and rarer in Africa and America (Marcadier et al. 2015; Bell et al. 1973; Ellestad-Sayad et al. 1978).
The gold standard for the diagnosis of CSID is to demonstrate the deficiency of disaccharidase enzyme level and/or its activity level in duodenal biopsies (Treem et al. 2012). This method is invasive and is not available in Turkey. The carbon-13 (C13) labelled sucrose breath test and hydrogen breath test are noninvasive tests that measure C13 labelled CO2 and breath hydrogen in exhaled air, respectively, after oral sucrose loading. Nevertheless, a troubling abdominal pain and diarrhea may be encountered in patients after sucrose is given for these tests (Robayo-Torres et al. 2018). Furthermore, false positives can be seen in small bowel diseases other than CSID.
The screening test for homozygous or compound heterozygous mutations in the SI gene, which encodes the sucrase–isomaltase enzyme protein on the 3rd chromosome, is a new alternative method for the diagnosis of CSID and is available in Turkey. To date, 55 different mutations have been identified; the four most common of these are G1073D, F1745C, V577G and R1124X (Chiruvella et al. 2021).
In this study, we aimed to identify a common mutation for CSID in the Turkish pediatric population admitted to the hospital with chronic diarrhea and to evaluate the demographic characteristics, clinical symptoms and response of the newly diagnosed patients for treatment.
Material and methods
Patients who were admitted to the Pediatric Gastroenterology Department of Dr Sadi Konuk Training and Research Hospital due to chronic diarrhea between May 2021 and January 2022 were evaluated with history, physical examination, stool and blood tests to rule out infectious diarrhea, celiac disease, inflammatory bowel diseases and food allergies and diagnosed with nonspecific chronic diarrhea in compliance with Rome 4 criteria. They also had stool culture, fecal calprotectin and stool parasites investigations, where no evidence of infection or inflammation was noted. There was no reducing agent in stool samples and all had a stool pH above 5. No increase in acute phase reactant, transglutaminase IgA or IgG levels was shown in blood tests. Patients with a history of inflammatory bowel disease, infectious colitis, celiac disease, and food allergy were not included in this study. All our patients underwent oesophagogastroduodenoscopy and their biopsy materials were also evaluated by the same pathologist. In the pathological examination, there was no villus atrophy, crypt hyperplasia, increased intraepithelial lymphocytes or any inclusion body. Since no test for measuring disaccharidase enzyme level or disaccharidase enzyme activity in the biopsy material was available in Turkey, we performed a next-generation sequencing (NGS) analysis for all of our patients.
SI gene mutation analyses were performed by sequencing the gene's coding exons and exon–intron boundaries. Genomic DNA was isolated from peripheral blood cells by standard techniques and amplicon-based target enrichment procedure was performed using the FAST SI NGS Sequencing Kit (Multigen, Turkey) to amplify the coding exons and exon–intron boundaries of the SI gene. All PCR products were sequenced on the Illumina MiniSeq system (San Diego, USA) using the Nextera XT sample preparation kit and Mid-Output cartridge (Illumnina, San Diego, USA). All sequencing results were visualized in IGV software and analysed using HGMD, ClinVar, Varsome, Franklin and GnomAD software and databases. Patients with genetic mutations were evaluated in terms of age, gender, laboratory findings, complaints and response to sacrosidase enzyme treatment. All patients underwent gastroduodenoscopy and duodenal biopsies. Families and patients were informed about genetic testing and their written consent was obtained. Our study was also approved by the ethics committee of of Dr Sadi Konuk Training and Research Hospital. We clearly explained the patients as how to use the sucrosidase enzyme and asked them to write down the frequency of defecation by keeping a diary during the use of the enzyme. In addition, we explained them how to evaluate stool consistency according to the Bristol mayer scale. We obtained written informed consent from all patients.
