
Abstract
Acromesomelic dysplasia, type Maroteaux is a disorder characterized by disproportionate short stature predominantly affecting the middle and distal segments of the upper and lower limbs. It is an autosomal recessive disorder due to mutation in NPR2 gene which impairs skeletal growth. To screen the mutations in the gene NPR2, all of its coding exons and splice junction sites were PCR amplified from genomic DNA of affected individuals of four families and sequenced. Four homozygous mutations in four different families were identified. These include three novel mutations including a deletion frameshift mutation (p.Cys586Ter), one nonsense mutation (p.Arg479Ter), one missense mutation (p.Val187Asp) and one reported missense mutation (p.Tyr338Cys). The study describes phenotypes of Indian patients and expands the mutation spectrum of the disorder.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acromesomelic dysplasia, type Maroteaux (AMDM) is a skeletal dysplasia affecting the middle and distal segments of the upper and lower limbs. It is a rare disorder with a prevalence of about 1 in 1,000,000 birth (Langer and Garrett 1980). The birth weight and length of upper and lower limbs are normal. It is usually detected at the age of one when the skeletal growth is slow (Bartels et al. 2004). Radiographic findings include abnormal growth plates and short bones in the limbs detectable by two years of age. Involvement of the axial skeleton differentiates it from acromesomelic dysplasia of Hunter–Thompson type which is caused by mutations in GDF5 gene. Recently, it has been found that the height of the parents are less than the average (Borrelli et al. 1983) and mutation in heterozygote form is the cause of small percentage of cases with short stature (Wang et al. 2015). NPR2 gene was mapped to chromosome 9p13.3 by Kant et al. (1998). It encodes natriuretic peptide receptor B which produces cytoplasmic cyclic guanosine monophosphate (GMP) from guanosine triphosphate (GTP) on binding to C-type natriuretic peptide (CNP). Natriuretic peptides are peptide hormones which on binding to its receptor regulates cardiac growth, blood pressure and endochondral ossification. It is already known that CNP has a role in the skeletal growth. Thus, the mutation in NPR2 gene leading to loss of function impairs skeletal growth and is responsible for AMDM. Its gain of function leads to the overgrowth syndrome (Miura et al. 2014). In the report presented here, we have investigated four Indian families with individuals showing typical features of AMDM. This is the first mutation-proven case study from India.
Materials and methods
Clinical examination and sample collection
All children with AMDM and their available parents were examined and anthropometric measurements were taken. Informed consents were obtained from the parents of the clinically confirmed cases for the molecular genetic analysis of the proband and the family members and also consent was taken for printing their photographs. Blood samples from affected individuals and their parents were collected in EDTA vacutainer.
Molecular analysis
Genomic DNA from the blood samples was extracted using QIAamp DNA blood extraction kit. Sequencing of NPR2 gene was done followed by bioinformatic analysis. Various softwares like SIFT (http://sift.jcvi.org), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2) and mutation taster (http://www.mutationtaster.org) were used to predict the pathogenicity of variants identified. The following are the clinical details of the patients.
Case 1
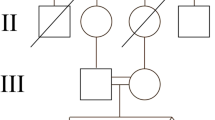
A four year boy presented with short stature and short stubby fingers which was noticed at one year of age. There was no history of any hearing or vision abnormality. He had achieved normal developmental milestones for his age. There was third degree consanguinity in the family (figure 1a). On examination, height was 87 cm (−3.5 standard deviation (SD)), weight was 12.5 kg (−2.5 SD) and head circumference was 47.5 cm (1.6 SD). He had frontal prominence with a flat nasal bridge and had mild pectus carinatum. The arm span was 75 cm (suggesting short limb dwarfism) and upper segment / lower segment was 1.28 (expected: 1.25). He had brachydactyly and broad great toes. Movements at elbows were normal. Radiological findings showed midface hypoplasia. Forearm bones were not curved, but the lower end of ulna was short. Metacarpals and phalanges were short and broad. Vertebrae showed wedging posteriorly as dorsal parts of vertebrae were shorter than ventral parts (figure 1b). Iliac wings were flared. There was a similar history in his aunt’s (father’s sister) family also. Three offspring of aunt were having short stature with short stubby fingers. There was a history of fourth degree consanguinity in aunt. None of the other affected members were examined. Both parents were short; father was 153 cm (−4.75 SD), mother was 148 cm (−2 SD); the mid-parental height was 157 cm (at ∼−3.5 SD).

