
Abstract
Gallbladder cancer (GBC) is one of the most fatal malignancies of the biliary tract system and is ranked sixth among the neoplasms of the gastrointestinal tract. Gallstone disease (GSD) is considered the major risk factor for GBC. However, the underlying molecular mechanism of GBC pathogenesis from different stages of GSD is not yet clearly understood. We analyzed transcriptomic datasets of GBC with reference to GSD of three different follow-up periods, i.e., GBC vs. GSD3 (1–3 years), GBC vs. GSD5 (5–10 years), and GBC vs. GSD10 (more than 10 years). We identified overlapping and specific molecular signatures in GBC compared with GSD at three different follow-up periods. Using integrative network biology approaches, such as protein–protein interaction network analysis, transcriptional regulatory network analysis, and miRNA–target gene network analysis, we have identified a few hub genes. The hub genes identified from GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10 were directly or indirectly associated with cancer progression and initiation from GSD. Functional enrichment analysis indicated significant correlation between GSD and GBC pathogenesis. The identified hub genes can be used for future targeted validation to develop potential diagnostic, prognostic, or therapeutic biomarkers in GBC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Gallbladder cancer (GBC) is one of the most fatal malignancies of biliary tract cancers, where malignant cells form in the tissues of the gallbladder (Hundal and Shaffer 2014; Muhammad et al. 2018). Globally it accounts for around 80–90% of all the biliary tract cancers, and ranks sixth among gastrointestinal cancers (Hundal and Shaffer 2014; Song et al. 2020). As reported by the 2018 GLOBOCAN data, GBC accounts for around 1.7% of cancer-related deaths globally (Rawla et al. 2019). The incidence rate of GBC shows very high geographical, racial, and socioeconomic variations, suggesting the potential role of different environmental as well as genetic factors associated with the development and progression of this cancer (Hundal and Shaffer 2014; Sharma et al. 2017; Muhammad et al. 2018).
GBC does not exhibit any specific clinical symptoms. This causes difficulty in diagnosing the disease at an early stage. It is often diagnosed at an advanced stage (Letelier et al. 2012; Hundal and Shaffer 2014). Most of the time, GBC is incidentally diagnosed in patients undergoing cholecystectomy for the treatment of cholecystitis or cholelithiasis (Muhammad et al. 2018). According to different epidemiological and pathological investigations, patients with gallstones have a higher risk of GBC than healthy individuals. Gallstone disease (GSD) is considered as the major risk factor of GBC, affecting ~20% of the adult population worldwide and also present in more than 85% of GBC patients (Letelier et al. 2012; Hundal and Shaffer 2014; Jinghan Wang et al. 2020). Gallstones cause local mucosal irritation and chronic inflammation. This process has been speculated to activate intracellular enzymes involved in promoter methylation of some potential genes, and also produces some inflammatory mediators in the tissue microenvironment. Such events may result in alteration of the transcriptomic and genomic landscape, contributing to early-stage carcinogenesis in GBC (Letelier et al. 2012; Hundal and Shaffer 2014; Muhammad et al. 2018; Jinghan Wang et al. 2020). However, the detailed molecular mechanism associated with the transition of GSD to GBC is not yet understood. The available tumor markers for diagnosis of GBC do not have high specificity, and therefore cannot be detected until the advanced stages of the disease (Sharma et al. 2017). Understanding of the molecular mechanism behind the transition of GSD to GBC will help in the identification of crucial molecular markers for its early detection and treatment.
The complex interactions of molecular and environmental factors may initiate GBC pathogenesis in a progressive manner, which could lead to the dysregulation of multiple processes such as cell cycle, DNA repair, apoptosis, as well as immune responses (Knox 2010). Integrative analysis of multi-dimensional data using systems biology-based approaches will provide the basis for understanding the complex molecular mechanisms responsible for carcinogenesis of the gallbladder. Network biology is an integrative systems biology approach that can help us understand the complex molecular mechanisms responsible for GBC pathogenesis (Furlong 2013), and for the development of personalized treatment protocols (Chand and Alam 2012; Masoudi-Nejad and Wang 2015). Different types of networks such as protein–protein interaction (PPI) networks, gene regulatory networks, metabolic pathways, and various signaling pathways interact in a conjugated manner to define the fate of cellular behavior (Barabási and Oltvai 2004). The results generated from such integrative analysis of complex biological networks help to determine the specific roles of differentially regulated molecules, pathways, or processes in different cellular conditions especially in a multifactorial disease such as cancer (Barabási and Oltvai 2004). Here, we analyzed a transcriptomic dataset of 10 gallbladder cancer samples with respect to their adjacent 10 normal tissue samples, and 30 gallstone disease tissue samples in three different follow-up periods. An integrative network-based analysis was carried out on the differentially expressed genes (DEGs) obtained to identify the overlapping and unique molecular signatures. We performed differential gene expression analysis, functional enrichment analysis, PPI network analysis, module analysis, and regulatory network analysis of the specific DEGs identified from GBC vs. gallstone disease with three different follow-up periods to identify significant hub genes and hub transcription factors (TFs). Moreover, we also carried out miRNA–hub genes network analysis, hub gene signaling network analysis, and evaluation of genomic alteration of the hub genes.