Results
Between May 2021 and January 2022, 94 children with chronic nonspecific diarrhea were tested for SI gene mutations. All the patients were of Turkish origin and 11.7% had one of the genetic mutations related to CSID. We identified one novel, homozygous frameshift mutation and 10 heterozygous mutations. Two cases were from the same family and nine cases were from different families. One of the cases was diagnosed on the basis of the index case in the family and was found to have similar symptoms to the index case. The mean age of the patients was 79 months (18–1992 months). The mean age of onset of diarrhea was 4.5 months (0–12 months). In addition to diarrhea that started with sucrose-containing food, all patients except cases 1 and 5 had growth retardation. The characteristics of other patients are listed in table 1.
Genetic testing
Case 1 _ case 4: In these cases (they are siblings), a heterozygous c.853G>T (p.E285X) variant was detected. In her sister (case 4), however, this variant was detected in a homozygous pattern. The c.853G>T (p.E285X) variant detected in the patients is listed in the HGMD mutation database (CM1926357) (Capalbo et al. 2019). There is one pathogenic submission about it in the ClinVar mutation database, which was also classified as ‘pathogenic’ by Varsome and Franklin Genoox software. After taking sucrase enzyme extract for three consecutive months, the patient's complaints of diarrhea, abdominal pain, bloating, and vomiting were resolved. The defecation frequency decreased to 1–2 times a day in this patient.
Case 2: The heterozygous c.1730T>G (p.V577G) variant detected in the patient is listed in the HGMD mutation database (CM060473) (Haberman et al. 2017; Ceyhan-Birsoy et al. 2019) and the ClinVar mutation database contains many pathogenic and possibly pathogenic submissions about it. This variant is classified as ‘pathogenic’ by Varsome and Franklin Genoox software. After taking sucrase enzyme extract for three consecutive months, the patient's diarrhea, bloating and vomiting complaints were resolved. In this patient, the daily defecation frequency decreased to 1–2 times and a significant weight gain was achieved with an increase in height.
Case 3: The heterozygous c.4239C>T (p. D1413=) variant detected in the patient is not listed in the HGMD mutation database. There is a submission called as ‘variant of unknown significance’ (VUS) in the ClinVar mutation database. This variant has been classified as ‘probably benign’ by Varsome and ‘VUS’ by Franklin Genoox. The population frequency of this new variant is 1/3689 in the gnomAD database. This variant was considered VUS and response to treatment was evaluated to reclassify this VUS. After taking sucrase enzyme extract for three consecutive months, the patient's complaints of diarrhea, abdominal pain, bloating, and vomiting resolved. The patient’s daily defecation frequency decreased to 1–2 times and a significant weight gain was achieved.
Cases 4 and 5: Cases 4 and 5 are exactly identical and hence they are clubbed in this paragraph. The heterozygous c.5403C>T (p. N1801=) variant detected in the patient is not listed in the HGMD mutation database. There are two submissions (one VUS and one benign) in the ClinVar mutation database. This variant is classified as ‘VUS’ by Varsome and ‘probably benign’ by Franklin Genoox. The population frequency of this new variant is 1/1853 in the gnomAD database. This variant was accepted as VUS and response to treatment was evaluated to reclassify this VUS. After the patient took sucrase enzyme extract for three consecutive months, the complaint of diarrhea and bloating resolved. The patient’s daily defecation frequency decreased to once, which was followed by a significant weight gain.
Case 6: The heterozygous c.2923T>C (p.Y975H) variant detected in the patient is listed in the HGMD mutation database (CM1826958) (Garcia-Etxebarria et al. 2018). There are two submissions (one VUS and one benign) in the ClinVar mutation database. This variant is classified as ‘benign’ by Varsome and ‘probably benign’ by Franklin Genoox. However, the incidence of the disease was higher than expected. After the ACMG criteria changed BS1, these estimators predicted this variant as VUS. The population frequency of this variant is 1/238 in the gnomAD database. This variant was considered VUS and response to treatment was evaluated to reclassify this VUS. After taking sucrase enzyme extract for three consecutive months, the patient's complaints of diarrhea and abdominal pain resolved. The patient’s daily defecation frequency decreased to 1–2 times and a significant weight gain was achieved.
