
Abstract
A number of genetic maps for Fusarium wilt resistance in chickpea have been reported in earlier studies, however QTLs identified for Fusarium wilt resistance were unstable. Hence, the present study aims to map novel molecular markers and to identify QTLs for Fusarium wilt resistance in chickpea. An intraspecific linkage map of chickpea (Cicer arietinum L.) was constructed using F 10–F 11 recombinant inbred lines (RILs) derived from a cross between K850 and WR315 segregating for H 2 locus. A set of 31 polymorphic simple sequence repeat (SSR) markers obtained by screening 300 SSRs and were used for genotyping. The linkage map had four linkage groups and coverage of 690 cM with a marker density of 5.72 cM. The RILs were screened for their wilt reaction across two seasons in wilt sick plot at International Crop Research Institute for Semi-Arid Tropics (ICRISAT), Hyderabad, India. Five major quantitative trait loci (QTLs) were detected in both seasons for late wilting (60 days after sowing). A stable QTL (GSSR 18-TC14801) for wilt resistance was identified in both the seasons, and the QTL explained a variance of 69.80 and 60.80% in 2007 and 2008 rabi respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chickpea (Cicer arietinum, 2 n= 2 x= 16) is a major cool season legume crop of India with a genome size of ∼738 Mbp (Jain et al. 2013; Varshney et al. 2013). Though about 65% of the world acreage and 68% production are accounted in India, the production is still not adequate to meet the domestic demand due to its low productivity (850 kg/ha). The major causes for low productivity of chickpea are low yield potential and susceptibility of improved present day cultivars to various biotic and abiotic stresses (Gowda et al. 2011). Fusarium wilt, caused by Fusarium oxysporum ciceri(FOC), can cause an annual yield losses of up to 10–15%, under epidemics can be devastating and may cause 100% yield loss under favourable conditions. Among eight races reported till now (Jimenez-Diaz et al. 1993), race 1A, is more prevalent in peninsular India in epidemic form. Genetics of resistance to race 1A (FOC1) suggested that two to three major independent loci H 1, H 2 and H 3 (Upadhyaya et al. 1983a, 1983b; Singh et al. 1987; Brinda and Ravikumar 2005) govern wilt resistance in chickpea. Dominant alleles at both H 1 and H 2 loci cause early wilting and dominance at any one locus (either H 1 or H 2) lead to late wilting, while homozygous recessive at H 1 and H 2 loci confer complete resistance (Brinda and Ravikumar 2005). Even though high level of resistance to FOC race 1A is available in the cultivated species, the progress in the development of high-yielding wilt-resistant varieties is very slow. Since, the selection for wilt resistance in segregating population and maintenance of wilt sick plot is difficult task, hence marker-assisted selection (MAS) will be an efficient strategy to accelerate pyramiding of different resistance genes for Fusarium wilt resistance in chickpea. In chickpea, a large number of molecular markers, molecular maps and QTLs linked to Fusarium wilt resistance are available (Millan et al. 2010; Nayak et al. 2010; Gujaria et al. 2011; Thudi et al. 2011; Choudhary et al. 2012; Hiremath et al. 2012; Barman et al. 2014; Patil et al. 2014). However, these reported QTLs are derived from mapping studies with limited population size and the identified QTLs were not validated across environments and in different genetic background. Hence, it is important to explore novel markers to develop linkage maps and to identify stable QTLs linked to Fusarium wilt resistance for reliable selection of resistant genotypes from segregating populations.
In this context, the study was conducted to develop a linkage map using genic and genomic simple sequence repeat (SSR) markers to tag H 2 locus and to identify QTLs linked to wilt resistance for race 1A, which is prevalent in peninsular India.
Materials and methods
Plant material and DNA extraction
The experimental material consist of a set of 141 intraspecific F 10–F 11 recombinant inbred lines (RILs) segregating for (H 2 locus) wilt resistance derived by single seed descent method from intraspecific cross between K850 (susceptible late wilter, h 1 h 1 H 2 H 2) and WR315 (resistant, h 1 h 1 h 2 h 2) (Upadhyaya et al. 1983a). Good quality genomic DNA was extracted from vegetative buds and young leaves of RILs along with their parental genotypes by following cetyl trimethyl ammonium bromide (CTAB) method of extraction with slight modifications (Doyle and Doyle 1987). DNA quantification was done by nanodrop (Eppendorf Biospectrometer) and stored in −20 ∘C freezer for genotyping.