2 Methodology
2.1 Retrieval of transcriptomic data
The RNA-seq dataset of GBC and GSD samples were obtained from the European Nucleotide Archive (ENA) database in Sequence Read Archive (SRA) format with the accession number SRP226150. The dataset contained a total of 50 samples obtained through surgical resection. The data comprised 10 GBC tissues, 10 adjacent normal tissue samples, and 30 GSD tissue samples from three different follow-up periods of 1–3 years (GSD3), 5–10 years (GSD5) and more than 10 years (GSD10). The Illumina HiSeq 2500 platform was used to generate the paired end reads of these 50 samples (Jinghan Wang et al. 2020). Using this published dataset, we carried out a detailed integrative analysis with various benchmarked network-based approaches to identify systems-level molecular signatures in GBC and the three different follow-up periods of GSD.
2.2 Transcriptomic data analysis and identification of differential overlapping and specific molecular signatures
The retrieved RNA-seq datasets in SRA format were converted into FastQ reads. The FastQ reads were pre-processed using an in-house RNA seq data analysis pipeline. Pre-processing is an important step to either remove or trim the adapter, poly N, as well as the low-quality reads. FastQC and fastp tools were used for the quality check (QC) of the reads and adapter trimming, respectively (Chen et al. 2018; de Sena Brandine and Smith 2019). The pre-processed high-quality reads after quality control were mapped against the reference human genome Homo sapiens (GRCh38) using HISAT2 (version 2.2.1)(Kim et al. 2019). The aligned or mapped reads were then quantified using the FeatureCounts tool to obtain the gene expression profile of each sample as a single-count matrix file (Liao et al. 2014). The counts matrix file obtained from RNA-seq analysis was used to identify the differentially expressed genes (DEGs) using DESeq2 package in R (Love et al. 2014). The DESeq2 tool gives the log2 fold changes and absolute gene expression levels relative to each sample by calculating the ratio of each read count to the logarithmic mean value of all the read counts for each gene across all the samples. The lists of significant DEGs were generated separately for GBC vs. adjacent normal (DEG list 1), and GBC vs. GSD with three different follow-up periods. We have considered a P-adjusted value ≤0.05 and |Log2 fold change| ≥1 for identifying DEGs. The overlapping and specific DEGs between GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10 were identified using Venny tool 2.1.0 (Oliveros 2007). The unique DEGs identified for GSD3 (DEG list 2), GSD5 (DEG list 3), and GSD10 (DEG list 4) were further used for downstream integrative analysis. The overall methodology has been described in figure 1.

A schematic representation of the overall methodology used in this study.
2.3 Functional annotation and pathway analysis
We carried out functional annotation and pathway enrichment analysis using the unique DEG lists (DEG lists 2, 3, and 4) identified from three follow-up periods. Functional annotations provided an overview of associations of the DEGs with biological processes, pathways, and disease phenotypes. We used two independent tools, i.e., DAVID tool (v6.8) (Dennis et al. 2003) and BINGO (a Cytoscape plugin), to determine the enriched biological processes associated with the unique DEGs. The enriched pathways associated with unique DEGs were identified from the KEGG database. The threshold of p-value <0.05 and gene counts >5 were considered for selecting the enriched biological processes and KEGG pathways.
2.4 PPI-based network analysis and screening of hub genes
The STRING database, version 11.5 (http://www.stringdb.org/), was used to construct the PPI network with unique DEGs identified from the DEG lists 2, 3, and 4 (Suratanee and Plaimas 2018). In the PPI network topology, the nodes represented the seed proteins (seed DEGs) and the edges represented the interactions between the DEGs. The PPI networks were analyzed using Cytoscape version 3.8 (Shannon et al. 2003). The plugin CytoHubba in cytoscape was used for topological analysis of the PPI networks and subsequent identification of hub genes (Chin et al. 2014). The hub genes for each disease group were identified through an ensemble approach by taking the consensus of five topological parameters, viz., maximum clique centrality (MCC), maximum neighborhood component (MNC), degree, edge percolated component (EPC), and betweeness centrality (Chin et al. 2014). The five top-ranked genes were considered to be the potential candidate genes for each of the conditions. Furthermore, highly connected gene modules from the PPI networks were detected using the Molecular Complex Detection (MCODE) algorithm (Pruitt et al. 2001). MCODE scores ≥4 and the number of nodes >4 were set as cutoff criteria with the default parameters (degree cutoff ≥2, node score cutoff ≥2, K-core ≥2, and max depth =100) (Roy et al. 2021).
2.5 Transcription regulatory-based network analysis and screening of hub transcription factors
Transcription factors (TFs) are the key regulators in the transcription process which influence overall gene expression by binding to the start site of the promoter region. For the construction of transcriptional regulatory networks, 1KB upstream FASTA sequence of the specific DEGs identified from GBC compared with GSD with different follow-up periods (GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10) were extracted using Regulatory Sequence Analysis Tools (RSAT) (Thomas-Chollier et al. 2008). Experimentally determined benchmarked position weight matrices (PWMs) for all the TFs were obtained from the CIS-BP database (Weirauch et al. 2014). PWM is a mathematical model describing the binding specificity of a TF. PWMs were used to scan cis-regulatory sequences of a gene for determining the enrichment of the defined patterns which were significantly more similar to the PWM than to the background models (Stormo 2000). A widely used benchmark matrix scan tool in MEME suite (v3.4.0) was used for PWM scanning by considering p-value cutoff of 10-4 (Bailey et al. 2009). Finally, the transcriptional regulatory networks (TRNs) with prediction scores were visualized in the form of interactive networks.
2.6 Prediction of hub genes–microRNA network analysis
MicroRNAs (miRNAs) belong to class of small noncoding RNAs that play a crucial role in cancer development by acting either as oncogenes and/or tumor suppressor genes. We performed hub gene–miRNA network analysis to identify potential hub gene–miRNA interactions. To identify hub gene–miRNA interactions, we used the miRTar database that stores experimentally validated miRNA–gene interaction data. Signaling network analysis of hub genes was performed using SIGNOR 2.0 database (http://signor.uniroma2.it/) to identify key signaling pathways. The cBioPortal database (https://www.cbioportal.org/) was used to identify genetic alterations associated with the identified hub genes.
