
Abstract
While the acquisition of drug resistance is often accompanied by fitness costs, Mycobacterium tuberculosis has developed mechanisms to overcome these costs in the form of compensatory mutations. In an attempt to dissect strain-specific differences in biological fitness, 10 M. tuberculosis genomes, representing F15/LAM4/KZN, Beijing, F11 and F28 genotypes were sequenced on the Illumina MiSeq platform. Drug-susceptible F15/LAM4/KZN strains differed by 43 SNPs, demonstrating that heterogeneity exists even among closely-related strains. We found unique, nonsynonymous single-nucleotide polymorphisms (SNPs) in the sigA and grcC1 genes of multidrug resistant (MDR) and XDR F15/LAM4/KZN strains, respectively. The F28 MDR strain harboured a novel ubiA mutation in combination with its embB M306I mutation, which may be related to ethambutol resistance. In addition, it possessed a low-frequency rpoC mutation, suggesting that this strain was in the process of developing compensation. In contrast, no compensatory mutations were identified in Beijing and F11 MDR strains, corroborating its low in vitro fitness. Clinical strains also harboured unique SNPs in a number of important genes associated with virulence, highlighting the need for future studies which examine the correlation of genetic variations with phenotypic diversity. In summary, whole-genome sequencing revealed the presence of fitness-compensatory mutations in F15/LAM4/KZN and F28 genotypes which predominate in MDR and/or extensively drug resistant (XDR) forms in KwaZulu-Natal, South Africa.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Since the discovery of the first antituberculosis (TB) drug about 70 years ago, Mycobacterium tuberculosis has progressively evolved from monodrug resistant to multidrug resistant (MDR), extensively drug resistant (XDR) and recently, totally drug-resistant (TDR) forms, in various parts of the world (WHO 2014). Anti-TB drugs, which are generally designed to target essential, highly conserved genes, may be impeded by chromosomal mutations which alter the target site (Sandgren et al. 2009). The acquisition of drug resistance is often associated with a fitness ‘cost’ as mutations may affect the normal function of target genes, thereby reducing the growth rate of M. tuberculosis (Andersson and Levin 1999; Billington et al. 1999; Gillespie 2002). Moreover, strains carrying identical resistance-encoding mutations may differ in their ability to spread from person-to-person, demonstrating the significance of strain genetic background in the context of fitness (Gagneux et al. 2006; Fenner et al. 2012). Compensatory mutations however, may help restore the fitness of drug-resistant strains (Sherman et al. 1996; de Vos et al. 2013).
The World Health Organisation (WHO) estimates that one out of every 100 people develop active TB disease in South Africa every year (WHO 2014). As a result of poor TB control programmes and treatment strategies, M. tuberculosis strains have been found to disseminate in MDR (resistance to isoniazid and rifampicin) and XDR (MDR in addition to resistance to any fluoroquinolone and at least one injectable drug) forms in South Africa (Pillay and Sturm 2007; Victor et al. 2007; Cox et al. 2010; WHO 2014; Cohen et al. 2015). Moreover, the appearance of XDR-TB is strongly associated with specific strain genotypes (Muller et al. 2013; Cohen et al. 2015), including the F15/LAM4/KZN (KZN) genotype which caused the largest XDR-TB outbreak, in Tugela Ferry, KwaZulu-Natal (Gandhi et al. 2006). Our recent study demonstrated increased fitness for KZN strains in laboratory culture, contrasting with Beijing and F11 MDR strains which were characterized by impeded growth and metabolic profiles (Naidoo and Pillay 2014). Competitive fitness assays also showed consistently lower fitness indices for most drug-resistant strains when compared with susceptible competitor strains (Naidoo and Pillay 2014). In support of this, preliminary sequencing of selected resistance-conferring genes revealed the presence of low or no fitness cost mutations in KZN and F28 strains and high fitness cost mutations in Beijing and F11 strains, however further genetic differences between these strains remain to be determined.
Despite the recent advances in understanding the biological cost of drug resistance, this subject remains incompletely studied among clinical strains. This is in part due to the difficulty in assessing the effects of strain-specific variation on TB pathogenesis. The introduction of next-generation sequencing technology may help decipher the functional and clinical consequences of M. tuberculosis genetic diversity, displayed in the form of single-nucleotide polymorphisms (SNPs), small insertions and deletions (indels) and gene rearrangements (Coscolla and Gagneux 2014). In this study, we correlated whole-genome sequencing (WGS) data with previous experimental fitness data of 10 clinical strains from South Africa. Comparative genomic analysis revealed the existence of significant sequence diversity among clinical strains and helped uncover novel mutations—some of which may aid in the amelioration of fitness costs.
Materials and methods
Clinical M. tuberculosis strains
Ten M. tuberculosis strains were selected for WGS analysis. This included four susceptible, four MDR and two XDR strains for which drug susceptibility testing, spoligotyping and IS6110-RFLP typing were performed previously (Naidoo and Pillay 2014). Strains were classified into four families, namely: F15/LAM4/KZN (\(n = 4\)), Beijing (\(n = 2\)), F11/LAM3 (\(n = 2\)) and F28/S (\(n = 2\)). Mycobacteria were grown in Middlebrook 7H9 broth (Difco) containing 10% oleic albumin dextrose catalase (OADC) (BD Biosciences), 0.5% glycerol (Sigma) and 0.05% Tween-80 (Sigma) at 37\({^{\circ }}\)C, until an optical density (OD\(_{600})\) of 1.