Phenotyping of RILs for Fusarium wilt reaction
The RILs were phenotyped for wilt reaction, based on the evaluation made over two seasons rabi 2007 (F 7) and 2008 rabi (F 8) in wilt sick plot at International Crop Research Institute for Arid and Semi-arid Tropics (ICRISAT), Patancheru, Hyderabad, India (Sharma et al. 2005; Ravikumar et al. 2007). The wilt incidence was measured on the basis of number of wilted plants to the total number of plants in each row and expressed as per cent wilt incidence. The average of wilt incidence was calculated based on values from two replications. The percentage wilting was recorded on 30 days after sowing (DAS) and 60 DAS (Haware and Nene 1982). The death due to wilting was confirmed by observing the discolouration of the xylem by uprooting the plants.
SSR marker analysis
A set of 300 novel microsatellite markers (table 1 in electronic supplementary material at http://www.ias.ac.in/jgenet/) developed at National Institute of Plant Genome Research (NIPGR), New Delhi, along with earlier reported SSR markers were used to survey parental polymorphism (Jhanwar et al. 2012; Kujur et al. 2013). The genic SSRs were developed from transcriptome sequence of various tissues of chickpea variety ICC4958, using next generation sequencing platforms (Garg et al. 2011). The genomic markers are based on the draft genome sequence of Desi chickpea (Jain et al. 2013).
The parental lines K850 and WR315 were tested for polymorphisim with SSR markers in 15 μL reactions using thermal cycler (Mastercycler gradient, Eppendorf, Hamburg, Germany). The PCR products from each tube along with 5 μL of loading dye were separated electrophoretically using 3.0% agarose gels containing 0.05 μg/mL ethidium bromide. The amplification products were examined under UV light and photographed using Alpha digidoc 1000 system (Alpha Innotech Corporation, USA) gel documentation system (figures 1 and 2). The polymorphic markers were used to genotype all the RILs and the data generated was recorded was subsequently used for mapping studies.

Segregation pattern of genic marker (EST-SSR 3) across chickpea RILs and parents K850 (K) and WR315 (W).

Segregation pattern of genomic marker (GSSR 50) across chickpea RILs and parents K850 (K) and WR315 (W).
Linkage and QTL analysis
The segregation of markers was tested for goodness of fit to the expected ratio of 1:1 using chi-square (χ 2) test (P < 0.05). Linkage mapping was performed using G-Mendel 2.0 programme of iMAS 1.0.1 (integrated marker assisted selection) developed by ICRISAT, Hyderabad. A minimum LOD of 2.5 and maximum recombination fraction of 0.4 were set as threshold value for linkage group determination. The marker ordering is validated by Monte Carlo and bootstrap methods. Haldane mapping function was used to convert recombination fraction into map distances in centimorgans (cM).
QTL analysis was performed using QTL Cartographer ver. 2.5 (Basten et al. 1994). The composite interval mapping was used to identify putative Fusarium wilt resistance QTLs in every linkage group considering the wilt data of 2007 and 2008. A walk speed of 2 cM and a minimum LOD score of 3.0 were used for declaring the presence of a putative QTL. The presence of putative QTL in an interval was tested by using a critical value for LOD threshold of 3.0 as determined by WQTL Cartographer using the Bonferroni chi-square approximation (Zeng 1994).
Results
Phenotyping of RILs for Fusarium wilt reaction
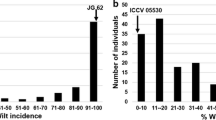
In the present investigation, 141 RILs derived from the cross between WR315 (resistant parent) and K850 (late wilter) were phenotyped for wilt incidence. At 60 DAS during 2007 and 2008 seasons, the distribution of RILs showed continuous distribution with respect to per cent of wilt incidence in both seasons (figure 3).

Frequency distribution of RILs for Fusarium wilt incidence at 60 DAS during 2007 and 2008 rabi seasons.