(a) Pedigree of family 1. (b) Clinical photographs and radiographs of case 1. Midface hypoplasia, low set ears, broad great toes, acral shortening. Platyspondyly with short posterior ends of vertebrae, flared iliac wings and short ulna.
Case 2
A one and a half-year old boy of consanguineous parents (third degree) was referred for evaluation of short stature (figure 2a). There was no satisfactory height gain after birth as noticed by the parents. The social, language and fine motor development were within normal limits. His mother’s height was also short and the father was not examined. On examination, his weight was 8 kg (−2.5 SD), length was 67 cm (−4.5 SD) and head circumference was 47 cm (+ 0.4 SD). He had prominent forehead, midface hypoplasia and low set ears. The extension at elbow was limited and great toes were broad. Radiographs showed bent radius and ulna, ovoid vertebrae with shorter dorsal height than the ventral height (figure 2b). Iliac wings were flared.

(a) Pedigree of family 2. (b) Clinical photographs and radiographs of case 2. Midface hypoplasia, prominent fore head, low set ears, elbow deformities, ovoid vertebrae and curved forearm bones are acromesomelic.
Case 3
A 11-year old boy presented with disproportionate short stature. Parents were nonconsanguineous (figure 3a), had apparently normal development and cognitive function. On examination, his height was 95 cm (−8 SD) and head circumference was 54 cm (1.0 SD) and weight was 15 kg (−5 SD). He had broad nasal bridges, slight, thick lips, low set ears and midface hypoplasia and great toes were broad. Limbs were short and had lumbar lordosis. Elbow showed restriction of terminal extension. Radiographs showed thick ribs and relatively long clavicles. Radiograph of thoracolumbar spine shows platspondyly with increased intervertebral spaces in lumbar region. Vertebral bodies are flattered posteriorly. Short metacarpals and phalanges give them the appearance of being broad (figure 3b). Lower ends of ulna were short and slanting. Iliac wings are flared with notch above acetabular roof with sclerosis of acetabular roof.

(a) Pedigree of family 3. (b) Clinical photographs and radiographs of case 3. Acromesomelic shortening, midface hypoplasia, low set ears, platyspondyly and posterior wedging of vertebrae. Note short metacarpals, phalanges, short ulna and slanting distal end.
Case 4
A one-year old girl was brought by her parents with a complaint of short stature noticed since early infancy. Parents were consanguineous (figure 4a). She had apparently normal development and cognitive function. On examination, her length was 61 cm (−5 SD) and head circumference was 44.5 cm (−0.4 SD) and weight was 5.2 kg (−5 SD). On examination, the child had prominent forehead. Extension of elbow was restricted. There was mesomelic and acromelic shortening. Radiographs showed shortened metacarpals and phalanges and the epiphysis was cone-shaped (figure 4b). Radiograph of the forearm also showed shortened radius and ulna. Spine showed anterior beaking of the vertebrae in the thoracolumbar region. There was similar complaint of short stature in her elder brother who is 7 years old. The elder brother’s height was 84 cm (−8 SD), weight was 14.2 kg (−4.4 SD) and head circumference 51 cm (−1.3 SD). Parents were consanguineous, were first cousins and were also short, father’s height was 156.3 cm (0.01 SD) and mother’s height was 145 cm (−3.8 SD).

(a) Pedigree of family 4. (b) Clinical photographs and radiographs of case 4. Midface hypoplasia, prominent fore head, brachydactyly, elbow deformities, ovoid vertebrae and curved forearm bones are acromesomelic.