Case 7: Compound heterozygous c.2737-1G>C/c.315G>T (p.W105C) variants were detected in the patient. The c.315G>T (p.W105C) variant is listed in the HGMD mutation database (CM171097) (Gericke et al. 2017). There are two submissions (one probable pathogenic and one VUS) in the ClinVar mutation database. This variant was classified as ‘probably pathogenic’ by Varsome and ‘probably pathogenic’ by Franklin Genoox. The population frequency of this variant is 1/31,369 in the gnomAD database. The c.2737-1G>C variant was not listed in the HGMD and ClinVar mutation databases. This variant has been classified as ‘pathogenic’ by Varsome and ‘probably pathogenic’ by Franklin Genoox (with PM3 modification). The population frequency of this new variant is 1/85,040 in the gnomAD database. Segregation analyses were performed and the detected variants were shown to be in the trans position. After the patient took sucrase enzyme extract for three consecutive months, the complaint of diarrhea resolved. The patient’s daily defecation frequency decreased to one, which was followed by a significant weight gain and height increase.
Case 8: The heterozygous c.4951G>A (p.V1651I) variant detected in the patient is not listed in the HGMD mutation database. There are two submissions (one VUS and one benign) in the ClinVar mutation database. This variant is classified as ‘VUS’ by Varsome and ‘probably benign’ by Franklin Genoox. The population frequency of this new variant is 1/796 in the gnomAD database. This variant was considered VUS and response to treatment was evaluated to reclassify this VUS. After the patient took sucrase enzyme extract for three consecutive months, the complaint of diarrhea resolved. The patient’s daily defecation frequency decreased to 1–2 times. There has been also a substantial weight gain.
Case 9: Heterozygous c.3331A>G (p.I1111V)-c.1020+12G>A variants were detected in the cis position after the genetic workup. The c.3331A>G (p.I1111V) variant is not listed in the HGMD mutation database. There is a submission as VUS in the ClinVar mutation database. This variant is classified as ‘VUS’ by Varsome and ‘VUS’ by Franklin Genoox. The population frequency of this variant is 1/17,932 in the gnomAD database. No variant was listed in the c.1020+12G>A HGMD and ClinVar mutation databases. This variant is classified as ‘VUS’ by Varsome and ‘VUS’ by Franklin Genoox. The population frequency of this new variant is 1/83,198 in the gnomAD database. Segregation analyses were performed and the detected variants were shown to be in the cis position. This variant was considered VUS and response to treatment was evaluated to reclassify this VUS. After taking sucrase enzyme extract for three consecutive months, the patient's complaints of diarrhea and abdominal pain resolved. The patient’s daily defecation frequency decreased to 1–2 times. There was also a significant weight gain and height increase.
Case 10: A heterozygous c.119-6C>T variant was detected in the patient and it was not listed by HGMD and ClinVar databases. This variant is classified as ‘VUS’ by Varsome and ‘VUS’ by Franklin Genoox. The population frequency of this new variant is 1/83,174 in the gnomAD database. This variant was considered as VUS, and response to treatment was evaluated to reclassify this VUS. After the patient received sucrase enzyme extract for three consecutive months, the complaints of diarrhea, abdominal pain and bloating resolved. The patient’s daily defecation frequency decreased to 1–2 times and a significant weight gain was observed.
Case 11: The heterozygous c.2395A>G (p.I799V) variant detected in the patient is not listed in the HGMD mutation database. There are two submissions (one VUS and one probable benign) in the ClinVar mutation database. This variant is classified as ‘VUS’ by Varsome and ‘probably benign’ by Franklin Genoox. The population frequency of this new variant is 1/428 in the gnomAD database. This variant was considered VUS and response to treatment was evaluated to reclassify this VUS. After taking sucrase enzyme extract for three consecutive months, the patient's complaints of diarrhea, abdominal pain, and bloating improved. The patient’s daily defecation frequency decreased to one and a significant weight gain was achieved.