Linkage map construction
Among 300 SSRs screened for parental polymorphism, a total of 280 (93.3%) SSRs produced scorable amplicons, of which 31 (10.3%) primers were found to be consistently polymorphic between the parental genotypes. The 31 polymorphic primers consist of 23 genomic SSRs and eight genic SSRs. Among the 23 polymorphic genomic SSRs, 15 were novel and remaining eight were mapped in earlier studies using different mapping populations, whereas all the eight genic SSRs used in the present study were novel and were not mapped in any of the earlier studies.
All the markers analysed except four showed expected 1:1 ratio (data not shown). In linkage analysis 23 marker were mapped to four linkage groups covering a total genetic distance of 690 cM with an average marker density of 30 cM. The length of the linkage group ranged from 12.8 (LG 2) to 588.60 cM (LG 1). The number of markers mapped per linkage group varied from 2 (LG 2) to 14 (LG 1). The highest marker density was observed in LG 2 with an average marker density of 6.4 cM and the least marker density of 42.04 cM was observed in LG 1 (table 1).
QTLs for Fusarium wilt resistance
The current linkage map was used to detect QTLs using the data obtained on wilt reaction of RILs for two years. A total of five major QTLs (LOD ≥ 3.0) for late wilting (60 DAS) of which, two QTLs (GSSR 18-TC14801 and GSSR 11-EST SSR 3) in 2007 rabi and three QTLs (TR 24-EST SSR 21, EST SSR 21-EST SSR 65 and GSSR 18-TC14801) were identified in 2008 rabi (table 2). The QTLs GSSR 18-TC14801 and GSSR 11-EST SSR 3 explained phenotypic variance of 69.80 and 68.80% with an additive gene effects of −25.0 and 24.4, respectively. Similarly, three QTLs such as TR 24-EST SSR 21, EST SSR 21-EST SSR 65 and GSSR 18-TC14801, explained phenotypic variance of 56.80, 56.20 and 60.80% with an additive gene effects of 21.98, 21.94 and −22.33, respectively (figure 4).

Genetic linkage map depicting marker position and QTLs associated with Fusarium wilt resistance in chickpea.
Discussion
Resistance to F. oxysporiumrace 1 is reported to be controlled by 2–3 genes (Upadhyaya et al. 1983a, 1983b; Singh et al. 1987; Brinda and Ravikumar 2005). The presence of two major genes H 1 and H 2 in dominant homozygous conditions cause early wilting and any one in dominant condition causes late wilting (Brinda and Ravikumar 2005). Mayer et al. (1997) first reported linkage of an allele-specific marker (CS27A) to H 1 locus of FOC 1. However, the mapping population segregate for only H 2 locus and late wilting and the QTLs identified in this study targets the locus other than H 1 locus.
The continuous distribution of RILs with respect to per cent of wilt incidence in both the seasons indicate involvement of many minor genes along with major genes governing the trait. At 60 DAS, majority of RILs (>60%) showed more than 50% wilt incidence which confirms genotypic constitution of the parents involved such as WR315 (h 1 h 1 h 2 h 2) and K850 (h 1 h 1 H 2 H 2). The disease reaction of RILs showed continuous variation suggesting polygenic inheritance of wilt reaction along with major genes (Upadhyaya et al. 1983a, 1983b).
Linkage map construction
The per cent polymorphism observed in the present study (10.3%) is in accordance with majority of earlier studies namely, Tullu et al. (1998) (14%), Radhika et al. (2007) (9.5 and 11.57%), Gowda et al. (2009) (13.45%), Nayak et al. (2010) (16.7%) and Sabbavarapu et al. (2013) (20.8%). However, when compared with reports by Ratnaparkhe et al. (1998) (38%) and Patil et al. (2014) (21.30%), the per cent polymorphism obtained in the present study was rather low (10.3%). The possible reason could be the use of intraspecific mapping population and more number (84.3%) of genic markers (which are developed from conserved regions of the genome) employed in the present study, unlike interspecific mapping population and ISSR markers used by Ratnaparkhe et al. (1998).
Segregation distortion observed in the mapping population (12.9%) was relatively less compared to that reported by Tullu et al. (1999), Winter et al. (2000), Radhika et al. (2007) and Flandez-Galvez et al. (2003) in chickpea. The observed segregation distortion is in favour of the maternal alleles (K850) which was in agreement with Flandez-Galvez et al. (2003). Segregation distortion affects the estimation of map distances and the order of markers and, hence, affect linkage map construction and QTL analysis. Though, Winter et al. (2000) observed different amounts of segregation distortion for different classes of markers, segregation distortion was less related to the class of affected markers than to the genomic region where they resided.