Whole-genome sequencing
Genomic DNA was isolated using the CTAB-lysozyme method (Larsen et al. 2007) and purified using the Genomic DNA Clean & Concentrate \(\hbox {Kit}^{\mathrm{TM}}\) (Zymo Research, Pretoria, South Africa). Whole-genome sequencing was performed on the Illumina MiSeq Sequencer (Illumina) at Inqaba Biotec (Pretoria, South Africa). An Illumina-provided tagmentation kit (Nextera) was used for library preparation. Paired-end massively parallel sequencing (\(2 \times 300\) bp) was carried out with a MiSeq v3 sequencing kit.
Read mapping
Raw sequence reads were trimmed to remove adaptor sequences and low quality sequences using the CLC Genomics Workbench (ver. 7.5.1; QIAGEN, Aarihus, Denmark). The ‘Map Reads to Reference’ function was used to assemble paired reads from all strains to the M. tuberculosis H37Rv reference genome (NCBI accession number N000962) using the following parameters: similarity fraction \(=\) 0.8, length fraction \(=\) 0.5, mismatch cost of 2, deletion cost of 3 and insertion cost of 3. Similarly, KZN strains were mapped against KZN-4207 (NCBI accession number CP001662), a previously sequenced susceptible strain. In addition, resequenced MDR and XDR–KZN strains, V1435 and KZN605, were mapped against previous versions by the Broad Institute (www.broad.mit.edu/annotation/genome/mycobacterium_tuberculosis_spp) (NCBI accession number CP001658 and number NC_018078, respectively). Beijing strains were mapped against HN878 (NCBI accession number NZ_CM001043) and F11 strains were mapped against the F11 reference genome (NCBI accession number NC_009565). Since no reference genome was available at the time for F28, sequences were mapped against H37Rv only. All statistical information was derived from the resulting table generated by the software. The ‘k-mer based tree construction’ function in the CLC Genomics Workbench was used for phylogram construction. Regions with low coverage were filled from the reference sequence (H37Rv) and quality scores were used for conflict resolution.
SNP analysis and confirmation
Drug resistance-conferring mutations were characterized using publicly available databases including TBDreaMDB and the Broad Institute Tuberculosis Drug Resistance Mutation Database, together with recently published reports. Variants were called on the basis of high quality (Phred score of Q30) and more than 70% of the reads reflecting the SNP. Verification of a subset of SNPs was accomplished by PCR using Q5 High-Fidelity DNA Polymerase (NEB) and primers listed in supporting data (table 1 in electronic supplementary material at http://www.ias.ac.in/jgenet/). Sanger sequencing was performed on the ABI 3500XL Genetic Analyzer using forward primers. Bioedit software and chromatograms were used to analyse sequences for the presence of wild type or mutant peaks. Nonsynonymous SNPs (nSNPs) were categorized into functional groups according to Tuberculist (Lew et al. 2011). A selection of genes encoding virulence factors was inspected for nSNPs, across the four strain families (Smith 2003). We used the ‘InDels and Structural Variants Tool’ on paired reads mapped to H37Rv with the following parameters: P value threshold \(=\) 0.0001, maximum number of mismatches \(=\) 2 and minimum number of reads \(=\) 2.
Minimum inhibitory concentration (MIC) testing of isoniazid and ethambutol
Strains were cultured on Middlebrook 7H11 agar (Difco) and adjusted to a McFarland no. 1 turbidity standard in phosphate buffered saline with Tween-80. Serially-diluted suspensions were inoculated onto Middlebrook 7H10 agar containing 10% OADC, 0.5% glycerol and drug concentrations ranging from 0.0625 to 16 mg/L for isoniazid (INH) and 0.4687 to 120 mg/L for ethambutol (EMB). Strains were classified as having low-level or high-level resistance in the presence of 0.2 mg/L or 1 mg/L of INH, respectively. Two critical breakpoints of 5 and 7.5 mg/L were tested for EMB.

Phylogenetic relationships of clinical strains. The tree was constructed by the neighbour-joining method using the CLC Genomics Workbench.