All our patients had diarrhea. The frequency of defecation started to decrease in the first week after sucrosidase treatment was started. We made our patients use sucrosidase for at least three months for treatment response. Stool frequency of all patients decreased and stool consistency improved beginning from the first week of sucrosidase treatment. All of them gained weight while the frequency of defecation decreased to 1–2 per day. The stool consistency changed to type 4 according to the Bristol Mayer scale.
Discussion
CSID is generally regarded as a rare inherited disorder caused by sucrase–isomaltase gene mutations (Weijers et al. 1960). However, the true prevalence of CSID in the Turkish population is still unknown. A case series of only four cases of CSID has been reported so far in the Turkish population, possibly due to underdiagnosis or misdiagnosis (Karakoyun et al. 2015). In two different studies by Gupta et al. (1999), Roberto et al. (2007), ~1000 pediatric duodenal biopsy specimens with normal histologic findings, which belong to children who underwent gastroscopy for different reasons were examined. When an enzymatic activity of < 10% or an enzyme level being < − 1 SDS was evaluated in favour of disaccharidase deficiency, frequency of disaccharidase deficiency was found to be 11% respectively (Gupta et al. 1999). In our study population, the frequency of CSID disease was 11.7%.
When the literature is reviewed, it has been observed that the frequency of CSID is higher in males than females (Zhou et al. 2021). In our study, seven of 11 newly-diagnosed patients were male, consistent with the literature.
In this study, the frequency of symptoms in 65 CSID patients was analysed, it was found that 95% of the patients had diarrhea, 85% had gas and bloating, and 66% had abdominal pain (Treem et al. 2009). In our study, all of our patients had diarrhea. 65% of the patients had gas pain, 55% had abdominal pain and bloating, and 81% had growth retardation.
In a review of the literature, it was claimed that the diagnosis of CSID was made long after the symptoms started in patients, and the reason for this was to reduce the consumption of elementary formula or carbohydrate-containing food given by misdiagnosing. Moreover, it has been also stated that the disaccharides that are not broken down in the small intestines and pass to the colon have the effect of a prebiotic for the colonic bacteria (8th Starch Digestion Consortium 2012). We found that the complaints of nine of our patients started before the postnatal 7th month, although we did not have a patient diagnosed before the age of 1 year. We diagnosed only two of our patients before they were 2 years old. Five of our patients were diagnosed between the ages of 2–6, and four of them over the age of 7 years.
In the study of Karnsaku et al. (2002), duodenal biopsy samples of 44 dyspeptic children were examined and it was found that 22 of the patients had at least one or more of the sucrose, isomaltase and lactase deficiencies (Karnsaku et al. 2002). Frequency of dyspeptic complaints among our newly-diagnosed patients was 55% (6/11).
In a study around 6-year follow-up of CSID patients, it was determined that only 10% of the patients' complaints improved when they were fed with a sucrose-restricted diet, and 60–75% of the patients continued to suffer (Antonowicz et al. 1972). This made us think that dietary restriction is not sufficient and that enzyme replacement therapy is required.
Studies have shown that as the amount of sucrose taken in the diet increases, gas, bloating and diarrhea increase, and sucrose taken together with sacrosidase enzyme replacement does not cause these complaints (8th Starch Digestion Consortium 2012). In all of our patients, diarrhea, bloating, nausea-vomiting, and abdominal pain were completely resolved while taking sacrosidase enzyme treatment and no sucrose restriction was needed. Moreover, weight gain was achieved in all of our patients.
It has been reported in the literature that diarrhea does not improve despite a sucrose-free diet, and the frequency of defecation does not increase in patients who continue to take sucrose with sucrosidase treatment. We did not recommend a sucrose free diet to our patients and they continued their normal diet. They received only sucrosidase as medical treatment.