Linkage group 1 (LG 1) of present study composed of 14 markers and spanning a length of 588.60 cM may correspond to LG II of map of Radhika et al. (2007) as it contains STMS loci TR24, TA186 and TA72. However, it may also correspond to LG IV of Millan et al. (2010) and Sabbavarapu et al. (2013) due to the presence of a STMS marker TA72. The LG 4 composed of four markers and comprising a length of 50.30 cM may equated to LG I of map of Radhika et al. (2007) since both the maps shared three STMS markers (TR59, TA42 and TR29) in common. This group may also correspond to LG V of Sabbavarapu et al. (2013) and Millan et al. (2010) due to the presence of STMS markers TR59 and TR29 respectively. However, this group may also be equated with LG IV of Patil et al. (2014) due to the presence of a STMS marker TR29. This clearly indicated the lack of sufficient number of markers for joining the corresponding linkage groups and the other reason could be attributed to the use of novel markers in the present study for the development of linkage map. Among 31 SSR markers, 23 SSRs (eight genic and 15 genomic) were informatively mapped on four linkage groups, of which, 16 were novel markers and were mapped for the first time in chickpea. Hence, it is an addition to already existing integrated map by Nayak et al. (2010).
QTLs for Fusarium wilt resistance
All the five QTLs reported in the study were for late wilting (60 DAS) and no QTLs were identified for early wilting (30 DAS) since the experimental material used in the study were derived from a cross between a late wilter (K850) and a resistant (WR315) parent and hence, the RILs segregate for H 2 locus alone. The QTLs identified in the study were major with R 2 more than 50% and LOD > 4. It is interesting to note that a QTL identified in 2007 rabi and another QTL identified in 2008 rabi were apparently in the same locus, having the same flanking markers GGSR 18 and TC 14801 with slightly different positions. However, difference observed between phenotypic variance and LOD can be accounted for differences in environmental conditions between two trial seasons. The presence of common flanking markers indicates the same genomic regions. In addition, negative additive effect for the both QTLs indicated the favourable alleles were donated by female parent. Hence, it can be considered as relatively stable and apparently common QTL. The similar report of presence of apparently common QTL for Ascochyta blight resistance in chickpea was reported by Stephens et al. (2014). However, as the distance between flanking markers spanning the identified stable QTL (GGSR 18 and TC 14801) is more, therefore, further efforts are required to fine map the QTL region in order to identify more closely-linked flanking markers for successful introgression of the QTL using markers-assisted selection (MAS) and map-based cloning of identified Fusarium resistant QTL.
The remaining three QTLs namely (GSSR 11-EST SSR 3) in 2007 rabi and (TR 24-EST SSR 21 and EST SSR 21-EST SSR 65) in 2008 rabi were also major QTLs with high additive effect indicated the contributing alleles were from male parent WR315. The QTLs found during 2008 rabi (TR 24-EST SSR 21 and EST SSR 21-EST SSR 65) were very close to each other while the second QTL (GSSR 11-EST SSR 3) of 2007 was at the other end of linkage group 1.
The major QTL (GSSR 18-TC14801) present in LG 1 for Fusarium wilt resistance which was contradictory with majority of earlier reports (Ratnaparkhe et al. 1998; Tekeoglu et al. 2000; Winter et al. 2000; Millan et al. 2006; Sharma and Muehlbauer 2007; Cobos et al. 2009; Gowda et al. 2009; Millan et al. 2010; Barman et al. 2014; Patil et al. 2014; Varshney et al. 2014) where the QTLs for FOC 1 were mapped on LG 2 and few studies reported the QTL on other linkage group, Cobos et al. (2005) reported a QTL on LG 3 and Sabbavarapu et al. (2013) reported two novel QTLs for Fusarium wilt race 1 on LG 6 in a different mapping population derived from same resistant parent (WR315).