THP-1 macrophage infection with M. tuberculosis strains
F15/LAM4/KZN strains were cultured in Middlebrook 7H9 broth (Difco) supplemented with 10% oleic acid albumin dextrose (OADC) enrichment (Becton Dickinson), 0.5% glycerol and 0.05% Tween-80 until an optical density (\(\hbox {OD}_{600})\) of 0.6–1. The THP-1 human derived macrophage cell line (ATCC TIB-202) was propagated in RPMI-1640 (Lonza) containing 10% foetal bovine serum (Biowest) (RPMI-C) at \(37{^{\circ }}\hbox {C}\) with 5% \(\hbox {CO}_{2}\). Cells were enumerated by trypan blue exclusion prior to reconstitution with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) for overnight differentiation into macrophages. Cells were seeded at \(2 \times 10^{5}\) cells/mL in 24-well cell culture plates (Porvair). Infection media (RPMI-C) was freshly prepared immediately before use. Bacterial cultures were pelleted by centrifugation at 2000 \(\times \) g for 10 min and resuspended in 1 mL of RPMI-C. Bacteria were passaged 10 times through a 21-gauge needle (attached to a 1-mL syringe) and diluted to \(2 \times 10^{5}\) bacteria per mL in RPMI-C. Following overnight incubation, monolayers were washed twice with warm PBS to remove nonadherent cells. Macrophages were infected, in triplicate wells, at a multiplicity of infection (MOI) of 1:1. After 4 h of incubation at \(37{^{\circ }}\hbox {C}\) (5% \(\hbox {CO}_{2})\), monolayers were washed thrice with warm PBS to remove extracellular bacteria and fresh media was added to day 5 wells. For colony forming unit (CFU) enumeration at 4 h and 5 d postinfection, macrophages were lysed for 20 min with 0.1% Triton X-100. Serially-diluted lysates were plated, in triplicate, onto Middlebrook 7H10 medium (Difco) containing 10% OADC and 0.5% glycerol and incubated for 3 weeks at \(37{^{\circ }}\hbox {C}\). Three independent macrophage experiments were performed for each strain. To confirm the MOI during each experiment, bacterial inocula was plated onto Middlebrook 7H10 medium and incubated for 3 weeks. Ethical approval was obtained from the Biomedical Research Ethics Committee, University of KwaZulu-Natal, South Africa (BE258/13).
Results
Whole-genome sequencing
Mapping reports detailing summary statistics of read distribution are described in supporting data (tables 2 and 3 in electronic supplementary material). Sequencing reads have been deposited in the NCBI SRA database under the accession number SRP067784. Sequencing depth in strains ranged from 20 to 60 X with average read length of 230 bp (table 1). Phylogenetic relationships among clinical strains are shown in figure 1.
Mapping of F15/LAM4/KZN strains to KZN-4207
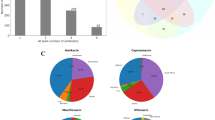
Sequence reads from KZN strains mapped uniquely to 4,394,985 bases (99%) of the KZN-4207 reference genome (table 2 in electronic supplementary material). The susceptible strain, KZN-V9124 had 43 nSNPs relative to KZN-4207, of which 37 were unique and six were shared with the drug-resistant strains (figure 2a). Seven nSNPs were common to the MDR (KZN-V1435) and XDR strains (KZN-605 and KZN-X162) of which four had no definitive link to drug resistance. Drug-resistant strains had previously reported mutations in resistance-conferring genes (Ioerger et al. 2009). Mapping of the XDR strains to KZN-4207 revealed one unique mutation in KZN605 and three in KZN-X162. Of these, one mutation (L129M) identified in the grcC1 gene (Rv0562) of X162, encoding polyprenyl–diphosphate synthase, was not detected in previously-sequenced KZN genomes (figure 2a). Fifteen frameshift mutations were identified in V1435 (MDR) with respect to Broad’s sequence (table 4 in electronic supplementary material), together with a nSNP in sigA, albeit with low read frequency (<70%). Sanger sequencing of sigA revealed the presence of two peaks at the target nucleotide site, confirming it was not a false positive. Fewer differences were evident between KZN605 and the previously-sequenced genome. These included five frameshift mutations (four in conserved hypothetical genes and one in a gene encoding a RifB protein) and two nSNPs in conserved hypothetical genes (table 5 in electronic supplementary material).

Venn diagrams depicting the distribution of nonsynonymous SNPs in clinical strains. (a) F15/LAM4/KZN strains relative to KZN-4207, (b) Beijing strains relative to HN878, (c) F11 strains relative to F11 reference genome and (d) large indels (100–500 bp) in F15/LAM4/KZN strains relative to H37Rv reference genome. DS, drug-susceptible; MDR, multidrug-resistant; XDR, extensively drug-resistant.
Mapping of Beijing strains to HN878
A total of 120 and 135 nSNPs were detected in susceptible and MDR Beijing strains respectively, of which 65 were shared (figure 2b). The majority of shared mutations (46%) occurred within hypothetical proteins, underscoring the need for functional assignment to yet uncharacterized genes. Unique nSNPs in the susceptible strain included those in moaA1 and rpfB, whilst the MDR strain harboured nSNPs in sigK and double mutations in murD.
Mapping of F11 strains to F11 reference
A comparison of the susceptible and MDR F11 strains to the F11 reference genome revealed 54 and 52 nSNPs, respectively (figure 2c). A total of 30 and 28 nSNPs were uniquely found in the susceptible and MDR strain, respectively. Of particular interest, was a Val104Leu mutation detected in the hrp1 gene (Rv2626c) of the susceptible strain. This gene is a member of the dormancy regulon and is strongly upregulated under hypoxic conditions (Sharpe et al. 2008). The MDR strain harboured a mutation in vapC38 (Rv2494) which encodes a toxin previously shown to have increased expression during nutrient starvation, with plausible importance in the establishment of latent infection (Albrethsen et al. 2013).

Functional categorization of mutated genes within different strain families. Number of nSNPs in each functional group is depicted above bars.
Drug resistance and efflux pumps
Reads from the 10 strains were uniquely mapped to an average of 4,341,256 bases (98.4%) of the H37Rv reference genome (table 3 in electronic supplementary material). A total of 50 mutations spanning 25 genes associated with drug resistance, were identified (table 2).
