
Abstract
Atherosclerosis is a common cardiovascular disease in which the arteries are thickened due to buildup of plaque. This study aims to identify programmed cell death (PCD)-related biomarkers and explore the crucial regulatory mechanisms of atherosclerosis. Gene expression profiles of atherosclerosis and control groups from GSE20129 and GSE23746 were obtained. Necroptosis was elevated in atherosclerosis. Weighted gene co-expression network analysis (WGCNA) was conducted in GSE23746 and GSE56045 to identify PCD-related modules and to perform enrichment analysis. Two necroptosis-related genes (IRF9 and STAT1) were identified and considered as biomarkers. Enrichment analysis showed that these gene modules were mainly related to immune response regulation. In addition, single-cell RNA sequencing data from GSE159677 were obtained and the characteristic cell types of atherosclerosis were identified. A total of 11 immune cell types were identified through UMAP dimension reduction. Most immune cells were mainly enriched in plaque samples, and STAT1 and IRF9 were primarily expressed in T-cells and macrophages. Moreover, the roles of IRF9 and STAT1 were assessed and found to be significantly upregulated in atherosclerosis, which was associated with increased risk of atherosclerosis. This study provides a molecular feature of atherosclerosis, offering an important basis for further research on its pathological mechanisms and the search for new therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Atherosclerosis is a chronic immune-inflammatory, progressive, and age-related disease characterized by lipid-rich plaques in arterial walls (Xiang et al. 2022). Despite advances in cardiology treatments, atherosclerosis continues to be a leading cause of death worldwide. Atherosclerotic plaques severely restrict oxygen-rich blood flow to the heart, brain, and other organs, leading to serious problems such as stroke, myocardial infarction, peripheral vascular disease, or even death (Chen et al. 2021). The pathogenesis of atherosclerosis involves multiple factors, including genetics, lifestyle, and endocrine disorders (Dabravolski et al. 2022). To better understand the pathogenesis of atherosclerosis and strategies for prevention and treatment, many researchers have explored the biological features of the disease from various physiological process and molecular perspectives.
Programmed cell death (PCD) is a highly regulated cellular response that controls cell fate in multicellular organisms under various cellular stresses and/or external stimuli. The most common forms of cell death are apoptosis, necroptosis, pyroptosis, ferroptosis, and cuproptosis (Kari et al. 2022; Xiong et al. 2023). Recent research has confirmed the implications of PCD in atherosclerotic lesions (Lin et al. 2021). Regulated proteins produced during cell death processes lead to late-stage plaque instability, ultimately resulting in plaque rupture and other unpredictable clinical events (Uyy et al. 2022). Given the relationship between PCD and atherosclerosis, treating atherosclerosis from a PCD perspective seems to be a more effective anti-atherosclerotic therapy. However, PCD features, gene expression, potential mechanisms, and biomarkers of the immune microenvironment in atherosclerosis remain to be elucidated.
Recently, the dysregulation of immune cells in plaques has been discovered through the use of single-cell RNA sequencing (scRNA-seq) analysis (Fernandez et al. 2019; de Winther et al. 2023). Patients’ plaques exhibit unique CD4+ T-cell subpopulations and characteristics of T-cell activation and differentiation (Kong et al. 2022). In chronic inflammation, CD4+ T-cells can secrete large amounts of Th1-cell cytokines, TNF-α, and IFN-γ, which are effective atherogenic cytokines (Tay et al. 2019). The number of CD8+ T-cells in advanced atherosclerotic plaque increases and promotes atherosclerosis (Engelen et al. 2022).
Therefore, identifying PCD-related biomarkers that play a significant role in the process of atherosclerosis may promote the development of promising therapeutic strategies. By comparing the gene expression profiles of atherosclerosis patients and healthy controls, we identified a set of necroptosis-related genes and potential biomarkers among them. In addition, we also evaluated the expression of markers at the single-cell level. In summary, this study provides a PCD-related molecular feature of atherosclerosis, offering an important basis for further research on its pathological mechanisms and the search for new therapeutic targets.