All the QTLs reported in the present study for wilt resistance against race 1 were located on LG 1 and explained a higher phenotypic variation ranging from 53.34 to 71.41% and had a higher LOD score of 8.90%. The QTLs detected were not reported so far in any of the earlier studies, hence, the QTLs may be considered as novel. In earlier studies, locating the additional QTLs on other linkage groups (LG 2, LG 3 and LG 6) accounted for a lesser proportion of genetic variation compared to the current study. This variation could be due to various factors, such as use of different methodology, different genetic background, different phenotypic techniques (sick plot versus sick pot), different types of markers and interspecific versus intraspecific crosses.
The identification of closely-linked markers or stable QTLs for the Fusariumwilt resistance genes, will facilitate introgression of resistant genes from Fusariumwilt resistant cultivars carrying individual genes into commercially competitive chickpea varieties, as witnessed in the recent study by Varshney et al. (2014). In earlier studies, efforts were also made to map QTLs/genes and markers flanking QTLs for different Fusarium wilt races. For instance, markers flanking Foc0 locus (OPJ20 600 and TR59), Foc1 locus (TA110 and H 3 A 12), Foc2 locus (H 3 A 12 and TA96), Foc3 locus (TA96 and TA194), Foc4 locus (TA96 and CS27) and Foc5 locus (TA59 and TA96), determining resistance to race 0 (Cobos et al. 2005), race 1 (Gowda et al. 2009), race 2 (Gowda et al. 2009), race 3 (Sharma et al. 2004; Gowda et al. 2009), race 4 (Winter et al. 2000; Sharma et al. 2004) and race 5 (Cobos et al. 2009) respectively, were reported.
Conclusion
A set of 31 novel molecular markers were mapped in the present study and stable QTL (GSSR 18–TC14801) across seasons for Fusariumwilt resistance was identified. Since the distance between the flanking markers is high, validation of the identified stable QTL and further efforts to fine map the region to identify more closely-linked markers is needed so that the identified stable QTL can be effectively made use in chickpea breeding programme.
References
Barman P., Handique A. K. and Tanti B. 2014 Tagging STMS markers for Fusarium wilt race-1 resistance in chickpea (Cicer arietinum, L.). Ind. J. Biotechnol. 13, 370–375.
Basten C., Weir B. and Zeng Z. 1994 Z map—a QTL cartographer. In Proceedings of the 5th world congress on genetics applied to livestock production: computing strategies and software. Vol. 22, 5th World Congress on Genetics Applied to Livestock Production, pp. 65. Organizing Committee Guelph, Ontario, Canada.
Brinda S. and Ravikumar R. L. 2005 Inheritance of wilt resistance in chickpea: a molecular marker analysis. Curr. Sci. 88, 12–19.
Choudhary S., Gaur R., Gupta S. and Bhatia S. 2012 EST derived genic molecular markers: development and utilization for generating an advanced transcript map of chickpea. Theor. Appl. Genet. 124, 1449–1458.
Cobos M., Fernandez M., Rubio J., Kharrat M., Moreno M., Gil J. and Millan T. 2005 A linkage map of chickpea (Cicer arietinum L.) based on populations from Kabuli × Desi crosses: location of genes for resistance to Fusarium wilt race 0. Theor. Appl. Genet. 110, 1347–1353.
Cobos M., Winter P., Kharrat M., Cubero J., Gil J., Millan T. and Rubio J. 2009 Genetic analysis of agronomic traits in a wide cross of chickpea. Field Crops Res. 111, 130–145.
Doyle J. J. and Doyle J. L. 1987 A rapid DNA isolation procedure for small amounts of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Flandez-Galvez H., Ades P., Ford R., Pang E. and Taylor P. 2003 QTL analysis for ascochyta blight resistance in an intraspecific population of chickpea (Cicer arietinum L.). Theor. Appl. Genet. 107, 1257–1264.
Garg R., Patel R. K., Tyagi A. K. and Jain M. 2011 De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res. 18, 53–63.
Gowda S. J. M., Radhika P., Mhase L. B., Jamadagni B. M., Gupta V. S. and Kadoo N. Y. 2011 Mapping of QTLs governing agronomic and yield traits in chickpea. J. Appl. Genet. 52, 9–21.
Gowda S. J. M., Radhika P., Kadoo N., Mhase L. and Gupta V. 2009 Molecular mapping of wilt resistance genes in chickpea. Mol. Breed. 24, 177–184.