Our results show that the prevalence of CSID in children with chronic nonspecific diarrhea is quite high. The gastrointestinal symptoms associated with CSID usually emerge after an infant is exposed to sucrose and starch. In our patients, the symptoms of the disease were diarrhea, abdominal pain, bloating, vomiting and growth retardation, and the severity of these symptoms varied. This clinical heterogeneity is associated with residual sucrase–isomaltase enzyme activity resulting from different genetic mutations in CSID, amount of carbohydrate consumption, gastric emptying, and small bowel transit time.
A reduced biopsy disaccharidase enzyme activity has become the gold standard for the diagnosis of CSID. However, biopsy is invasive and requires rapid freezing and transfer of samples to the analytical laboratory. Alternative modalities include the sucrase breath test. Unfortunately, C13 sucrose test or sucrase-isomaltase enzyme activity measurements are not available for fresh intestinal biopsy samples in our country. This shortcoming may partly explain the diagnostic challenge in Turkey. In the last decade, NGS has become a leading player in the diagnosis of rare diseases. NGS technology is available in Turkey, and Alaska Native Medical Center (ANMC) guidelines recommend sequencing the SI gene for the diagnosis of CSID.
In this study, we identified one homozygous mutation and 10 heterozygous carriers. The inheritance pattern of CSID is either based on single mutations in both alleles (homozygous pattern) or is caused by compound heterozygosity. Recently, however, heterozygous carriers with typical symptoms of CSID have been described. Although homozygous and compound heterozygous inheritance traits are well documented in patients with diagnosed CSID, there are a few reports of CSID patients with heterozygous genotypes. However, it is also important to note that normal genetic testing does not rule out a genetic diagnosis of CSID, as not all mutations are identified. ANMC also recommends considering full sequencing of the SI gene if the analysis for known mutations is negative and symptoms still indicate possible CSID.
Our results show that patients with heterozygous VUS variants have similar symptoms and respond better to sacrosidase treatment than those with pathogenic variants. However, literature data on the genotype–phenotype correlation of most CSID variants remain inconclusive, despite several extensively studied pathogenic variants. It is concluded that the normal allele in heterozygotes should express an SI molecule capable of metabolizing sacarose. However, there are some heterozygous cases in the literature presented with low enzymatic sucrase or maltase levels and symptoms of carbohydrate malabsorption.
This study has several limitations. Firstly, it is retrospective review of the experience reported by a pediatric gastroenterologist from a single centre. Another limitation is the small sample size and a relatively short follow-up period.
Conclusion
Finally, we were unable to perform histopathological correlation of disaccharidase deficiency diagnosed with genetic testing in the pediatric population. Although long-term follow-up outcomes of the treatment were not available, sacrosidase therapy was well tolerated and effective in both homozygous and heterozygous patients without any adverse effects observed. Per-meal sacrosidase treatment resulted in relief of gastrointestinal symptoms such as diarrhea, vomiting, and abdominal pain within three months in both heterozygous and homozygous patients. All patients gained weight with treatment. The high rate of CSID (11.7%) in children with chronic nonspecific diarrhea means that this patient group should be evaluated meticulously. Larger-scale future studies with long-term data are needed to investigate the use of sacrosidase in children with heterozygous symptomatic VUS variants.
References
Antonowicz I., Lloyd-Still J. D., Khaw K. T. and Shwachman H. 1972 Congenital sucrase-isomaltase deficiency. Observations over a period of 6 years. Pediatrics 49, 847–853.
Bell R., Draper H. and Bergan J. G. 1973 Sucrose, lactose, and glucose intolerance in northern Alaskan Eskimos. Am. J. Clin. Nutr. 26, 1185–1190.
Capalbo A., Valero R. A., Jimenez-Almazan J., Pardo P. M., Fabiani M., Jime´nez D. et al. 2019 Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLoS Genet. 15, e1008409.
Chiruvella V., Cheema A., Arshad H. M. S., Chan J. T. and Yap J. E. L. 2021 Sucrase-isomaltase deficiency causing persistent bloating and diarrhea in an adult female. Cureus 13, e14349.
Ceyhan-Birsoy O., Murry J. B., Machini K., Lebo M. S., Yu T. W., Fayer S. et al. 2019 interpretation of genomic sequencing results in healthy and Ill newborns: results from the BabySeq project. Am. J. Hum. Genet. 104, 76–93.