3 Results
3.1 Identification of differentially expressed genes in GBC and GSDs
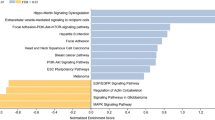
Differential gene expression analysis using DESeq2 identified four significant lists of DEGs for GBC vs. adjacent normal (DEG list 1), GBC vs. GSD3 (DEG list 2), GBC vs. GSD5 (DEG list 3), and GBC vs. GSD10 (DEG list 4) conditions. DEG list 1 contained 985 genes, of which 248 were upregulated, and 737 were downregulated. The total number of upregulated and downregulated DEGs (DEG lists 1, 2, 3, and 4) has been summarized in table 1. The complete lists of DEGs for all the comparisons along with the log2 fold change values and the corresponding adjusted p-values have been presented in the supplementary dataset. The results from DEG list 1 show that the significant DEGs identified in GBC were mostly downregulated (figure 2A). The downregulated DEGs were mainly enriched in important cell signaling pathways such as cAMP signaling, AMPK signaling pathway, PPAR signaling, and adipocytokine signaling pathways (figure 2B).

Overview of gene expression profile and cellular pathways identified in GBC compared with adjacent normal. (A) Volcano plot and hierarchical clustering showing the expression significant DEGs in GBC compared with that of adjacent normal. (B) The bubble plot represents the top 10 key pathways associated with significant DEGs in GBC.
3.2 Overlapping and unique DEGs in GBC compared to GSD with different follow-up periods
The objective of our work was to identify common and overlapping molecular signatures between GBC and GSD for understanding the possible mechanisms through which GSD progress to GBC. There were 3102 overlapping genes identified among the DEG lists 2, 3, and 4. GSD3 had 824, GSD5 had 499, and GSD10 had 446 unique DEGs (figure 3A). The heatmap visualization of the significant unique DEGs reflected variation in the expression pattern of DEGs identified in each GSD follow-up period as compared with the DEGs from GBC (figure 3B). This suggested that the differential expression pattern of these genes in GSD might manifest into a wide pathological spectrum, and thereby could contribute to GBC pathogenesis.

Differential gene expression profiles of overlapping and unique signatures. (A) Venn diagram showing the number of unique and overlapping DEGs between GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10. The bar plot represents key pathways associated with overlapping DEGs. (B) Heatmap plot for significant unique DEGs identified in between GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10.
3.3 Functional enrichment and pathways analysis of specific DEGs identified in GSD3, GSD5, and GSD10 follow-up periods
Functional enrichment and pathway enrichment analysis were performed for the identification of significant biological processes (table 2) and pathways (table 3) in the identified unique lists of DEGs. The enrichment analysis from both DAVID and BINGO showed that the DEGs in GSD3 and GSD10 were largely associated with immune response regulation and cell adhesion processes such as collagen organization. However, the unique DEGs in GSD5 were associated with distinct biological processes such as ion-transport channel-related processes. This suggested that among the GSD cases with different follow-up periods, there were potential molecular signatures which might contribute to GBC progression from GSD. The pathways associated with GSD3 were also enriched in cell adhesion pathways such as extracellular matrix organization, whereas the GSD5 DEGs were mainly linked with endocannabinoid signaling, leukocyte transendothelial migration, and neuroactive ligand-receptor interaction.
3.4 Construction of PPI networks and screening of significant hub genes/proteins associated with GBC progression
The specific DEGs identified from GBC compared with that of GSD with different follow-up periods (GSD3, GSD5, and GSD10) were used to construct the PPI networks. The queried DEGs with an effective binding score >0.4 were used to build the PPI networks. The effective binding score represents how likely the interactions between nodes are true. In PPI networks, nodes and edges represent proteins and interactions, respectively, and the nodes with high degree are considered as hub genes/proteins. The interactive PPI networks were analyzed and visualized using Cytoscape v3.8.2 (figure 4A). The detailed statistics of the PPI networks analysis are given in supplementary table 1.

PPI network construction from the unique DEGs. (A) PPI networks of unique DEGs identified from GBC vs. GSD3, GBC vs. GSD5, and GBC vs. GSD10. The red triangle represents the hub genes identified based on the consensus of five topological algorithms. (B) Significant modules extract from the PPI networks. The red triangle in the module network represents hub genes.
CytoHubba, a Cytoscape plugin, was used to identify the hub DEGs from the PPI networks generated using the unique DEGs identified from DEG lists 2, 3, and 4. Five topological parameters (MCC, MNC, Degree, EPC, and Betweenness) were considered to identify the predicted hub DEGs. The 20 top-ranked DEGs identified from these five algorithms were considered for further evaluation (supplementary figure 1). The predicted hub DEGs from each of the topological parameter were intersected for the identification of consensus significant hub DEGs in the PPI networks (table 4). The identified hub genes in the PPI networks were mostly downregulated (supplementary table 2).
Functionally enriched significant modules in the PPI networks were identified using the Molecular Complex Detection (MCODE) algorithm (figure 4B). The significant module for GSD3 was associated with cell adhesion and collagen fibril organization, the GSD5 module was associated with ion transport and metabolic pathways, and the module identified for GSD10 was linked with immune system regulation.
