
Abstract
Alzheimer’s disease (AD) is a progressive neurological disorder characterized by cognitive decline. This study was undertaken to evaluate the effects of selegiline (SEL) against AD-induced cognitive deficits and explore the possible involved mechanisms. AD was induced by unilateral intracerebroventricular (U-ICV) injection of 5 μg of amyloid beta1-42 (Aβ1-42), and oral administration of SEL (0.5 mg/kg/day) was performed for 30 consecutive days. Aβ injection resulted in spatial cognitive decline, as demonstrated by a decrease in the time spent in the target zone on the probe day (P < 0.01) in the Barnes maze test (BMT). This spatial cognitive decline was associated with disrupted synaptic plasticity, as indicated by reductions in both components of hippocampal long-term potentiation (LTP), namely population spike amplitude (P < 0.001) and field excitatory postsynaptic potential (P < 0.001). On the other hand, the injection of Aβ resulted in oxidative stress by decreasing total thiol group (TTG) content and increasing malondialdehyde (MDA) levels in the rat plasma (P < 0.001). Additionally, the number of healthy cells in the hippocampal dentate gyrus (DG) and CA1 regions was reduced in AD rats (P < 0.001). However, oral administration of SEL improved spatial cognitive decline in the Aβ-induced AD rats. The results suggest that improvement of neuroplasticity deficiency, regulation of oxidant/antioxidant status, and suppression of neuronal loss by SEL may be the mechanisms underlying its beneficial effect against AD-related spatial cognitive impairment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a progressive neurological disorder and the main cause of cognitive decline, affecting mental capacity and cognitive function [1]. Several risk factors such as increasing age, genetic factors, head injuries, and environmental factors are implicated in the development of this multifactorial disease [2]. The pathology of AD is studied to understand different mechanisms, such as abnormal metabolism of tau protein, amyloid beta (Aβ), inflammatory response, cholinergic dysfunction, and oxidative stress [3]. Aβ plays a major role in neuronal toxicity and impairment of neuronal function, so the accumulation of Aβ plaques in the hippocampus, amygdala, and cerebral cortex can lead to axonal damage, dendritic loss, synaptic loss, and cognitive impairments [4]. Oxidative stress and Aβ are interconnected and positively affect each other’s increase. The brain’s vulnerability to oxidative stress, caused by decreased antioxidants or accumulation of free radicals within cells, is considered a critical detrimental factor in AD. AD begins years before its symptoms appear, and antioxidant therapy can be an important therapeutic target to combat this disease [5]. However, to date, there is no definitive treatment that completely halts the progression of AD. Therefore, finding effective treatments for AD can be highly valuable.
It has been shown that monoamine oxidase B (MAO-B) expression increases in AD brains’ cortex and hippocampus compared to healthy brains [6]. Furthermore, there is a significantly more than three-fold increase in active MAO-B in reactive astrocytes surrounding Aβ deposits [7]. This excessive expression of MAO-B in astrocytes is believed to contribute to the improper breakdown of monoamine neurotransmitters (dopamine, norepinephrine, epinephrine, and serotonin) and the heightened production of free radicals and hydrogen peroxide (H2O2), potentially promoting the observed neurodegenerative processes in AD [8]. This phenomenon appears to be an early occurrence in AD that persists throughout the progression of the disease [9]. These findings suggest that the inhibition of MAO-B through drugs may offer a new strategy to alleviate the pathological symptoms of AD [10].
Selegiline (SEL), a selective and irreversible inhibitor MAO-B, is clinically used to slow the progression of Parkinson’s disease (PD) [11]. SEL exhibits a wide spectrum of biological properties, including anti-inflammatory [12], anti-apoptotic [13, 14], neurotrophic [15, 16], and cognitive-ameliorating effects [17, 18]. Previous studies have demonstrated SEL’s ability to scavenge reactive oxygen species (ROS) and suppress lipid peroxidation in various neurodegenerative diseases and insult models, including PD [19], cerebral ischemia [20], scopolamine-induced memory deficits [21], and 3-nitropropionic acid (3-NP)-induced neurotoxicity [22]. Additionally, SEL has shown neuroprotective properties in conditions such as traumatic brain injury (TBI) [23], cerebral ischemia [20, 24], schizophrenia [25], PD [26], and major depressive disorder [27]. Given SEL’s capability to increase levels of monoamine neurotransmitters, particularly dopamine [28], it offers a potential avenue for improving cognitive deficits associated with AD. Preclinical and clinical trials have been conducted to evaluate SEL’s efficacy in treating cognitive deficits associated with AD [29,30,31,32,33]. However, the underlying mechanisms involved in SEL’s therapeutic effects on AD-associated cognitive deficits remain poorly understood.