For INH resistance, we analysed katG, ndh, accD6, kasA, Rv1592c and mabA. Drug-resistant KZN and F28 strains harboured the canonical katG Ser315Thr mutation with variable INH MICs (table 3). Although the Beijing-MDR strain had a different substitution at codon 315, it maintained a comparably high INH MIC. In contrast, the F11 MDR strain possessed a rare katG mutation which corresponded to the lowest INH MIC detected in this study. Mutations detected in ndh, accD6, kasA and Rv1592c did not correlate well with INH resistance as these were present in susceptible strains (table 2). A recently characterized, \(\textit{mabA}^{\mathrm{g609a}}\) silent mutation (Ando et al. 2014) was detected in our Beijing MDR strain. Drug-resistant KZN strains harboured a mutation (T1673432A)-8 upstream of mabA (data not shown).
Rifampicin-resistant strains exhibited mutations across five codons in rpoB (table 2). Interestingly, KZN-V1435 had a secondary rpoB mutation which was reflected by 61% of the reads. Sanger sequencing indicated the presence of double nucleotide peaks (G and A) at position 1460, confirming it was a true variant (data not shown). Both susceptible and MDR Beijing strains harboured a synonymous SNP (sSNP) in rpoB (3225T>C). The MDR-F28 strain possessed an rpoC compensatory mutation previously associated with the Ser450Leu rpoB mutation (de Vos et al. 2013), however only 42% of the reads reflected this SNP. Resequencing of rpoC revealed the presence of two peaks at nucleotide position 1448 (data not shown), confirming that it was a true variant.
M. tuberculosis possesses three Emb homologs of which embA and embB are cotranscribed; embC on the other hand is cotranscribed with dprE1, Rv3791 (dprE2) and Rv3792 (aftA) (Goude et al. 2008). Moreover, mutations in ubiA have been shown to increase EMB MICs in wild-type strains and strains with mutated embB codons (Safi et al. 2013). Four embB mutations were observed in KZN, F11 and F28 resistant strains corresponding to different levels of EMB resistance (table 3). The susceptibility of strains V9124, B910, R490 and R104 to EMB was confirmed by MICs equivalent to 1.875 mg/L. Despite having an embB mutation, the F11 MDR strain presented with a lower EMB MIC than the Beijing MDR strain without an embB mutation.
For fluoroquinolone resistance, one of five gyrA mutations correlated with ofloxacin resistance (A90V), while others were common to both susceptible and resistant strains. All SNPs identified in our examination of gyrB, thyA, ddlA and gidB were present in both susceptible and susceptible-resistant strains, thus ruling out their association with drug resistance.
Previous studies have shown associations between drug efflux pumps and pathogenicity/virulence (Piddock 2006; Bina et al. 2009). Of the 36 SNPs occurring in 20 genes encoding drug efflux pumps, 31 (86%) were present in susceptible strains (table 4), suggesting that drug resistance is not always acquired by genetic modification of efflux pumps. Mutations in drrA and ctpB (G23S), a gene believed to encode a putative copper transporter (Knapp et al. 2015), were uniquely present in the drug-resistant KZN strains.
Functional categorization of SNPs
Genes harbouring nSNPs relative to H37Rv were functionally categorized according to Tuberculist. The vast majority of nSNPs, regardless of strain family, were in genes encoding cell and cell wall processes followed by the intermediary metabolism and respiration group, and conserved hypotheticals (figure 3). Closer examination of 13 genes encoding known virulence factors revealed clustering of strain genotypes with mutations in specific genes, i.e. mmaA4 for KZN strains; glcB, fadD26, fadD28, plcA, plcC, mbtB and lipF for Beijing strains; narG and mmpL7 for F11 strains; and plcB for F28 strains (table 5). While Beijing, F11 and F28 harboured distinct mutations in mas (Rv2940c), the KZN group was unique in that it lacked mutations in this gene.
Indels
Indels represent a significant source of phenotypic variability and may enhance the pathogenesis of infectious agents (Liu et al. 2014). Small (<100 bp) and large (100–500 bp) insertions and deletions were analysed in strains relative to H37Rv (data not shown). In total, 562 small indels and 207 large indels were identified in all strains, of which 96 were in PE-PPE-PGRS regions, a significant source of antigenic variability (Sampson 2011). Within the KZN group, two indels, i.e. a large deletion (321 bp) and insertion (124 bp), were shared among all four strains in Rv0145 and Rv2112c, respectively (figure 2d). Susceptible strains, KZN-V9124 and F28-R104 harboured the highest (57) and lowest number (8) of large indels, respectively. A higher degree of overlap was observed within the Beijing group, with 15 shared indels spanning PPE-PE-PGRS regions, hypothetical genes and transmembrane proteins.
Mycobacterial growth in THP-1 macrophages
We used THP-1 macrophages to assess differences in intracellular growth in F15/LAM4/KZN strains. Mycobacterial uptake was determined by expressing the number of CFU at 4 h as a percentage of the initial inoculum, while growth indices were calculated as log CFU/mL on day 5 divided by log CFU/mL at 4 h. Strains did not statistically differ in uptake efficiencies (data not shown). The MDR strain, V1435, had the highest overall intracellular growth at 5 d postinfection (\(P=0.001\) vs V9124; \(P=0.002\) vs X162), while the XDR strain, KZN605 had higher intracellular growth than V9124 (\(P=0.005\)) and X162 (\(P=0.009\)) (figure 4).