3.5 Analysis of TRNs and identification of potential TFs
The TRNs were constructed through PWM scanning, followed by identification of transcription factor binding sites (TFBS) on the target DEGs. The TFs were considered as source nodes, and non-TF DEGs were considered as target nodes for each condition (figure 5). The topological analyses of the TRNs such as assortativity and shortest path length were calculated using igraph, an R package (Csardi and Nepusz 2006). The top 10 highly connected TFs were identified based on degree centrality (table 5). The topology of the TRN obtained from GSD3 was the largest, with 663 nodes and 4896 edges. Zinc finger Family (ZNF) proteins were the commonly enriched regulatory hubs in all the three GSD follow-up periods. ZNF genes act as tumor suppressor and oncogenes. They regulate the key pathways and processes of cancer initiation, development, as well as progression. Some of the key pathways and processes are apoptosis, metastasis, and regulation of transcription, protein degradation mediated by ubiquitin–proteasome pathway, etc.

Transcriptional regulatory network of the unique DEGs. The red node represents the hub genes identified based on degree centrality, and the small yellow nodes represent target DEGs. The TF–TG interactions are identified through PWM scanning of 1 kbp upstream sequences of significant DEGs that assign a probability score of the transcription factor binding to the target gene.
3.6 Prediction of miRNA interactions with the hub genes
The miRTar database was used to identify the miRNA regulators associated with the hub genes. The Network Analyzer tool in Cytoscape was used to analyze the miRNA regulatory network connections (figure 6A; supplementary table 3). In GSD3, COL1A1 had the highest number of interacting miRNAs, indicating its potential role in regulating crucial miRNAs associated with cancer progression. In GSD5, 3 out of the 8 hub genes were found to interact with miRNAs, viz., HBEGF, KIF5a, and GABRG2. In GSD10, GAPDH was found to have the highest number of miRNA connections, followed by CD3E and EGR2. However, no miRNAs were found to be associated with IL17A.

Hub gene analysis. (A) Hub gene–miRNA interactive networks. The small green nodes and big red nodes are the interacting miRNAs and hub genes, respectively. (B) Signaling network showing complex, associated proteins and signaling pathways associated with hub genes. The green nodes represent associated proteins; light blue nodes indicate signaling pathways; circled blue nodes represents hub genes; squared blue nodes represents signaling complex, and yellow nodes indicate protein families.
3.7 Identification of key signaling complex associated with hub genes
We identified the key signaling complex associated with the hub genes using the SIGNOR 2.0 database (figure 6B). The hub genes COL1A1 identified from the GBC vs. GSD3 PPI network analysis were associated with the ECM interaction signaling pathway through A11/b1 and A2/b1 integrin complexes. The ECM interaction pathway is known to be one of the hallmarks of cancer. The significant hub genes from GBC vs. GSD5 were associated with MAPK signaling pathways. The hub genes identified from GBC vs. GSD10 were largely associated with TCR signaling and PI3K signaling pathways. These pathways have been reported to be deregulated in many cancers (Sanchez-Vega et al. 2018).
3.8 Mining genomic alterations of the DEGs/hub genes from external datasets
Genomic alterations such as mutations and copy number variations (CNVs) associated with hub genes were evaluated from TCGA-GBC data and other TCGA datasets of gastrointestinal cancers such as esophageal cancer, stomach cancer, liver cancer, colorectal cancer, and pancreatic cancer using the cBioPortal database (figure 7). It was been observed that hub gene amplification is prominent in other gastrointestinal cancers, whereas the hub genes in GBC patients are associated with mutations. The OncoPrint tool of cBioportal showed 36% of patients’ cases to have genetic alterations such as amplification, deletion, and several mutations.

Genomic alterations associated with the hub genes in gastrointestinal cancers.
4 Discussion
Gallbladder cancer is known to be the most fatal malignancy of biliary tract cancer and it ranks sixth among the neoplasms of the gastrointestinal tract (Hundal and Shaffer 2014; Song et al. 2020). Among all the different risk factors, gallstones are considered as the major risk factor, as in most cases the cancer is incidentally diagnosed while the patient is undergoing treatment for gallstones or cholelithiasis (Hundal and Shaffer 2014). Identification of molecular markers for early diagnosis is very important to reduce the death risk of this cancer. Hence, our objective was to identify crucial molecular signatures that lead to the progression of GSD to GBC.
We carried out an integrative network-based analysis of transcriptomic datasets to compare and identify key molecular signatures in GBC with reference to GSD of different follow-up periods. Differential gene expression analysis and hierarchical clustering analysis showed significant variation in gene expression pattern among the unique DEGs identified from GSD with three different follow-up periods with respect to the GBC samples. The significant hub genes and TFs identified from GBC with reference to GSD of all the three follow-up periods were directly or indirectly associated with a few important processes and pathways known to be involved in cancer development and progression.