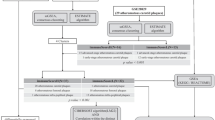
2 Materials and methods
2.1 Data collection
Gene expression profiling data were collected from the GSE20129 (Huang et al. 2011), GSE23746, and GSE56045 (Reynolds et al. 2014) datasets. GSE20129 included whole blood samples of 48 carotid atherosclerosis patients and 71 non-atherosclerosis controls. GSE23746 included monocytes in the peripheral blood of 76 carotid atherosclerosis patients and 19 non-atherosclerosis controls. GSE56045 included monocytes in the peripheral blood of 1202 atherosclerosis patients. GSE159677 (Alsaigh et al. 2022) included the scRNA-seq data of atherosclerotic core (AC) plaques and patient-matched proximal adjacent (PA) tissue from 3 atherosclerosis patients. Genes in PCD were acquired from the Molecular Signature Database (MsigDB) database (https://www.gsea-msigdb.org).
2.2 Differential expression and enrichment analysis
Differentially expressed genes between atherosclerosis patients and controls were analyzed in the GSE20129 and GSE23746 datasets using the R package limma (Ritchie et al. 2015). Differentially expressed genes (DEGs) were selected according to set significance thresholds (p<0.05). The enriched scores of PCD were calculated using gene set variation analysis (GSVA) (Hanzelmann et al. 2013).
2.3 Weighted gene co-expression network analysis
In the GSE20129 and GSE56045 datasets, a co-expression network was constructed using the R package WGCNA (Langfelder and Horvath 2008). Pearson’s correlation coefficients between gene expression data were calculated, resulting in a gene correlation matrix. The gene correlation matrix was transformed into a weighted network. The optimum β-value was selected by the scale-free topology criterion method to ensure that the network was scale-free. A gene cluster tree was generated by calculating the topological overlap between genes using a weighted similarity matrix, followed by hierarchical clustering analysis. Then, gene modules were determined by setting the dendrogram cut height (0.25). Each module was a subset of genes that had similar patterns in expression profiles. Module eigengene (ME) was computed for each module, calculating the association between ME and traits. Then, enrichment analysis for modules was performed using the R package clusterProfiler (Wu et al. 2021), including the Gene Ontology (GO) or Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
2.4 scRNA-seq data analysis
scRNA-seq data analysis was used to remove low-quality cells with gene expression numbers fewer than 500 from GSE159677. Gene expression data were normalized using log normalization to reduce technical bias. Highly variable genes (greater than 2000) were then selected. Dimensionality reduction was further performed to facilitate data visualization using the uniform manifold approximation and projection (UMAP). Each cell subpopulation was assigned a corresponding cell type by known cell type-specific marker genes.
2.5 Evaluation of marker genes
Receiver operating characteristic (ROC) curves were used to evaluate the ability of biomarkers to differentiate between disease and control groups. The R package pROC (Robin et al. 2011) was used to plot ROC curves and to calculate the area under the ROC curve (AUC) values. A nomogram was then constructed with the R package rms (Zhang et al. 2019) using logistic regression. The calibration curve was used to compare the consistency between predicted and actual probabilities.
2.6 Sample collection
Blood samples were obtained from 10 patients with atherosclerosis and 10 healthy controls from the Xinjiang Production and Construction Corps Hospital (table 1). All patients gave informed consent, and this study was approved by the ethics committee of the Xinjiang Production and Construction Corps Hospital (No. 202201401).
2.7 Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA extraction was performed using the PAXgene Blood RNA Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s instructions. The concentration and purity of RNA were determined by NanoDrop. Reverse transcription reactions were performed using a superscript IV reverse transcriptase kit (Invitrogen, CA, USA). The qRT-PCR experiments were performed using SYBR Green qPCR Master Mix (Invitrogen, CA, USA). Relative expression levels of target genes were calculated using the 2-ΔΔCt method with β-actin as the internal reference gene. The primers used for the experiment are as follows: STAT1: F,5′-CAGCTTGACTCAAAATTCCTGGA-3′, R,5′-TGAAGATTACGCTTGCTTTTCCT-3′; IRF9: F,5′-GCCTACAAGGTGTATCAGTTG-3′, R,5′-TGCTGTCGCTTTGATGGTACT-3′; β-actin: F,5′-ACGTGGACATCCGCAAAG-3′, R,5′-TGGAAGGTGGACAGCGAGGC-3′.