Therefore, this study aimed to investigate the effects of chronic oral administration of SEL on spatial cognitive ability in an Aβ1-42-infused AD rat model. Additionally, this study aimed to identify specific underlying mechanisms, including hippocampal synaptic plasticity and oxidative stress.
Materials and Methods
Animals and Experimental Design
Forty adult male Wistar rats were obtained from the animal house of Hamedan University of Medical Sciences (Hamedan, Iran), with a weight range of 210 to 230 g. The rats were housed in an animal room with a controlled temperature of 22 ± 2 ℃, relative humidity of 60 ± 5%, and a 12-h light–dark cycle, with free access to food (47% carbohydrate, 5% fat, 23% protein, 5% fiber, 20% water, vitamins, and minerals) and water. Animal care and experimental procedures were conducted in accordance with NIH guidelines and approved by the ethics committee of Bu-Ali Sina University–Hamedan (ethical code IR.BASU.REC.1400.001). After a 7-day adaptation, the rats were randomly divided into five groups of eight as follows:
-
I.
Control group: Rats received 5 ml/kg/day of 0.9% normal saline via oral gavage (P.O.) for 30 days.
-
II.
PBS group: Rats received a stereotaxic unilateral intracerebroventricular (U-ICV) injection of phosphate-buffered saline (PBS; 5 μL/rat) plus normal saline (5 ml/kg/day; P.O. for 30 days).
-
III.
SEL group: Rats received SEL (0.5 mg/kg/day; P.O. for 30 days).
-
IV.
AD group: Rats received a stereotaxic U-ICV injection of 5 μL of Aβ1-42 (1 μg/μL) plus normal saline (5 ml/kg/day; P.O. for 30 days).
-
V.
AD + SEL (AS) group: Rats received SEL (0.5 mg/kg/day; P.O. for 30 days) after a stereotaxic U-ICV injection of 5 μL of Aβ1-42 (1 μg/μL).
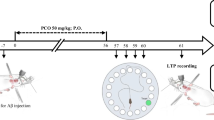
A solution of SEL was prepared immediately before use and administered once daily for 30 consecutive days via oral gavage. The dose of SEL used in this study was chosen based on available literature [34,35,36,37,38,39]. Numerous studies have reported that administration of SEL at the dose of 0.5 mg/kg in animal models has beneficial effects on behavioral, electrophysiological, and neurochemical features [34,35,36,37,38,39]. Figure 1 shows the experimental timeline. Following 1 week of adaptation to the laboratory environment, Aβ solution injection was performed. Subsequently, the rats had 7 days of recovery. The Barnes maze test (BMT) was assessed from the 31st 34th (n = 8). Hippocampal long-term potentiation (LTP) was evaluated on the 35th day (n = 8). The biochemical assessments (n = 8) and histological analysis (n = 4) were also examined on the same day.

The rats underwent a unilateral intracerebroventricular (U-ICV) injection of a 5-μL solution of Aβ1-42 (1 mg/mL). Selegiline was administered at a dose of 0.5 mg/kg/day; P.O. for 30 days. Subsequently, spatial memory testing and LTP recording were performed. At the end of the experiment, plasma biomarkers (MDA and TTG) were measured, and hematoxylin and eosin staining were performed on the hippocampal tissue
AD Induction
Based on previous studies [40,41,42,43], a 5-μL solution of Aβ (1 mg/mL) was administered to induce AD in rats. Therefore, Aβ peptide1-42 rat (product No/SKU SCP0038-1MG, Sigma–Aldrich, USA) was dissolved in PBS solution. Before U-ICV injection, the Aβ solution was incubated at 37 °C for 4 days. Amyloid fibrils are produced during this process, which are neurotoxic [41, 44].