The hub genes identified from unique DEGs of GBC vs. GSD3 are SERPINH1, COL1A1, TPT1, and THBS1. The hub genes are linked with cell adhesion and collagen fibril organization processes. Cell adhesion molecules play an important role in regulating epithelial-to-mesenchymal transition (EMT) and influence malignant transformation and metastasis (Janiszewska et al. 2020). The Serpin Family H Member 1 (SERPINH1) gene is aberrantly expressed in different cancers: in gastric cancer it is involved in metastasis and EMT via the Wnt/β-catenin signaling pathway, and regulates the expression of the proteins of the extracellular matrix (ECM) to promote breast cancer (Tian et al. 2020). The COL1A1 gene encodes type 1 collagen, which is a major structural component of the ECM known to be involved in EMT. EMT allows epithelial cells to adopt a more mesenchymal state to enhance cellular migration, which thereby helps in the metastasis of cancer. Upregulation of COL1A1 promotes tumor metastasis by regulating the WNT/planar cell polarity (PCP) signaling pathway (Zhang et al. 2018). THBS1, or thrombospondin1, plays a key role in cellular communication, both cell-to-cell and cell-to-ECM interactions (Hu et al. 2021). Earlier studies reported that THBS1 was upregulated due to aberrant DNA methylation in various types of human cancer including breast cancer, gastric cancer, oral cancer, etc., to promote proliferation, invasion, and migration (Zhang 2021). TPT1, or Tumor Protein Translationally-Controlled 1, is an anti-apoptotic protein-coding gene which is involved in various cellular pathways like cell proliferation, growth, apoptosis, metabolism, and stabilization of microtubules during cell division (Zhang et al. 2021). It is also known to be involved in cancer progression and is differentially expressed in many types of human cancer. Studies revealed that TPT1 is upregulated in colon cancer and prostate cancer (Hosseinzadeh et al. 2020). In the case of epithelial ovarian cancer, TPT1 promotes tumor growth and metastasis via the TPT1/PI3K/AKT signaling pathway (Wu et al. 2019).
The hub genes identified from the unique DEGs in GBC vs. GSD5 were CX3CR1, GRM1, HBEGF, KIF5A, HEY2, GABRG2, GJA1, and GJA5. These hub genes are mainly involved in different types of cellular pathways including inflammation, cell growth and development, intracellular organelle transport, and cellular interaction. Inflammation in GBC due to GSD leads to the release of some carcinogenic molecules that ultimately result in tumor growth and development. Hub genes identified from the unique DEGs of GBC vs. GSD5 were associated with key cancer-related pathways such as inflammatory response, cellular interaction, as well as cell growth and proliferation, suggesting a higher risk of cancer progression. CX3CR1 or C-X3-C Motif Chemokine Receptor 1 is a transmembrane protein involved in the regulation of immune response, cell adhesion, inflammation, etc. But it is aberrantly expressed in many types of cancers like gastric cancer, breast cancer, pancreatic cancer, lung cancer, etc. (Marchesi et al. 2010; Wei et al. 2015). CX3CR1 overexpression in gastric cancer promotes migration, proliferation, and survival of tumors (Wei et al. 2015). However, in the case of glioma, neuroblastoma, and other non-neural-origin cancers, overexpression of CX3CR1 helps in the trans-endothelial migration and metastasis of cancer (Marchesi et al. 2010; Wei et al. 2015). GRM1 or Glutamate Metabotropic Receptor 1, is a G-protein-coupled receptor for glutamate that plays a crucial role in synaptic plasticity and the development of the cerebellum. GRM1 can hydrolyze phosphoinositide through phospholipase C activation. Apart from various neurological disorders, GRM1 is also known to be involved in human cancers like breast cancer, skin cancer, etc. Overexpression of GRM1 in melanocytes promotes tumor growth and progression through activation of PI3K/AKT and MAPK signaling pathways (Wangari-Talbot et al. 2012; Wen et al. 2014). However, GRM1 also involved in other cancer-related pathways like neuro-active ligand-binding receptor interaction, FOXO signaling pathway, etc. The growth factor HB-EGF, or heparin-binding epidermal growth factor-like growth factor, is one of the ligands of the epidermal growth factor receptor (EGFR) that mediates its function via ERBB1/HER1 (also EGFR) and ERBB4/HER4. According to various studies, HBEGF is highly expressed in hepatocellular carcinoma, breast cancer, colon cancer, prostate cancer, and ovarian cancer, where it can help in the growth, proliferation, and progression of tumors (Miyamoto et al. 2004; Miyata et al. 2012). KIF5A, or kinesin family member 5A, is a member of the kinesin family protein, mainly expressed in neurons. It acts as a microtubular motor protein in axonal transport (Brenner et al. 2018). It has been observed from various studies that the kinesin proteins were aberrantly expressed in different types of human cancers including breast cancer, prostate cancer, lung cancer, bladder cancer, etc. Kinesins mediate the process of tumorigenesis by promoting cell growth and proliferation (Rath and Kozielski 2012; Tian et al. 2019). GJA1, or Gap Junction Protein Alpha 1, and GJA5, or Gap Junction Protein Alpha 5, are members of the connexin family of proteins that are involved in cellular communication. In gastric cancer, higher expression of GJA1 leads to shorter overall survivability of patients (Zhao et al. 2019). HEY2, a bHLH transcription factor with YRPW motif 2, is a transcription-factor-encoding gene of the hairy and enhancer-of-split-related (HESR) family. Expression of HEY2 is regulated by the Notch signal transduction pathway and TGF-β signaling pathway, which are mostly dysregulated in various human cancers (Liu et al. 2017). According to previous studies, HEY2 is highly expressed in different cancers like esophageal squamous cell carcinoma and non-small-cell lung carcinoma, where it can promote metastasis, cancer cell self-renewal, angiogenesis, EMT, as well as tumor proliferation (Forghanifard et al. 2015; Liu et al. 2017; Cheng et al. 2018). In hepatocellular carcinoma, upregulation of HEY2 plays an important role in cancer progression through the TGF-β/Smad signaling pathway by inhibiting TGF-β-induced growth arrest (Wang et al. 2019). The GABRG2 gene encodes a subunit of the gamma-aminobutyric acid type A receptor. Although this gene is most commonly involved in the function of the central nervous system, a recent study has suggested it as a novel oncogene promoting tumor invasion and metastasis (Jin et al. 2017). In thyroid cancer, higher expression of GABRG2 promotes tumor metastasis to lymph node (Jin et al. 2017). GABRG2 is also found to be highly expressed in colon adenocarcinoma (Yan et al. 2020).