2.8 Western blot
Total protein was extracted using a whole blood protein extraction kit (Solarbio, Beijing, China) according to the manufacturer's instructions. Protein concentration was measured using the BCA kit ‘Enhanced BCA Protein Assay Kit’ (Beyotime, Shanghai, China). An equal amount of protein sample (15 μL) was loaded and separated in SDS-PAGE. The separated protein was then transferred onto a PVDF membrane. The membranes were incubated in 5% skimmed milk for 2 h at room temperature. The membranes were then incubated with the first antibodies targeting STAT1, IRF9, and β-actin (Proteintech, Wuhan, China) overnight at 4°C. After washing, HRP-labeled antibodies (Proteintech, Wuhan, China) and the membranes were incubated for 2 h at room temperature. Electrogenerated chemiluminescent (ECL) reagents (Beyotime) were added to capture the signals. β-actin was used as the internal reference protein, and quantitative analysis was performed using ImageJ software (National Institutes of Health, Bethesda, USA).
2.9 Statistical analysis
GraphPad Prism 9.3.0 and R software version 3.6.0 were used for statistical analyses. Data are presented as means±standard deviation. Student’s t-test was used to assess statistical significance. A p-value <0.05 was considered statistically significant.
3 Results
3.1 Assessment of PCD in atherosclerosis
We analyzed the Database of Essential Genes (DEGs) between atherosclerosis patients and controls in the GSE20129 and GSE23746 datasets. We selected 1878 DEGs in GSE20129 (figure 1A) and 4675 DEGs in GSE23746 (figure 1B) according to the significance threshold (p<0.05). We calculated enrichment scores for PCD using GSVA and found that necroptosis was elevated in atherosclerosis in both datasets (figure 1C and D).

Differentially expressed genes and PCD in atherosclerosis. (A) Volcano plot of differentially expressed genes between atherosclerosis and controls in GSE20129. Red represents upregulation (p<0.05) and green represents downregulation (p<0.05). (B) Volcano plot of differentially expressed genes between atherosclerosis and controls in GSE23746. Red represents upregulation (p <0.05) and green represents downregulation (p<0.05). (C) Enrich scores of PCD in atherosclerosis and controls were calculated by GSVA in GSE20129. * p<0.05, ** p<0.01. (D) Enrich scores of PCD in atherosclerosis and controls were calculated by GSVA in GSE23746. * p<0.05, ** p<0.01.
3.2 Co-expression network identifies necroptosis-related modules
A co-expression network was constructed separately using WGCNA in the GSE20129 and GSE56045 datasets. Eight co-expression modules were identified in GSE20129 (figure 2A) and 16 co-expression modules were identified in GSE56045 (figure 2B). The correlation analysis revealed that the red module in GSE20129 had the highest positive correlation with necroptosis (figure 2C) and the black module in GSE56045 had the highest positive correlation with necroptosis (figure 2D). Therefore, we compared the genes in these two modules and identified 35 overlapping module genes (figure 2E). Interestingly, we found 2 necroptosis genes (IRF9 and STAT1) in the overlapping module genes and considered them as marker genes.

Biological roles in which necroptosis-related module genes participate. Clustering dendrogram of modules in the GSE20129 (A) and GSE56045 (B). Different colors represent different modules. Correlation analysis between module eigengene and traits in GSE20129 (C) and GSE56045 (D). Red represents positive correlation and green represents negative correlation. (E) Intersection of red module genes in GSE20129 and black module genes GSE56045. (F) GO functions of overlapping module genes enrichment. BP, biological progression; CC, cellular composition; MF, molecular function. (G) KEGG pathways of overlapping module genes enrichment.
Enrichment analysis showed that the overlapping module genes were mainly related to the GO term ‘response to interferon-gamma’ (figure 2F). In the KEGG enrichment results, we found that the overlapping module genes were significantly enriched in Herpes simplex virus 1 infection, NOD-like receptor signaling pathway, and Kaposi sarcoma-associated herpesvirus infection (figure 2G).