Gujaria N., Kumar A., Dauthal P., Dubey A., Hiremath P., Prakash A. B. et al. 2011 Development and use of genic molecular markers (GMMs) for construction of a transcript map of chickpea (Cicer arietinum L.). Theor. Appl. Genet. 122, 1577–1583.
Haware M. and Nene Y. 1982 Races of Fusarium oxysporum f. sp. ciceri. Plant Dis. 66, 809–910.
Hiremath P. J., Kumar A., Penmetsa R. V., Farmer A., Schlueter J. A., Chamarthi S. K. et al. 2012 Large-scale development of cost-effective SNP marker assays for diversity assessment and genetic mapping in chickpea and comparative mapping in legumes. Plant Biotechnol. J. 10, 716–724.
Jain M., Misra G., Patel R. K., Priya P., Jhanwar S., Khan A. W. et al. 2013 A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.). Plant J. 74(5), 715–723.
Jhanwar S., Priya P., Garg R., Parida S. K., Tyagi A. K. and Jain M. 2012 Transcriptome sequencing of wild chickpea as a rich resource for marker development. Plant Biotechnol. J. 10, 690–702.
Jimenez-Diaz R., Alcala-Jimenez A., Hervas A. and Traperocasas J. L. 1993 Pathogenic variability and host resistance in the Fusarium oxysporum f. sp. ciceris Cicer arietinum pathosystem. In Proceedings of the third European Seminar on Fusarium mycotoxins, taxonomy, pathogenicity and host resistance, pp. 87–94. Plant Breeding and Acclimatization Institute, Radzikov, Poland.
Kujur A., Bajaj D., Saxena M. S., Tripathi S, Upadhyaya H. D., Gowda C. L. et al. 2013 Functionally relevant microsatellite markers from chickpea transcription factor genes for efficient genotyping applications and trait association mapping. DNA Res. 20, 355–373.
Mayer M., Tullu A., Simon C., Kumar J., Kaiser W., Kraft J. and Muehlbauer F. 1997 Development of a DNA marker for Fusarium wilt resistance in chickpea. Crop Sci. 37, 1625– 1637.
Millan T., Clarke H. J., Siddique K. H., Buhariwalla H. K., Gaur P. M., Kumar J. et al. 2006 Chickpea molecular breeding: new tools and concepts. Euphytica 147, 81–90.
Millan T., Winter P., Jungling R., Gil J., Rubio J., Cho S. et al. 2010 A consensus genetic map of chickpea (Cicer arietinum L.) based on 10 mapping populations. Euphytica 175, 175–185.
Nayak S. N., Zhu H., Varghese N., Datta S., Choi H. K., Horres R. et al. 2010 Integration of novel SSR and gene-based SNP marker loci in the chickpea genetic map and establishment of new anchor points with Medicago truncatula genome. Theor. Appl. Genet. 120, 1415–1428.
Patil B. S., Ravikumar R. L., Bhat J. S. and Soregaon D. 2014 Molecular mapping of QTLs for resistance to early and late Fusarium wilt in chickpea. Czech. J. Genet. Plant Breed. 50, 171–176.
Radhika P., Gowda S., Kadoo N., Mhase L., Jamadagni B., Sainani M. et al. 2007 Development of an integrated intraspecific map of chickpea (Cicer arietinum L.) using two recombinant inbred line populations. Theor. Appl. Genet. 115, 209–217.
Ratnaparkhe M., Santra D., Tullu A. and Muehlbauer F. 1998 Inheritance of inter-simple-sequence-repeat polymorphisms and linkage with a Fusarium wilt resistance gene in chickpea. Theor. Appl. Genet. 96, 348–352.
Ravikumar R., Patil B., Soregaon C. and Hegde S. 2007 Genetic evidence for gametophytic selection of wilt resistant alleles in chickpea. Theor. Appl. Genet. 114, 619–626.
Sabbavarapu M. M., Sharma M., Chamarthi S. K., Swapna N., Rathore A., Thudi M. et al. 2013 Molecular mapping of QTLs for resistance to Fusarium wilt (race 1) and Ascochyta blight in chickpea (Cicer arietinum L.). Euphytica 193, 121–138.
Sharma K. D. and Muehlbauer F. J. 2007 Fusarium wilt of chickpea: physiological specialization, genetics of resistance and resistance gene tagging. Euphytica 157, 1–14.