For AD induction, each rat was anesthetized by intraperitoneally (I.P.) injection of a mixture of ketamine (100 mg/kg) and xylazine (10 mg/ kg). Stereotaxic (Dual Lab Standard Stereotaxic apparatus; Stoelting Co., Wood Dale, IL, USA) surgery was performed according to Paxinos and Watson’s rat brain atlas [45]. The head was shaved, and a midline sagittal incision was made in the scalp. A tiny hole was drilled carefully up to the level of the dura mater in the skull over the ventricular region (coordinates relative to bregma: medial–lateral (M/L), 1.5 mm and anterior–posterior (A/P), − 0.9 mm). Hamilton syringe needle was slowly directed down to beneath the surface of the cortex for the U-ICV injections, into the right lateral ventricle (coordinates relative to the skull: dorsal–ventral (D/V), 3.2 mm) [45]. A 5-μL of Aβ solution was administered at a 1 μL/min rate. The PBS group received 5 μL of PBS (10 mM), the same volume as the Aβ injection (Fig. 1).
Barnes Maze Test (BMT)
The BMT provides a means to measure spatial learning and memory, similar to the Morris water maze (MWM). However, the BMT imposes less stress on the rats than the MWM, which involves swimming in water [46]. The BMT is positioned 90 cm above the ground. It consists of a gray circular platform with a diameter of 120 cm, divided into four hypothetical equal zones (northeast, northwest, southeast, and southwest). There are 18 holes with a diameter of 10 cm around the circular platform. At the bottom of one of the holes, a black box (20 cm3) is placed and considered the target hole. In the testing room, various spatial cues are visible in four directions for the animal on the platform (Fig. 1). This test aims to measure the ability to learn and remember the location of the target hole and box using the installed cues on the walls. The rat is initially placed in the center of the platform so that the movement of all rats is recorded equally from the center of the platform. An 80-dB noise acts as an escape stimulus for the animal, emitted by a hidden audio device 40 cm away from the circular platform. This loud sound stops by finding the target hole and the rat entering the target box to stimulate the animal to enter the box. The bell rings again if the rat moves away from the target hole. After the rat spends 20 s in the escape box, it is taken out of the BMT. This learning phase of the test was repeated for 3 consecutive days as learning days (3 trials per day, with a 60-min interval between each repetition, each trial for 180 s). In these stages, the learning process of the animal was measured based on the time spent (escape latency) and distance traveled in the maze until entering the target box, as well as the number of search errors (checking non-target holes). On the fourth day (Probe), the target box was removed and the activity of the rat was recorded for 180 s without interrupting the sound. The time spent in the target zone and the number of search errors were recorded. All sessions were recorded with a camera and video tracking software.
Long-Term Potentiation (LTP)
The urethane solution (1.5 g/kg, I.P.) was injected for deep anesthesia and then the rat was placed in the stereotaxic apparatus. The locations of the dentate gyrus (DG coordinates were AP, − 3.8 mm and ML, 2.3 mm relative to bregma; DV, 2.7 to 3.2 mm relative to the surface of the skull) and perforant pathway (PP coordinates were AP, − 8 mm and ML, 4.3 mm relative to bregma; DV, 3.2 mm relative to the surface of the skull) were determined using the Paxinos atlas, and two holes were drilled in the designated points on the skull (Fig. 1). The stimulating and recording electrodes (stainless steel with Teflon cover, 125 μm bare diameter, 175 μm coated diameter, A.M. Systems Inc., USA) were moved gently to the PP and DG, respectively (Fig. 2a). Single 0.1 ms biphasic square wave pulses at the frequency of 0.1 Hz were used for stimulation (eProbe software protocol in baseline stimulation: delay, 20,000 µs; pulse duration, 200 µs; pulse cycle, 100 µs, train: one; trial numbers, 10; trial period, 10 s). The baseline stimulation intensity for each rat was calculated based on the input–output (I/O) curve. This curve was plotted by recording the amplitude of population spikes (PS) at varying intensities, and 40% of the maximum response was considered as the baseline stimulation intensity (Fig. 2c). When the response was stable in a 10–20 min control period, LTP was induced using a high-frequency stimulation (HFS) protocol of 400 Hz (eProbe software protocol in HFS: delay, 0 µs; pulse duration, 200 µs; pulse cycle, 2500 µs, train, 20; trial numbers, 10; trial period, 10 s).