3.3 Major cell types in atherosclerosis
After dimension reduction by UMAP, we identified 29 cell clusters in GSE159677 (figure 3A). To determine the specific type of these cell clusters, we utilized known cell type-specific marker genes to assign corresponding cell types to each cell subset (figure 3B). Eleven cell types were finally identified (figure 3C). By comparing with different sample types, we found that most immune cells were mainly enriched in plaque samples (figure 3D). Additionally, STAT1 was mainly expressed in CD4+ T-cells, CD8+ T-cells, and macrophages, and IRF9 was mainly expressed in CD4+ T-cells and CD8+ T-cells (figure 3E).

Single-cell atlas of atherosclerosis. (A) UMAP plot of cell clusters from 6 samples. (B) Violin plots showing expression levels of marker genes. (C) UMAP plot of 11 cell types. (D) UMAP plot showing the distribution of cells in different types of samples. AC, atherosclerotic core; PA, proximal adjacent. (E) UMAP plot showing the expression of genes in cells.
3.4 Evaluation of biomarkers
We evaluated the roles of IRF9 and STAT1 in atherosclerosis. We found that compared with the control group, both IRF9 and STAT1 were significantly upregulated in atherosclerosis (figure 4A). ROC curve results showed that the AUC for IRF9 was 0.67 and for STAT1 it was 0.70, indicating the ability to distinguish between the disease and control groups (figure 4B). Nomogram results revealed that high expressions of IRF9 and STAT1 were associated with increased risk of atherosclerosis (figure 4C). Calibration curve results indicated high accuracy of model predictions (figure 4D). Importantly, by qRT-PCR experiments, we confirmed the high expression of mRNA levels of IRF9 and STAT1 in atherosclerotic patients compared with controls (figure 4E). The protein expressions of IRF9 and STAT1 were also higher in atherosclerotic patients compared with controls (figure 4F).

Diagnostic role of marker genes for atherosclerosis. (A) Expression of IRF9 and STAT1 in atherosclerosis and controls in GSE23746. *p<0.05, **p<0.01. (B) ROC curves of IRF9 and STAT1. AUC, area under the curve; CI, confidence interval. (C) Nomogram of IRF9 and STAT1 to predict the risk probability of atherosclerosis. (D) Calibration plot of the nomogram for predicting risk probability of atherosclerosis. (E) The mRNA levels of IRF9 and STAT1 in atherosclerosis and controls detected by qRT-PCR. **p<0.01. (F) The protein levels of IRF9 and STAT1 in atherosclerosis and controls detected by western blot. ***p<0.001.
4 Discussion
Emerging research indicates the significant role of PCD in atherosclerosis (Li et al. 2022a, b). Therefore, in this study, we explored PCD associated with pathogenesis of atherosclerosis. Based on comprehensive analysis of atherosclerosis gene expression data from four public datasets, necroptosis was found to be significantly upregulated in atherosclerosis. IRF9 and STAT1 serve as marker genes for risk prediction in atherosclerosis. Additionally, we identified the expression characteristics of marker genes in the atherosclerotic microenvironment, further verifying their critical roles in the pathogenesis of atherosclerosis.
Regulatory mechanisms related to necroptosis may be present during the development of atherosclerosis. Necroptosis is characterized by a typical inflammatory response (Chaouhan et al. 2022). In the pathological process of atherosclerosis, inflammation continuously occurs within the arterial wall, further exacerbating arterial injury and promoting lesion development (Zhe-Wei et al. 2018; Gui et al. 2022). Necrotic apoptotic cells persistently exist, releasing danger-associated molecular patterns and leading to continuous inflammation and the development of a necrotic core in atherosclerotic plaques at all stages of plaque expansion (Zhang et al. 2022). In late-stage atherosclerotic plaques, necroptosis is activated, and targeting the necroptosis pathway effectively reduces plaque vulnerability and lesion progression (Tang et al. 2011). Thus, studying the role of necroptosis in atherosclerosis is of great significance for understanding the mechanisms of disease onset.
In two independent datasets, co-expression modules related to necroptosis showed a certain degree of consistency. This provided strong evidence for our further exploration of the role of necroptosis in atherosclerosis. Notably, IRF9 and STAT1, two necroptosis-related genes, were identified in these overlapping modules, indicating that they may play a key role in the development and progression of atherosclerosis.