Ellestad-Sayad J., Haworth J. and Hildes J. 1978 Disaccharide malabsorption and dietary patterns in two Canadian Eskimo communities. Am. J. Clin. Nutr. 31, 1473–1478.
Garcia-Etxebarria K., Zheng T., Bonfiglio F., Bujanda L., Dlugosz A., Lindberg G. et al. 2018 Increased prevalence of rare sucrase-isomaltase pathogenic variants in irritable bowel syndrome patients. Clin. Gastroenterol. Hepatol. 16, 1673–1676.
Gericke B., Amiri M., Scott C. R. and Naim H. Y. 2017 Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 817–826.
Gupta S. K., Chong S. K. and Fitzgerald J. F. 1999 Disaccharidase Activities in children: normal values and comparison based on symptoms and histologic changes. J. Pediatr. Gastroenterol. Nutr. 28, 246–251.
Haberman Y., Di Segni A., Loberman-Nachum N., Barel O., Kunik V., Eyal E. et al. 2017 Congenital sucrase-isomaltase deficiency: a novel compound heterozygous mutation causing aberrant protein localization. J. Pediatr. Gastroenterol. Nutr. 64, 770–776.
Karakoyun M., Kilicoglu E., Sahan Y., Baran M., Unal F. and Aydogdu S. 2015 Our cases with sucrase isomaltase deficiency. J. Gastroenterol. Dig. Sys. 5, 354–358.
Karnsaku W., Luginbuehl U., Hahn D., Sterchi E., Avery S., Sen P. et al. 2002 Disaccharidase activities in dyspeptic children: biochemical and molecular investigations of maltase-glucoamylase activity. J. Pediatr. Gastroenterol. Nutr. 35, 551–556.
Marcadier J. L., Boland M., Scott C. R., Kheirie Issa K., Wu Z., McIntyre A. D. et al. 2015 Congenital sucrase-isomaltase deficiency: identification of a common Inuit founder mutation. Can. Med. Assoc. J. 187, 102–107.
Reinshagen K., Keller K. M., Haase B., Leeb T., Naim H. Y. and Zimmer K. P. 2008 Mosaic pattern of sucrase isomaltase deficiency in two brothers. Pediatr. Res. 63, 79–83.
Robayo-Torres C. C., Diaz-Sotomayor M., Hamaker B. R., Baker S. S., Chumpitazi B. P., Opekun A. R. et al. 2018 13C-labeled-starch breath test in congenital sucrase-isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 66, 61–64.
Roberto Q. C., Claudia R. T., Zihua A., Bruce H., Andrea Q. and Gary D. B. 2007 Luminal Substrate “Brake” on Mucosal Maltase-glucoamylase Activity Regulates Total Rate of Starch Digestion to Glucose. Journal of Pediatric Gastroenterology and Nutrition 45, 32–43.
Treem W. R. 2012 Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 55, 7–13.
Treem W. R. 2009 Congenital sucrase-isomaltase deficiency (CSID) in the era of Sucraid. J. Pediatr. Gastroenterol. Nutr. 53, 85–88.
Weijers H. A., de Va K. J., Mossel D. A. and Dicke W. K. 1960 Diarrhoea caused by deficiency of sugar-splitting enzymes. Lancet 2, 296–297.
Zhou J., Zhao Y., Qian X., Cheng Y., Cai H., Chen M. et al. 2021 Two novel mutations in the SI gene associated with congenital sucrase-isomaltase deficiency: a case report in China. Front. Pediatr. 9, 731–716.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Shrish Tiwari
Rights and permissions
About this article

Cite this article
Taskin, D.G., Civan, H.A., SarI, E.E. et al. Prevalence of congenital sucrase-isomaltase deficiency in Turkey may be much higher than the estimates. J Genet 102, 31 (2023). https://doi.org/10.1007/s12041-023-01428-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12041-023-01428-8