Intracellular growth of M. tuberculosis strains of the F15/LAM4/KZN genotype in THP-1 macrophages. Growth indices are expressed as the log CFU/mL on day 5 postinfection divided by the log CFU/mL at 4 h. One-way ANOVA was performed with Tukey’s HSD test, where \(P<0.05\) was considered significant. Data represents the mean ± SD of three independent experiments (**\(P<0.01\)). S, drug-susceptible; M, multidrug-resistant; X, extensively drug-resistant.
Discussion
We have previously shown that specific genotypes, i.e. F15/LAM4/KZN and F28 were better suited to drug-resistant forms than Beijing and F11 strains, in terms of in vitro fitness (Naidoo and Pillay 2014). Here, we addressed the role of resistance-conferring mutations and/or compensatory mutations in the fitness of these clinical strains. Mapping of the KZN susceptible strain (V9124) to the reference genome (KZN-4207) revealed a surprising number (\(n = 43\)) of nucleotide variations. This genetic dissimilarity between both susceptible strains most likely accounts for our previous observations, whereby resistant strains exhibited differential growth when paired with V9124 or KZN-V4207 in competitive growth assays (Naidoo and Pillay 2014).
The KZN-MDR strain, V1435, contained a novel, low-frequency sigA mutation that was absent in the KZN 1435 genome previously sequenced by the Broad Institute. Moreover, this mutation was undetected in other strains sequenced in this study. Since variants with at least 30% of Illumina reads reflecting a SNP can exist as subpopulations within the M. tuberculosis genome (Black et al. 2015), we resequenced the sigA gene. Double peaks were observed in the Sanger chromatogram, confirming it as a true variant. Overexpression of the sigA gene has been shown to enhance intracellular growth in macrophages and mouse lungs (Wu et al. 2004). Interestingly, V1435 which demonstrated comparable growth to V9124, KZN605 and X162 in laboratory culture (Naidoo and Pillay 2014), displayed increased intracellular replication compared to these strains in THP-1 macrophages, however the role of the sigA mutation in this context remains to be determined. Mapping of the XDR strain, KZN605 to a previous version revealed fewer genetic differences that were predominantly observed in hypothetical genes, thus their significance remains elusive. Overall, we speculated that these changes were due to adaptation during laboratory culture (although only two passages were performed prior to DNA isolation) or could reflect differences in read depth.
Comparative assessment of the KZN–XDR strains revealed the presence of some formerly reported, as well as a unique mutation in the grcC1 gene of X162, a mutation that was not reported in any of the previously-sequenced KZN-XDR strains (Ioerger et al. 2009). Intriguingly, this mutation was one of three grcC1 mutations detected in an outbreak strain which infected 69 patients in Bern, Switzerland (Stucki et al. 2015). Orthologues of this essential gene (in M. tuberculosis) are highly conserved across the mycobacteria family, advocating the potential significance of this gene in isoprenoid metabolism (Mann et al. 2012). Mutations that were shared, yet uniquely present in the KZN-XDR strains, were found in Rv2000 (L275P) and Rv3471c (D64E), as previously reported (Ioerger et al. 2009). Overexpression of Rv2000 in a recent study resulted in no change in susceptibility to EMB, kanamycin, rifampicin and streptomycin (He et al. 2015). Thus, their role in drug resistance or epistatic interactions remains to be determined. In addition to the canonical M306V embB mutation, both XDR strains harboured an accessory mutation in Rv3806c (ubiA), which functions to increase EMB resistance without imposing fitness deficits (Safi et al. 2013).
Because mutations in the rifampicin-resistance determining region (RRDR) are associated with fitness costs, the presence of intragenic mutations in highly transmissible strains is worthy of closer inspection. Drug-resistant KZN strains harboured previously-reported secondary (N568S) and tertiary (I1187T) mutations outside the RRDR (Ioerger et al. 2009) which represent putative compensatory mutations (Cohen et al. 2015). Further research with isogenic mutants may provide more insight regarding the level of compensation offered by such mutations in the evolution of fit drug-resistant strains.