Enrichment analysis results revealed the functions of overlapping module genes in various biological processes and signaling pathways. Th1-cells producing IFN-γ, CD8+ T-cells, and macrophages promote atherosclerosis (Li et al. 2017). Viral infection in KEGG enrichment results may exacerbate the progression of atherosclerosis by activating immune cells and inflammatory responses (Gopal et al. 2020). In addition to its association with viral effects, IFN-γ, as a proinflammatory cytokine, also promotes atherosclerosis mediated by CD4+ T-cells and CD8+ T-cells (Kyaw et al. 2017; Li et al. 2017). Nucleotide-binding oligomerization domain (NOD)-like receptor signaling pathways play an essential role in immune response and inflammation regulation (Ebersole et al. 2016). NOD-like receptors are a class of pattern recognition receptors within the immune system capable of recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) (Almeida-da-Silva et al. 2023). In atherosclerosis, NOD-like receptor signaling pathways may be involved in regulating the activation and migration of inflammatory cells, and promoting local inflammatory responses (Qiao et al. 2021; Li et al. 2022b).
On the other hand, single-cell data analysis results revealed the distribution and relationships of immune cell types in atherosclerotic plaques. Immune cells play a critical role in the development of atherosclerosis, exerting significant effects on plaque stability and rupture risk by regulating inflammatory responses and the local immune environment (Nie et al. 2022; Razeghian-Jahromi et al. 2022). This study showed that a large number of immune cells are enriched in plaque samples, indicating that local inflammation and immune cell infiltration play crucial roles in the development of atherosclerosis. These immune cells can produce various inflammatory factors, chemokines, and cytokines, promoting atherosclerotic plaque formation and progression through the regulation of endothelial cell function, smooth muscle cell migration, and extracellular matrix degradation (Gencer et al. 2021).
IRF9 and STAT1 are upregulated in atherosclerosis patients and have diagnostic value. This may imply that therapeutic strategies or drugs targeting IRF9 and STAT1 could have a positive impact on the treatment of atherosclerosis. IRF9 and STAT1 are essential regulatory factors in the immune system, involved in interferon signaling and the JAK-STAT signaling pathway (Al-Ahmadi et al. 2021; Markin et al. 2023). The complex formed by IRF9 and STAT1 controls IFN-γ responsiveness and is closely related to immune regulation and inflammatory responses (Piaszyk-Borychowska et al. 2019; Lu et al. 2021). The primary expression of STAT1 and IRF9 in immune cells such as CD4+ T-cells, CD8+ T-cells, and macrophages suggests that they may play a key role in regulating the function and activity of these cells. CD4+ T-cells and CD8+ T-cells participate in cell-mediated immune responses, while macrophages contribute to plaque inflammation through their phagocytic function and production of inflammatory factors (Herrero-Fernandez et al. 2019; Schafer and Zernecke 2020). Therefore, the role of IRF9 and STAT1 in atherosclerosis (Zhang et al. 2014; Cai et al. 2021) may be closely related to their functions in regulating inflammatory responses and cell death in biological processes.
However, the limitations of this study lie in the relatively small sample size, necessitating further research with larger sample sizes to verify these findings. Additionally, future studies should employ experimental models to further investigate the specific roles of IRF9 and STAT1 in the pathogenesis of atherosclerosis and their potential as therapeutic targets.
5 Conclusion
Using necroptosis-related IRF9 and STAT1 as biomarkers suggests that they may have diagnostic or prognostic value. By further validating the expression differences of these genes in atherosclerosis patients and control groups, we may be able to determine their application value in atherosclerosis diagnosis, disease monitoring, and risk assessment. At the same time, in-depth research into the roles of these two genes in the pathogenesis of atherosclerosis may help discover new therapeutic targets and provide new insights for improving disease treatment.
Data availability statement
Publicly available datasets analyzed in this study are available in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/gds) database (GSE20129, GSE23746, GSE56045, and GSE159677), and MsigDB database (https://www.gsea-msigdb.org).