The significant hub genes identified from the specific DEGs in GBC compared with GSD with follow-up period of more than 10 years were LCK, CCR7, CD3E, IKZF1, EGR2, GAPDH, and NR4A1. The majority of the identified hub genes were associated with immune response signaling pathways such as the T-cell receptor (TCR) signaling pathways. Chronic inflammation caused by gallstones is known to be the most potential risk factor in GBC development. Therefore, it is largely associated with immune cells and inflammatory mediators such as such as cytokines, chemokines, reactive oxygen species, prostaglandins (PGs), and growth factors which strongly influence the genetic and epigenetic aberrations in oncogenes and/or tumor suppressor genes (TSG) (Hussain and Harris 2007). We identified that DEGs in GBC vs. GSD10 were associated with immune cell regulatory processes. The T-cells are known to be the principal defensive components against tumors and pathogens. T-cell activation functions to regulate a wide array of metabolic pathways and any aberration in the T-cell signaling pathway can lead to oncogenesis (Franchina et al. 2018). Lymphocyte cell-specific protein-tyrosine kinase (LCK) is an important gene that is expressed on T-lymphocytes and natural killer cell, and plays a significant role in T-cell receptor signaling, which can affect the pathogenesis or metastasis of cancer (Kumar Singh et al. 2018; Weiße et al. 2021). LCK phosphorylates CD79a, which induces the distal signaling events involved in the addition of phosphate group to Syk, and thereby activates different signaling pathways such as PI3K/Akt, NF-kB, and ERK. These signaling pathways are known to be involved in cancer cell survival, proliferation, and also resistance to treatment of cancer (Fresno Vara et al. 2004; Kumar Singh et al. 2018). LCK is highly expressed in small-cell lung cancer, non-small-cell lung carcinoma, as well as lung cancer (Bommhardt et al. 2019). In cholangiocarcinoma, expression of LCK is related to the recurrence of tumors (Bommhardt et al. 2019). CCR7, or C-C motif chemokine receptor 7, encodes a G-protein-coupled receptor family protein that plays a crucial role in adaptive immune response through activation of B- and T-lymphocytes. CCR7 helps the tumor cell to escape immune surveillance and helps cancerous cells to survive by the activation of the PI3K/Akt signaling pathway (Legler et al. 2014). IL17A, or interleukin 17A, is a member of the interleukin 17 pro-inflammatory cytokine family produced by T-helper 17 (Th17) cells. Expression of IL17A has been found to be high in various tumor tissues, such as hepatocellular carcinoma, gastric cancer, etc. (Wu et al. 2014). IKZF1, or IKAROS Family Zinc Finger 1, is a zinc-finger DNA binding protein that acts as a transcription factor. It is involved in various biological processes such as immune system regulation and proliferation of hematopoietic cells, and also regulates cellular interaction via the Notch signaling pathway (Jedi et al. 2018). Epigenetic studies revealed that in colorectal cancer, IKZF1 was downregulated due to hypermethylation (Pedersen et al. 2015; Jedi et al. 2018). EGR2, or early growth response 2, is a sequence-specific DNA-binding protein which is a member of the Kruppel-like zinc finger transcription factor family (Bradley et al. 2008). EGR2 induced apoptosis through phosphatase and tensin homolog deleted on chromosome 10 or the PTEN growth suppressive signaling pathway (Unoki and Nakamura 2003). However, the negative regulation of EGR2 through miR-20a (a small noncoding RNA) promotes the growth of gastric cancer (Li et al. 2013).
From this study, we have observed that the identified hub genes and hub TFs were associated with different cellular processes and pathways directly or indirectly linked with cancer progression and metastatic invasion. The hub genes identified from each of the GSD follow-up periods were associated with distinct processes and signaling pathways. This suggested that GSD progresses to GBC through the dysregulation of multiple signal transduction pathways at different stages (initiation–progression–metastasis) with distinct pathological spectra. Hence, the identified common and unique molecular signatures between GSD and GBC reflect possible mechanisms through which GSD progressed to GBC. Further in-depth functional evaluation of the hub genes and TFs will be able to establish their association with specific stages of disease development and progression.
Dataset availability
The datasets used for this study are available at NCBI-GEO database (SRA Accession No: SRP226150).