HN878 was selected as a reference for Beijing strains on the basis of its pan-susceptibility and widespread inclusion in comparative studies (Manca et al. 2001). Both Beijing strains had gyrA Ser95Thr and katG R463L mutations, verifying that they belonged to principle genetic group 1 (Sreevatsan et al. 1997). The rpoB sSNP present in both Beijing strains was shown to be a phylogenetic marker rather than a predictor of rifampicin resistance (Comas et al. 2012). Synonymous SNPs are believed to have little effect on bacterial phenotype or fitness, however some sSNPs have been shown to play an important role in the generation of alternative transcriptional start sites (Coscolla and Gagneux 2014). In addition to the katG alteration, the Beijing MDR strain had a synonymous G609A mutation in the mabA gene. Commonly found in INH-resistant clinical isolates, this sSNP results in the amplification of transcriptional levels of inhA, and may represent an alternative mechanism of INH resistance in M. tuberculosis (Ando et al. 2014). Similarly, sSNPs in Rv3792 are believed to increase EMB MICs by upregulating embC, explaining the increased EMB MIC of the Beijing MDR strain relative to susceptible strains. Given that Ser315Asn katG mutants exhibit nearly half the catalase–peroxidase activity of Ser315Thr mutants (Unissa et al. 2011) and no compensatory mutations related to INH and rifampicin resistance were identified, the reduced in vitro fitness of the Beijing MDR strain is hardly surprising (Naidoo and Pillay 2014). While the Beijing genotype is highly transmissible in its susceptible form, it does not disseminate equally well in drug-resistant forms (notably XDR) in South Africa, and is most likely due to high fitness costs associated with drug resistance (van der Spuy et al. 2009; Ioerger et al. 2010). This is supported by the diversity of resistance-encoding mutations among XDR Beijing strains from South Africa (Ioerger et al. 2010), Russia (Casali et al. 2014) and Japan (Iwamoto et al. 2008), suggesting that mutations are independently acquired rather than clonally spread (Ioerger et al. 2009).
Clinical strains which harbour identical resistance-conferring mutations may demonstrate variable MICs (Kim et al. 2003). This was the case for KZN and F28 drug-resistant strains which had different INH MICs despite sharing the same katG S315T mutation. While the S315T mutation confers little or no fitness cost, other katG mutants may be compensated by ahpC mutations which ameliorate catalase–peroxidase activity (Sherman et al. 1996). Likewise, the fitness of rifampicin-resistant strains harbouring rpoB mutations may be restored by rpoC compensatory mutations (de Vos et al. 2013). Although both F11 and F28 MDR strains possessed a S450L mutation, only the F28 strain had an rpoC mutation, explaining its high fitness (Naidoo and Pillay 2014). The finding that only 42% of the reads reflected the rpoC SNP indicates that the genome was in the process of acquiring compensation in response to a changing environment (Black et al. 2015). We also found that 36% of the mapped reads from the F28 MDR strain reflected a newly-identified nSNP (A38S) in ubiA, which may augment EMB resistance, similar to KZN–XDR strains which harbour embB 306 and ubiA mutations, however, this requires further study. We surmise that the absence of compensatory ahpC and rpoC mutations to complement the rare katG and S450L rpoB mutations in the F11 MDR strain may explain its marked reduction in fitness, as evidenced by previous growth and metabolic assays (Naidoo and Pillay 2014). Although, the F11 MDR strain had a G406D embB mutation, this strain was susceptible to EMB at the critical breakpoint of 5 mg/L, substantiating that this mutation confers a low level of resistance.
We acknowledge that whole-genome sequencing of a larger number of strains would provide an improved framework for the detection of strain-specific SNPs associated with drug resistance and physiological fitness. However, due to financial constraints, we limited the number of strains sequenced to those used in our previous biological fitness and competitive assays. A selection of low-frequency alleles was verified by Sanger sequencing, all of which proved to be true variants. This supports a recent study by Black et al. (2015) which demonstrated that significant SNPs may be missed at higher frequency cut-offs. Genetic complementation assays are needed for delineating potential mechanisms underlying drug resistance, compensation and fitness of M. tuberculosis variants. The presence of subpopulations within the M. tuberculosis genome raises the question of how genetic heterogeneity shapes treatment outcomes, especially in highly endemic regions, i.e. South Africa, where selection of drug resistance is significantly influenced by TB control programmes (Muller et al. 2013).
During latent infection, M. tuberculosis enters a state of dormancy marked by heightened tolerance to anti-TB drugs and is thought to occur as a result of metabolic shutdown (Gomez and McKinney 2004; Gengenbacher and Kaufmann 2012) rather than resistance mutations (Garton et al. 2008). Mutations in genes related to intermediary respiration and metabolism are thus likely to influence drug resistance, and warrant further exploration in future studies.
Whole-genome sequencing provides an ideal platform for comparative analysis of dominant strains to better understand drug resistance and fitness-compensatory mutations. While there are certain limitations to experimental models, we and others have shown that laboratory studies are crucial in understanding the effects of resistance-encoding mutations on replicative fitness (Gagneux et al. 2006; Spies et al. 2013). Further studies evaluating the fitness of highly circulating strains may provide added insight on their epidemiological success and possibly reveal new ways to combat TB.
In conclusion, clinical strains possess considerable genetic diversity which may influence physiological fitness, i.e. growth characteristics, virulence and transmissibility. The results of this study corroborate our previous work which showed that (i) specific genetic backgrounds are better suited to coping with drug resistance, i.e. F15/LAM4/KZN and F28 strains, and (ii) resistance-conferring mutations with the lowest fitness costs are favoured in highly transmissible strains. Whole-genome sequencing also revealed the presence of novel SNPs in drug-resistant F15/LAM4/KZN strains, some of which may serve as fitness-compensatory mutations, and necessitate further investigation.
References
Albrethsen J., Agner J., Piersma S. R., Hojrup P., Pham T. V., Weldingh K. et al. 2013 Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Mol. Cell. Proteom. 12, 1180–1191.
Andersson D. I. and Levin B. R. 1999 The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2, 489–493.
Ando H., Miyoshi-Akiyama T., Watanabe S. and Kirikae T. 2014 A silent mutation in mabA confers isoniazid resistance on Mycobacterium tuberculosis. Mol. Microbiol. 91, 538–547.