References
Al-Ahmadi W, Webberley TS, Joseph A, et al. 2021 Pro-atherogenic actions of signal transducer and activator of transcription 1 serine 727 phosphorylation in LDL receptor deficient mice via modulation of plaque inflammation. FASEB J. 35 e21892
Almeida-da-Silva CLC, Savio LEB, Coutinho-Silva R, et al. 2023 The role of NOD-like receptors in innate immunity. Front. Immunol. 14 1122586
Alsaigh T, Evans D, Frankel D, et al. 2022 Decoding the transcriptome of calcified atherosclerotic plaque at single-cell resolution. Commun. Biol. 5 1084
Cai D, Liu H, Wang J, et al. 2021 Balasubramide derivative 3C attenuates atherosclerosis in apolipoprotein E-deficient mice: role of AMPK-STAT1-STING signaling pathway. Aging 13 12160–12178
Chaouhan HS, Vinod C, Mahapatra N, et al. 2022 Necroptosis: A pathogenic negotiator in human diseases. Int. J. Mol. Sci. 23 12714
Chen J, Zhang X, Millican R, et al. 2021 Recent advances in nanomaterials for therapy and diagnosis for atherosclerosis. Adv. Drug. Deliv. Rev. 170 142–199
Dabravolski SA, Markin AM, Andreeva ER, et al. 2022 Molecular mechanisms underlying pathological and therapeutic roles of pericytes in atherosclerosis. Int. J. Mol. Sci. 23 11663
de Winther MPJ, Back M, Evans P, et al. 2023 Translational opportunities of single-cell biology in atherosclerosis. Eur. Heart J. 44 1216–1230
Ebersole JL, Kirakodu S, Novak MJ, et al. 2016 Effects of aging in the expression of NOD-like receptors and inflammasome-related genes in oral mucosa. Mol. Oral Microbiol. 31 18–32
Engelen SE, Robinson AJB, Zurke YX, et al. 2022 Therapeutic strategies targeting inflammation and immunity in atherosclerosis: how to proceed? Nat. Rev. Cardiol. 19 522–542
Fernandez DM, Rahman AH, Fernandez NF, et al. 2019 Single-cell immune landscape of human atherosclerotic plaques. Nat. Med. 25 1576–1588
Gencer S, Evans BR, van der Vorst EPC, et al. 2021 Inflammatory chemokines in atherosclerosis. Cells 10 226
Gopal R, Marinelli MA and Alcorn JF 2020 Immune mechanisms in cardiovascular diseases associated with viral infection. Front. Immunol. 11 570681
Gui Y, Zheng H and Cao RY 2022 Foam cells in atherosclerosis: novel insights into its origins, consequences, and molecular mechanisms. Front. Cardiovasc. Med. 9 845942
Hanzelmann S, Castelo R and Guinney J 2013 GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14 7
Herrero-Fernandez B, Gomez-Bris R, Somovilla-Crespo B, et al. 2019 Immunobiology of atherosclerosis: a complex net of interactions. Int. J. Mol. Sci. 20 5293
Huang CC, Lloyd-Jones DM, Guo X, et al. 2011 Gene expression variation between African Americans and whites is associated with coronary artery calcification: the multiethnic study of atherosclerosis. Physiol. Genomics 43 836–843
Kari S, Subramanian K, Altomonte IA, et al. 2022 Programmed cell death detection methods: a systematic review and a categorical comparison. Apoptosis 27 482–508
Kong P, Cui ZY, Huang XF, et al. 2022 Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal. Transduct. Target Ther. 7 131
Kyaw T, Peter K, Li Y, et al. 2017 Cytotoxic lymphocytes and atherosclerosis: significance, mechanisms and therapeutic challenges. Br. J. Pharmacol. 174 3956–3972
Langfelder P and Horvath S 2008 WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9 559
Li M, Wang ZW, Fang LJ, et al. 2022a Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 13 467
Li XH, Liu LZ, Chen L, et al. 2022b Aerobic exercise regulates FGF21 and NLRP3 inflammasome-mediated pyroptosis and inhibits atherosclerosis in mice. PLoS One 17 e0273527
Li Y, Liu X, Duan W, et al. 2017 Batf3-dependent CD8alpha(+) dendritic cells aggravates atherosclerosis via Th1 cell induction and enhanced CCL5 expression in plaque macrophages. eBioMedicine. 18 188–198