References
Bailey TL, Boden M, Buske FA, et al. 2009 MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 37 202–208
Barabási AL and Oltvai ZN 2004 Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 5 101–113
Bommhardt U, Schraven B and Simeoni L 2019 Beyond TCR signaling: Emerging functions of Lck in cancer and immunotherapy. Int. J. Mol. Sci. 20 1–18
Bradley EW, Ruan MM and Oursler MJ 2008 Novel pro-survival functions of the kruppel-like transcription factor Egr2 in promotion of macrophage colony-stimulating factor-mediated osteoclast survival downstream of the MEK/ERK pathway. J. Biol. Chem. 283 8055–8064
Brenner D, Yilmaz R, Müller K, et al. 2018 Hot-spot KIF5A mutations cause familial ALS. Brain 141 688–697
Chand Y and Alam MA 2012 Network biology approach for identifying key regulatory genes by expression-based study of breast cancer. Bioinformation 8 1132–1138
Chen S, Zhou Y, Chen Y and Gu J 2018 Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 i884–i890
Cheng D, Bao C, Zhang X, et al. 2018 LncRNA PRNCR1 interacts with HEY2 to abolish miR-448-mediated growth inhibition in non-small cell lung cancer. Biomed. Pharmacother. 107 1540–1547
Chin CH, Chen SH, Wu HH, et al. 2014 cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8 S11
Csardi G and Nepusz T 2006 The igraph software package for complex network research. InterJournal Complex Systems 1695 http://igraph.sf.net
de Sena Brandine G and Smith AD 2019 Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Research 8 1874
Dennis G, Sherman BT, Hosack DA, et al. 2003 DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4 https://doi.org/10.1186/gb-2003-4-9-r60
Forghanifard MM, Taleb S and Abbaszadegan MR 2015 Notch Signaling target genes are directly correlated to esophageal squamous cell carcinoma tumorigenesis. Pathol. Oncol. Res. 21 463–467
Franchina DG, Dostert C and Brenner D 2018 Reactive oxygen species: Involvement in T cell signaling and metabolism. Trends Immunol. 39 489–502
Fresno Vara JÁ, Casado E, de Castro J, et al. 2004 P13K/Akt signalling pathway and cancer. Cancer Treat. Rev. 30 193–204
Furlong LI 2013 Human diseases through the lens of network biology. Trends Genet. 29 150–159
Hosseinzadeh O, Hekmat Z, Nekoufar S, et al. 2020 Evaluate the gene expression of TPT1, EDN3, and ANO7 in prostate cancer tissues and their relation with age, tumor stage and family history. Meta Gene 24 100671
Hu XY, Ling ZN, Hong LL, et al. 2021 Circulating methylated THBS1 DNAs as a novel marker for predicting peritoneal dissemination in gastric cancer. J. Clin. Lab. Anal. https://doi.org/10.1002/jcla.23936
Hundal R and Shaffer EA 2014 Gallbladder cancer: Epidemiology and outcome. Clin. Epidemiol. 6 99–109
Hussain SP and Harris CC 2007 Inflammation and cancer: An ancient link with novel potentials. Int. J. Cancer 121 2373–2380
Janiszewska M, Primi MC and Izard T 2020 Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 295 2495–2505
Jedi M, Young GP, Pedersen SK and Symonds EL 2018 Methylation and gene expression of BCAT1 and IKZF1 in colorectal cancer tissues. Clin. Med. Insights Oncol. 12 1179554918775064
Jin Y, Jin W, Zheng Z, et al. 2017 GABRB2 plays an important role in the lymph node metastasis of papillary thyroid cancer. Biochem. Biophys. Res. Commun. 492 323–330
Kim D, Paggi JM, Park C, Bennett C and Salzberg SL 2019 Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 907–915
Knox SS 2010 From “omics” to complex disease: A systems biology approach to gene-environment interactions in cancer. Cancer Cell Int. 10 1–13
Kumar Singh P, Kashyap A and Silakari O 2018 Exploration of the therapeutic aspects of Lck: A kinase target in inflammatory mediated pathological conditions. Biomed. Pharmacother. 108 1565–1571
Legler DF, Uetz-Von Allmen E and Hauser MA 2014 CCR7: Roles in cancer cell dissemination, migration and metastasis formation. Int. J. Biochem. Cell Biol. 54 78–82
Letelier P, Brebi P, Tapia O and Roa JC 2012 DNA promoter methylation as a diagnostic and therapeutic biomarker in gallbladder cancer. Clin. Epigenetics 4 11
Li X, Zhang Z, Yu M, et al. 2013 Involvement of miR-20a in promoting gastric cancer progression by targeting early growth response 2 (EGR2). Int. J. Mol. Sci. 14 16226–16239
Liao Y, Smyth GK and Shi W 2014 FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 923–930
Liu Z, Sanders AJ, Liang G, et al. 2017 Hey factors at the crossroad of tumorigenesis and clinical therapeutic modulation of hey for anticancer treatment. Mol. Cancer Therapeut. 16 775–786
Love MI, Anders S and Huber W 2014 Differential analysis of count data - the DESeq2 package. Genome Biol. 15 550
Marchesi F, Locatelli M, Solinas G, et al. 2010 Role of CX3CR1/CX3CL1 axis in primary and secondary involvement of the nervous system by cancer. J. Neuroimmunol. 224 39–44
Masoudi-Nejad A and Wang E 2015 Cancer modeling and network biology: Accelerating toward personalized medicine. Semin. Cancer Biol. 30 1–3
Miyamoto S, Hirata M, Yamazaki A, et al. 2004 Heparin-binding EGF-like growth factor is a promising target for ovarian cancer therapy. Cancer Res. 64 5720–5727
Miyata K, Yotsumoto F, Nam SO, Kuroki M and Miyamoto S 2012 Regulatory mechanisms of the HB-EGF autocrine loop in inflammation, homeostasis, development and cancer. Anticancer Res. 32 2347–2352
Muhammad JS, Khan MR and Ghias K 2018 DNA methylation as an epigenetic regulator of gallbladder cancer: An overview. Int. J. Surgery 53 178–183