Sharma K. D., Chen W. and Muehlbauer F. J. 2005 Genetics of chickpea resistance to five races of Fusarium wilt and a concise set of race differentials for Fusarium oxysporum f. sp. ciceris. Plant Dis. 89, 385–395.
Sharma K. D., Winter P., Kahl G. and Muehlbauer F. J. 2004 Molecular mapping of Fusarium oxysporum f. sp. ciceris race 3 resistance gene in chickpea. Theor. Appl. Genet. 108, 1243– 1256.
Singh H., Kumar J., Smithson J. B. and Haware M. P. 1987 Complementation between genes for resistance to race 1 of Fusarium oxysporum f. sp. ciceri in chickpea. Plant Pathol. 36, 539–543.
Stephens A., Lombardi M., Cogan N. O. L., Forster J. W., Hobson K., Materne M. and Kaur S. 2014 Genetic marker discovery, intraspecific linkage map construction and quantative trait locus analysis of Ascochyta blight resistance in chickpea (Cicer arietinum L.) Mol. Breed. 33, 297– 313.
Tekeoglu M., Tullu A., Kaiser W. J. and Muehlbauer F. J. 2000 Inheritance and linkage of two genes that confer resistance to Fusarium wilt in chickpea. Crop Sci. 40, 1247– 1256.
Thudi M., Bohra A., Nayak S. N., Varghese N., Shah T. M., Penmetsa R. V. et al. 2011 Novel SSR markers from BAC-end sequences, DArT arrays and a comprehensive genetic map with 1,291 marker loci for chickpea (Cicer arietinum L.). PLoS One 6, e27275.
Tullu A., Kaiser W., Kraft J. and Muehlbauer F. 1999 A second gene for resistance to race 4 of Fusarium wilt in chickpea and linkage with a RAPD marker. Euphytica 109, 43–47.
Tullu A., Muehlbauer F., Simon C., Mayer M., Kumar J., Kaiser W. and Kraft J. 1998 Inheritance and linkage of a gene for resistance to race 4 of Fusarium wilt and RAPD markers in chickpea. Euphytica 102, 227–267.
Upadhyaya H., Haware M., Kumar J. and Smithson J. 1983a Resistance to wilt in chickpea. I. Inheritance of late-wilting in response to race 1. Euphytica 32, 447–456.
Upadhyaya H., Smithson J., Haware M. and Kumar J. 1983b Resistance to wilt in chickpea. Further evidence for two genes for resistance to race 1. Euphytica 32, 749–755.
Varshney R. K., Song C., Saxena R. K., Azam S., Yu S., Sharpe A. G. et al. 2013 Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–256.
Varshney R., Mohan S., Gaur P., Chamarthi S., Singh V., Srinivasan S. et al. 2014 Marker-assisted backcrossing to introgress resistance to Fusarium wilt race 1 and Ascochyta blight in C 214, an elite cultivar of chickpea. Plant Genome 7, 278–289.
Winter P., Benko-Iseppon A. M., Huttel B., Ratnaparkhe M., Tullu A., Sonnante G. et al. 2000 A linkage map of the chickpea (Cicer arietinum L.) genome based on recombinant inbred lines from a C. arietinum × C. reticulatum cross: localization of resistance genes for Fusarium wilt races 4 and 5. Theor. Appl. Genet. 101, 1155–1167.
Zeng Z. B. 1994 Precision mapping of quantitative trait loci. Genetics 136, 1457–1468.
Acknowledgements
We thank Dr Mukesh Jain, National Institute of Plant Genome Research, New Delhi, for providing molecular markers used in the present study and the Department of Science and Technology (DST), New Delhi, for the financial support (GrantPS-170/2010).
Author information
Authors and Affiliations
Corresponding author
Additional information
[Jingade P. and Ravikumar R. L. 2015 Development of molecular map and identification of QTLs linked to Fusarium wilt resistance in chickpea. J. Genet. 94, xx–xx]
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
JINGADE, P., RAVIKUMAR, R.L. Development of molecular map and identification of QTLs linked to Fusarium wilt resistance in chickpea. J Genet 94, 723–729 (2015). https://doi.org/10.1007/s12041-015-0589-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12041-015-0589-7