Lin L, Zhang MX, Zhang L, et al. 2021 Autophagy, pyroptosis, and ferroptosis: New regulatory mechanisms for atherosclerosis. Front. Cell. Dev. Biol. 9 809955
Lu GF, Chen SC, Xia YP, et al. 2021 Synergistic inflammatory signaling by cGAS may be involved in the development of atherosclerosis. Aging 13 5650–5673
Markin AM, Markina YV, Bogatyreva AI, et al. 2023 The role of cytokines in cholesterol accumulation in cells and atherosclerosis progression. Int. J. Mol. Sci. 24 6426
Nie H, Yan C, Zhou W, et al. 2022 Analysis of immune and inflammation characteristics of atherosclerosis from different sample sources. Oxid. Med. Cell. Longev. 2022 5491038
Piaszyk-Borychowska A, Szeles L, Csermely A, et al. 2019 Signal integration of IFN-I and IFN-II with TLR4 involves sequential recruitment of STAT1-Complexes and NFkappaB to enhance pro-inflammatory transcription. Front. Immunol. 10 1253
Qiao L, Ma J, Zhang Z, et al. 2021 Deficient chaperone-mediated autophagy promotes inflammation and atherosclerosis. Circ. Res. 129 1141–1157
Razeghian-Jahromi I, Karimi Akhormeh A, Razmkhah M, et al. 2022 Immune system and atherosclerosis: Hostile or friendly relationship. Int. J. Immunopathol. Pharmacol. 36 1–14
Reynolds LM, Taylor JR, Ding J, et al. 2014 Age-related variations in the methylome associated with gene expression in human monocytes and T cells. Nat. Commun. 5 5366
Ritchie ME, Phipson B, Wu D, et al. 2015 limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids. Res. 43 e47
Robin X, Turck N, Hainard A, et al. 2011 pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12 77
Schafer S and Zernecke A 2020 CD8(+) T cells in atherosclerosis. Cells 10 37
Tang JJ, Li JG, Qi W, et al. 2011 Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 13 44–56
Tay C, Kanellakis P, Hosseini H, et al. 2019 B cell and CD4 T cell interactions promote development of atherosclerosis. Front. Immunol. 10 3046
Uyy E, Suica VI, Boteanu RM, et al. 2022 Regulated cell death joins in atherosclerotic plaque silent progression. Sci. Rep. 12 2814
Wu T, Hu E, Xu S, et al. 2021 clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2 100141
Xiang Q, Tian F, Xu J, et al. 2022 New insight into dyslipidemia-induced cellular senescence in atherosclerosis. Biol. Rev. 97 1844–1867
Xiong C, Ling H, Hao Q, et al. 2023 Cuproptosis: p53-regulated metabolic cell death? Cell. Death. Differ. 30 876–884
Zhang M, Zhu K, Pu H, et al. 2019 An immune-related signature predicts survival in patients with lung adenocarcinoma. Front. Oncol. 9 1314
Zhang SM, Zhu LH, Chen HZ, et al. 2014 Interferon regulatory factor 9 is critical for neointima formation following vascular injury. Nat. Commun. 5 5160
Zhang X, Ren Z, Xu W, et al. 2022 Necroptosis in atherosclerosis. Clin. Chim. Acta 534 22–28
Zhe-Wei S, Li-Sha G and Yue-Chun L 2018 The role of necroptosis in cardiovascular disease. Front. Pharmacol. 9 721
Acknowledgements
Not applicable.
Funding
This study was supported by the Research Plan Project of Xinjiang Production and Construction Corps Hospital of China (grant no. 2022004).
Author information
Authors and Affiliations
Contributions
WX designed the studies and wrote the draft manuscript. XG and LZ collected and analyzed the data. SS and NL conducted qRT-PCR. JL supervised the experiments and revised the manuscript. All the authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Corresponding editor: Dipankar Nandi
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xie, W., Gao, X., Zhao, L. et al. IRF9 and STAT1 as biomarkers involved in T-cell immunity in atherosclerosis. J Biosci 49, 84 (2024). https://doi.org/10.1007/s12038-024-00448-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-024-00448-5