Oliveros JC 2007 VENNY. An interactive tool for comparing lists with Venn diagrams (http://Bioinfogp.Cnb.Csic.Es/Tools/Venny/Index.Html)
Pedersen SK, Baker RT, McEvoy A, et al. 2015 A two-gene blood test for methylated DNA sensitive for colorectal cancer. PLoS One 10 1–14
Pruitt KD, Hogue CW, Groll M, et al. 2001 An automated method for finding molecular complexes in large protein interaction networks. Nucleic Acids Res. 29 137–140 https://academic.oup.com/nar/article-lookup/doi/10.1093/nar/29.1.137
Rath O and Kozielski F 2012 Kinesins and cancer. Nat. Rev. Cancer 12 527–539
Rawla P, Sunkara T, Thandra KC and Barsouk A 2019 Epidemiology of gallbladder cancer. Clin. Exp. Hepatol. 5 93–102
Roy N, Gaikwad M, Bhattacharrya DK and Barah P 2021 Identification of systems level molecular signatures from glioblastoma multiforme derived extracellular vesicles. J. Mol. Neurosci. 71 1156–1167
Sanchez-Vega F, Mina M, Armenia J, et al. 2018 Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173 321–337.e10
Sharma A, Sharma KL, Gupta A, Yadav A and Kumar A 2017 Gallbladder cancer epidemiology, pathogenesis and molecular genetics: Recent update. World J. Gastroenterol. 23 3978–3998
Shannon P, Markiel A, Ozier O, et al. 2003 Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 2498–2504
Song X, Hu Y, Li Y, et al. 2020 Overview of current targeted therapy in gallbladder cancer. Signal Transduct. Target. Ther. 5 https://doi.org/10.1038/s41392-020-00324-2
Stormo GD 2000 DNA binding sites: Representation and discovery. Bioinformatics 16 16–23
Suratanee A and Plaimas K 2018 Network-based association analysis to infer new disease-gene relationships using large-scale protein interactions. PLoS One 13 1–20
Thomas-Chollier M, Sand O, Turatsinze JV, et al. 2008 RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 36 https://doi.org/10.1093/nar/gkn304
Tian DW, Wu ZL, Jiang LM, et al. 2019 KIF5A promotes bladder cancer proliferation in vitro and in vivo. Dis. Markers https://doi.org/10.1155/2019/4824902
Tian S, Peng P, Li J, et al. 2020 SERPINH1 regulates EMT and gastric cancer metastasis via the Wnt/β-catenin signaling pathway. Aging 12 3574–3593
Unoki M and Nakamura Y 2003 EGR2 induces apoptosis in various cancer cell lines by direct transactivation of BNIP3L and BAK. Oncogene 22 2172–2185
Wang J, Zhu B, Zhang Y, et al. 2019 HEY2 acting as a co-repressor with smad3 and smad4 interferes with the response of TGF-beta in hepatocellular carcinoma. Am. J. Translat. Res. 11 4367–4381
Wang J, Xu C, Cheng Q, et al. 2020 RNA Sequencing revealed signals of evolution from gallbladder stone to gallbladder carcinoma. Front. Oncol. 10 https://doi.org/10.3389/fonc.2020.00823
Wangari-Talbot J, Wall BA, Goydos JS and Chen S 2012 Functional effects of GRM1 suppression in human melanoma cells. Mol. Cancer Res. 10 1440–1450
Wei LM, Cao S, Yu WD, Liu YL and Wang JT 2015 Overexpression of CX3CR1 is associated with cellular metastasis, proliferation and survival in gastric cancer. Oncol. Rep. 33 615–624
Weirauch MT, Yang A, Albu M, et al. 2014 Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158 1431–1443
Weiße J, Rosemann J, Müller L, et al. 2021 Identification of lymphocyte cell-specific protein-tyrosine kinase (LCK) as a driver for invasion and migration of oral cancer by tumor heterogeneity exploitation. Mol. Cancer 20 88
Wen Y, Li J, Koo J, et al. 2014 Activation of the glutamate receptor GRM1 enhances angiogenic signaling to drive melanoma progression. Cancer Res. 74 2499–2509
Wu W, Gao H, Li X, et al. 2019 LncRNA TPT1-AS1 promotes tumorigenesis and metastasis in epithelial ovarian cancer by inducing TPT1 expression. Cancer Sci. 110 1587–1598
Wu X, Zeng Z, Xu L, et al. 2014 Increased expression of IL17A in human gastric cancer and its potential roles in gastric carcinogenesis. Tumor Biol. 35 5347–5356
Yan L, Gong YZ, Shao MN, et al. 2020 Distinct diagnostic and prognostic values of γ-aminobutyric acid type A receptor family genes in patients with colon adenocarcinoma. Oncol. Lett. 20 275–291
Zhang L, Ye F, Zuo Z, et al. 2021 Long noncoding RNA TPT1-AS1 promotes the progression and metastasis of colorectal cancer by upregulating the TPT1-mediated FAK and JAK-STAT3 signalling pathways. Aging 13 3779–3797
Zhang X 2021 Upregulation of THBS1 is related to immunity and chemotherapy resistance in gastric cancer. J. Gen. Med. 14 4945–4957
Zhang Z, Wang Y, Zhang J, Zhong J and Yang R 2018 COL1A1 promotes metastasis in colorectal cancer by regulating the WNT/PCP pathway. Mol. Med. Rep. 17 5037–5042
Zhao X, Yu C, Zheng M and Sun J 2019 Prognostic value of the mRNA expression of gap junction α members in patients with gastric cancer. Oncol. Lett. 18 1669–1678
Acknowledgements
PB would like to acknowledge the Department of Biotechnology, India, for providing the Ramalingaswami Re-entry Fellowship grant.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors contributed to this manuscript declare no conflict of interests.
Additional information
Communicated by Susmita Roy.
Corresponding editor: Susmita Roy
This article is part of the Topical Collection: Emergent dynamics of biological networks.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Roy, N., Dihingia, B.R. & Barah, P. Integrative network-based approaches identified systems-level molecular signatures associated with gallbladder cancer pathogenesis from gallstone diseases. J Biosci 47, 31 (2022). https://doi.org/10.1007/s12038-022-00267-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-022-00267-6