Results
Mutation analysis
Mutation analysis of NPR2 gene revealed that all the patients were homozygous with different mutations in each family. A novel nonsense mutation was identified (p.Arg479Ter) in case 1 (figure 5a) and a novel deletion in case 2, which lead to frameshift mutation (p.Cys586Ter) (figure 5b). Both parents of case 1, case 4 and mother of case 2 and case 3 (father sample not available) were found to be carriers of the mutations. A missense novel mutation c.560T >A (p.Val187Asp) was identified in case 3 (figure 5c). Mother was found to be heterozygous for the same mutation (father sample was not available). One human genome mutation database (HGMD) reported mutation c.1013A >G (p.Tyr338Cys) was found in case 4 (figure 5d), which was also confirmed in the affected brother. Various bioinformatics tools like mutation taster, Polyphen-2 and SIFT also predicted it to be pathogenic. All were novel mutations, not reported in 1000 genomes, HGMD and human genome variation (HGV) database.

(a) Case 1 showing nonsense homozygous mutation p.R479X. Mother and father are carriers for the same mutation. (b) Case 2 showing novel homozygous [del C] frameshift mutation (p.Cys586X fs1). Mother is carrier for the same mutation. (c) Case 3 showing novel missense homozygous mutation (V187D). Mother is carrier for the same mutation. (d) Case 4 showing missense homozygous mutation (Y338C). Mother and father are carriers for the same mutation.
Discussion
Here, we describe four children with mutation proved acromesomelic dysplasia, Marteaux type. All four cases had characteristic clinical features like acromesomelic involvement including brachydactyly and broad great toes. Acromesomelic shortening, platyspondyly and broad great toes are the consistent features. One of the cases reported by Khan et al. (2012) had long face. All four cases in our study had mild midface hypoplasia and depressed and broad nasal bridge. Cases 2 and 3 had low set ears. Case 3 had thick lips and slight coarse appearance of face. All had markedly short hands and fingers with great toes. Mesomelic involvement varied. Except case 1, all had restriction of terminal extension of elbows. Case 2 had curved radius and ulna, while other two cases had short distal ends of ulna as an evidence of mesomelic involvement. Lower ends of ulna of case 3 were slanting. The spinal changes differentiate AMDM from the Hunter–Thompson type. The characteristic vertebral anomaly of platyspondyly and posterior wedging due to shorter height of dorsal side of vertebrae than that of ventral end was seen in all cases. In case 2, who was one and half-year of age had ovoid vertebrae and the posterior wedging was appreciable. Short stature becomes obvious at around one year of age in acromesomelic dysplasia and becomes pronounced with increasing age. The case 1, who was four-year old had height around −4.5 SD, while 11-year old child (case 3) had height of −8 SD, and case 4 was 1-year old with height of −5 SD indicating severe short stature as final height. Brother of case 4 (11-year old) also had very short stature (−8 SD).
It has been noticed that the carrier parents of individuals with AMDM are short. This were true in our cases as well as the heights of parents between −2 to −4 SD. Olney et al. (2006) showed that heterozygous mutations in NPR2 are associated with short stature. Similarly, Vasques et al. (2013) identified missense heterozygous NPR2 mutations (p.Ser76Pro, p.Arg263Pro and p.Arg819Cys) in three of 47 (6%) Brazilian children, initially classified as idiopathic short stature (ISS). Recently, Hisado-Oliva et al. (2015) also identified heterozygous NPR2 mutations in 3% cases of ISS. They found heterozygous NPR2 mutations in seven of 173 cases with features of Léri–Weill dyschondrosteosis (LWD) indicating that heterozygotes of NPR2 mutations not only have short stature, but also some mesomelic involvement suggesting phenotypic overlap with LWD. A Japanese group also identified two missense heterozygous NPR2 mutations (p.Arg110Cys and p.Gln417Glu) in two of 101 patients with short stature (Amano et al. 2014). Although, it has shown that this gene is also expressed in brain, adrenal gland, blood vessels and uterus (Tamura et al. 2000); no systems other than bones are involved in AMDM. In addition to AMDM caused by homozygous NPR2 mutations, recent publications indicated that dominant negative and gain-of-function heterozygous NPR2 mutations are responsible for short and tall stature phenotypes, respectively.
Two previous studies from Pakistan have reported mutations in nine Asian cases (Khan et al. 2012; Irfanullah et al. 2015). In five of them, there was a common mutation and a common haplotype suggesting founder effect. Being a rare disorder, presence of consanguinity in three of the four families is not unusual. Even though, there was no consanguinity in case 3, the proband was homozygous. This may be due to hidden consanguinity or a custom of inbreeding over centuries. This study highlights the consistency of phenotype as well as variability of mesomelic involvement and adds mild facial phenotype as additional feature and three novel mutations to the mutational spectrum causing AMDM.