Billington O. J., McHugh T. D. and Gillespie S. H. 1999 Physiological cost of rifampicin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 43, 1866–1869.
Bina X. R., Philippart J. A. and Bina J. E. 2009 Effect of the efflux inhibitors 1-(1-naphthylmethyl)-piperazine and pheny-arginine-beta-naphthylamide on antimicrobial susceptibility and virulence factor production in Vibrio cholera. J. Antimicrob. Chemother. 63, 103–108.
Black P. A., de Vos M., Louw G. E., van der Merwe R. G., Dippenaar A., Streicher E. M. et al. 2015 Whole genome sequencing reveals genomic heterogeneity and antibiotic purification in Mycobacterium tuberculosis isolates. BMC Genomics 16, 857.
Casali N., Nikolayevskyy V., Balabanova Y., Harris S. R., Ignatyeva O., Kontsevaya I. et al. 2014 Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286.
Cohen K. A., Abeel T., McGuire A. M., Desjardins C. A., Munsamy V., Shea T. P. et al. 2015 Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of Mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med. 12, 1–22.
Comas I., Borrell S., Roetzer A., Rose G., Malla B., Kato-Maeda M. et al. 2012 Whole genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identified compensatory mutations in RNA polymerase. Nat. Genet. 44, 106–110.
Coscolla M. and Gagneux S. 2014 Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 26, 431–444.
Cox H. S., McDermid C., Azevedo V., Muller O., Coetzee D., Simpson J. et al. 2010 Epidemic levels of drug resistant tuberculosis (MDR and XDR-TB) in a high HIV prevalence setting in Khayelitsha, South Africa. PLoS One 5, e13901.
de Vos M., Müller B., Borrell S., Black P. A., van Helden P. D., Warren R. M. et al. 2013 Putative compensatory mutations in the rpoC gene of rifampin resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 57, 827–832.
Fenner L., Egger M., Bodmer T., Altpeter E., Zwahlen M., Jaton K. et al. 2012 Effect of mutation and genetic background on drug resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 3047–3053.
Gagneux S., Burgo M. V., DeRiemer K., Encisco A., Munoz S., Hopewell P. C. et al. 2006 Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathol. 2, e61.
Gandhi N. R., Moll A., Sturm A. W., Pawinski R., Govender T., Lalloo U. et al. 2006 Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 368, 1–6.
Garton N. J., Waddell S. J., Sherratt A. L., Lee S., Smith R. J., Senner C. et al. 2008 Cytological and transcript analyses reveal fat and lazy persister-like bacilli in tuberculous sputum. PLoS Med. 5, e75.
Gengenbacher M. and Kaufmann S. H. 2012 Mycobacterium tuberculosis: success through dormancy. FEMS Microbiol. Rev. 36, 514–532.
Gillespie S. H. 2002 Evolution of drug resistance in Mycobacterium tuberculosis: clinical and molecular perspective. Antimicrob. Agents Chemother. 46, 267–274.
Gomez J. E. and McKinney J. D. 2004 M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis 84, 29–44.
Goude R., Amin A. G., Chatterjee D. and Parish T. 2008 The critical role of embC in Mycobacterium tuberculosis. J. Bacteriol. 190, 4335–4341.
He L., Wang X., Cui P., Juin J., Chen J., Zhang W. et al. 2015 ubiA (Rv3806c) encoding DPPR synthase involved in cell wall synthesis is associated with ethambutol resistance in Mycobacterium tuberculosis. Tuberculosis 95, 149–154.
Ioerger T. R., Koo S., No E., Chen X., Larsen M. H., Jacobs Jr W. R. et al. 2009 Genome analysis of multi- and extensively-drug-resistant tuberculosis from KwaZulu-Natal, South Africa. PLoS One 4, 1–9.
Ioerger T. R., Feng Y., Chen X., Dobos K. M., Victor T. C., Streicher E. M. et al. 2010 The non-clonality of drug resistance in Beijing genotype isolates of Mycobacterium tuberculosis from the Western Cape of South Africa. BMC Genomics 11, 670.
Iwamoto T., Yoshida S., Suzuki K. and Wada T. 2008 Population structure analysis of the Mycobacterium tuberculosis Beijing Family indicates an association between certain sublineages and multidrug-resistance. Antimicrob. Agents Chemother. 52, 3805–3809.
Kim S. Y., Park Y. J., Kim W. I., Lee S. H., Ludgerus Chang C., Kang S. J. et al. 2003 Molecular analysis of isoniazid resistance in Mycobacterium tuberculosis isolates recovered from South Korea. Diagn. Microbiol. Infect. Dis. 47, 497–502.
Knapp G. S., Lyubetskaya A., Peterson M. W., Gomes A. L. C., Ma Z., Galagan J. E. et al. 2015 Role of intragenic binding of Camp responsive protein (CRP) in regulation of the succinate dehydrogenase genes Rv0249c-Rv0247c in TB complex mycobacteria. Nucleic Acids Res. 43, 5377–5393.
Larsen M. H., Biermann K., Tandberg S., Hsu T. and Jacobs Jr W. R. 2007 Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 10A.2.1–10a.2.21.