References
Amano N., Mukai T., Ito Y., Narumi S., Tanaka T., Yokoya S. et al. 2014 Identification and functional characterization of two novel NPR2 mutations in Japanese patients with short stature. J. Clin. Endocrinol. Metab. 99, E713–E718.
Bartels C. F., Bükülmez H., Padayatti P., Rhee D. K., van Ravenswaaij-Arts C., Pauli R. M. et al. 2004 Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am. J. Hum. Genet. 75, 27–34.
Borrelli P., Fasanelli S. and Marini R. 1983 Acromesomelic dwarfism in a child with an interesting family history. Pediatr. Radiol. 13, 165–168.
Hisado-Oliva A., Garre-Vázquez A. I., Santaolalla-Caballero F., Belinchón A., Barreda-Bonis A. C., Vasques G. A. et al. 2015 Heterozygous NPR2 mutations cause disproportionate short stature, similar to Léri–Weill dyschondrosteosis. J. Clin. Endocrinol. Metab. 100, E1133–E1142.
Irfanullah U. M., Khan S. and Ahmad W. 2015 Homozygous sequence variants in the NPR2 gene underlying acromesomelic dysplasia Maroteaux type (AMDM) in consanguineous families. Ann. Hum. Genet. 79, 238–244.
Kant S. G., Polinkovsky A., Mundlos S., Zabel B., Thomeer R. T., Zonderland H. M. et al. 1998 Acromesomelic dysplasia Maroteaux type maps to human chromosome 9. Am. J. Hum. Genet. 63, 155–162.
Khan S., Ali R. H., Abbasi S., Nawaz M., Muhammad N. and Ahmad W. 2012 Novel mutations in natriuretic peptide receptor-2 gene underlie acromesomelic dysplasia, type Maroteaux. BMC Med. Genet. 13, 44.
Langer L. O. and Garrett R. T. 1980 Acromesomelic dysplasia. Radiology 137, 349–355.
Miura K., Kim O. H., Lee H. R., Namba N., Michigami T., Yoo W. J. et al. 2014 Overgrowth syndrome associated with a gain-of-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am. J. Med. Genet. A 164A, 156–163.
Olney R. C., Bükülmez H., Bartels C. F., Prickett T. C., Espiner E. A., Potter L. R. et al. 2006 Heterozygous mutations in natriuretic peptide receptor-B (NPR2) are associated with short stature. J. Clin. Endocrinol. Metab. 91, 1229–1232.
Tamura N., Ogawa Y., Chusho H., Nakamura K., Nakao K., Suda M. et al. 2000 Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 97, 4239–4344.
Vasques G. A., Amano N., Docko A. J., Funari M. F., Quedas E. P., Nishi M. Y. et al. 2013 Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature in patients initially classified as idiopathic short stature. J. Clin. Endocrinol. Metab. 98, E1636–E1644.
Wang S. R., Jacobsen C. M., Carmichael H., Edmund A. B., Robinson J. W., Olney R. C. et al. 2015 Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum. Mutat. 36, 474–481.
Acknowledgements
We sincerely thank the cooperation of patient’s families and acknowledge the Indian Council of Medical Research, New Delhi, for funding (BMS-63/8/2010).
Author information
Authors and Affiliations
Corresponding author
Additional information
Corresponding editor: S. Ganesh
[Srivastava P., Tuteja M., Dalal A., Mandal K. and Phadke S. R. 2016 Novel mutations in the transmembrane natriuretic peptide receptor NPR-B gene in four Indian families with acromesomelic dysplasia, type Maroteaux. J. Genet. 95, xx–xx]
Rights and permissions
About this article

Cite this article
SRIVASTAVA, P., TUTEJA, M., DALAL, A. et al. Novel mutations in the transmembrane natriuretic peptide receptor NPR-B gene in four Indian families with acromesomelic dysplasia, type Maroteaux. J Genet 95, 905–909 (2016). https://doi.org/10.1007/s12041-016-0715-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-016-0715-1