Lew J. M., Kapopoulou A., Jones L. M. and Cole S. T. 2011 TubercuList—10 years after. Tuberc. Edinb. Scotl. 91, 1–7.
Liu F., Hu Y., Wang Q., Li H. M., Gao G. F., Liu C. H. et al. 2014 Comparative genomic analysis of Mycobacterium tuberculosis clinical isolates. BMC Genomics 15, 469.
Manca C., Tsenova L., Bergtold A., Freeman S., Tovey M., Musser J. M. et al. 2001 Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc. Natl. Acad. Sci. USA 98, 5752–5757.
Mann F. M., Xu M., Davenport E. K. and Peters R. J. 2012 Functional characterisation and evolution of the isotuberclosinol operon in Mycobacterium tuberculosis and related mycobacteria. Front. Microbiol. 3, 368.
Muller B., Chihota V. N., Pillay M., Klopper M., Streicher E. M., Coetzee G. et al. 2013 Programmatically selected multidrug-resistant strains drive the emergence of extensively drug-resistant tuberculosis in South Africa. PLoS One 8, 1–9.
Naidoo C. C. and Pillay M. 2014 Increased in vitro fitness of multi- and extensively drug-resistant F15/LAM4/KZN strains of Mycobacterium tuberculosis. Clin. Microbiol. Infect. 20, O361–O369.
Piddock L. J. 2006 Multidrug-resistance efflux pumps? Not just for resistance. Nat. Rev. Microbiol. 4, 629–636.
Pillay M. and Sturm A. W. 2007 Evolution of the extensively drug-resistant F15/LAM4/KZN strain of Mycobacterium tuberculosis in KwaZulu-Natal, South Africa. Clin. Infect. Dis. 45, 1409–1414.
Safi H., Lingaraju S., Amin A., Kim S., Jones M., Holmes M. et al. 2013 Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-\(\upbeta \)-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 45, 1190–1197.
Sampson S. L. 2011 Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin. Dev. Immunol. 2011, 1–11.
Sandgren A., Strong M., Muthukrishnan P., Weiner B. K., Church G. M. and Murray M. B. 2009 Tuberculosis drug resistance mutation database. PLoS Med. 6, 0132–0136.
Sharpe M. L., Gao C., Kendall S. L., Baker E. N. and Lott J. S. 2008 The structure and unusual protein chemistry of hypoxic response protein 1, a latency antigen and highly expressed member of the DosR regulon in Mycobacterium tuberculosis. J. Mol. Biol. 383, 822–836.
Sherman D. R., Mdluli K., Hickey M. J., Arain T. M., Morris S. L., Barry 3rd C. E. et al. 1996 Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 272, 1641–1643.
Smith I. 2003 Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clin. Microbiol. Rev. 16, 463–496.
Spies F. S., von Groll A., Ribeiro A. W., Ramos D. F., Ribeiro M. O., Costa E. R. D. et al. 2013 Biological cost in Mycobacterium tuberculosis with mutations in the rpsL, rrs, rpoB and katG genes. Tuberculosis 94, 150–154.
Sreevatsan S., Pan X., Stockbauer K. E., Connell K. D., Kreiswirth B. N., Whittam T. S. et al. 1997 Restricted structural gene polymorphism in the Mycobacterium tuberculosis complex indicates evolutionary recent global dissemination. Proc. Natl. Acad. Sci. USA 94, 9869–9874.
Stucki D., Ballif M., Bodmer T., Coscolla M., Maurer A., Droz S. et al. 2015 Strain-specific single nucleotide polymorphism typing combined with targeted whole-genome sequencing. J. Infect. Dis. 211, 1306–1316.
Unissa A. N., Narayanan S. and Selvakumar N. 2011 Virulence in isoniazid-resistant clinical isolates of Mycobacterium tuberculosis from South India. Int. J. Mol. Clin. Microbiol. 1, 87–96.
van der Spuy G. D., Kremer K., Ndabambi S. L., Beyers N., Dunbar R., Marais B. J. et al. 2009 Changing Mycobacterium tuberculosis population highlights clade-specific pathogenic characteristics. Tuberculosis 89, 120–125.
Victor T. C., Streicher E. M., Kewley C., Jordaan A. M., van der Spuy G. D., Bosman M. et al. 2007 Spread of an emerging Mycobacterium tuberculosis drug-resistant strain in the Western Cape of South Africa. Int. J. Tuberc. Lung. Dis. 11, 195–201.
WHO 2014 Global Tuberculosis Report. http://www.who.int/tb/publications/global_report/en/
Wu S., Howard S. T., Lakey D. L., Kipnis A., Samten B., Safi H. et al. 2004 The principal sigma factor sigA mediates enhanced growth of Mycobacterium tuberculosis in vivo. Mol. Microbiol. 51, 1551–1562.
Acknowledgements
This study was supported by the National Research Foundation of South Africa (Grant no. 88959 and 90508) and the College of Health Sciences, University of KwaZulu-Natal.
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: Umesh Varshney
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Naidoo, C.C., Pillay, M. Fitness-compensatory mutations facilitate the spread of drug-resistant F15/LAM4/KZN and F28 Mycobacterium tuberculosis strains in KwaZulu-Natal, South Africa. J Genet 96, 599–612 (2017). https://doi.org/10.1007/s12041-017-0805-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-017-0805-8