The positions and traces of stimulating and recording electrodes on the DG and PP are represented in the transverse section of the hippocampus (a). Measurement of evoked potentials (b). Sample of an I/O curve of PS amplitude in hippocampal DG following PP stimulation (c)
Using the eLab system (ScienceBeam, Iran) and related computer software (eProbe), the PS amplitude and the slope of field excitatory postsynaptic potentials (fEPSP) were recorded at 5, 30, and 60 min after HFS in the granular cells layer of hippocampal DG following stimulation of the PP. Changes in PS amplitude and fEPSP slope were calculated during electrophysiological recordings, according to Eqs. 1 and 2, respectively (Fig. 2b):
where ΔV1 = The potential difference between two points e, as the peak of the first positive wave, and f, as the peak of the first negative deflection; ΔV2 = The potential difference between two points g, as the peak of the second positive wave and f as the peak of the first negative deflection; ΔT = Time difference between two points c and d; ΔV = The potential difference between two points c and d, that were between 20 and 80% of the first positive wave.
The values of the fEPSP slope and the PS amplitude at 5, 30, and 60 min were normalized relative to their baselines to measure the LTP magnitude (Eq. 3). Significant increase (P < 0.05) in PS amplitude and fEPSP slope (% change) was considered as a successful induction of LTP [41, 47, 48].
Biochemical Assay
Blood samples were collected from the hepatic portal vein. Each sample was centrifuged at a speed of 3500 rpm for 20 min, and then the clear plasma was divided into 100-µl aliquots and stored at − 80 ℃ until use. Malondialdehyde (MDA) and total thiol group (TTG) measurement kits were used according to the manufacturer’s protocols to calculate oxidative and antioxidant biomarkers levels in rat plasma.
Histology Analysis
After blood collection, four rats from each group were perfused with 0.9% normal saline and 10% formalin solution. Brains were fixed in a 10% formalin solution for 72 h. A 21-h protocol was performed in a tissue processor, and the brains were embedded in paraffin. Sections with a thickness of 5 µm were cut. After deparaffinization and rehydration, the sections were washed in distilled water. Hematoxylin–eosin staining was performed according to standard methods: staining with hematoxylin (8 min), rinsing in tap water, placing in 1% HCL and lithium carbonate (each for 30 s), staining with eosin (2 min), rinsing in tap water, rinsing in graded alcohol, and finally clearing in xylene. The number of healthy pyramidal cells in the hippocampus’s dentate gyrus (DG) and CA1 regions was counted.
Statistical Analysis
Data were analyzed and plotted using GraphPad Prism software, version 9 (GraphPad Software, San Diego, CA, USA). Normality of data was assessed using the Shapiro–Wilk test (P > 0.05). Analysis of variance (ANOVA) was performed, and if significant, post hoc analysis (based on equal variance of groups in Bartlett’s test and number of evaluated groups) was used. Data of BMT (distance moved, escape latency, and number of errors in learning days) and LTP (fEPSP slopes and PS amplitudes) related to the treatment (normal saline or SEL) and exposure (Aβ or non-Aβ) were compared and analyzed by Repeated-measures two-way ANOVA (two-way RM ANOVA) followed by Tukey’s post hoc test. The results of the mean percentage of potentiation between groups in LTP, time spent in the target zone on probe day of BMT, oxidative stress, and neuron counting data were compared using one-way ANOVA followed by Tukey’s or Bonferroni’s post hoc test. In all statistical analyses, P < 0.05 was considered significant, and the results were presented as mean ± standard deviation (mean ± SD).
Results
Body Weight
There was no significant difference in initial weight among the study groups (F (4, 35) = 1.005, P = 0.41). Additionally, no significant difference in final weight was observed at the end of the study (F (4, 35) = 2.12; P = 0.09, Fig. 3).

Effects of selegiline on body weights of AD rats. Data is presented as means ± SD of 8 animals per group (one-way ANOVA). ns, no significance
The Effects of SEL on the BMT in AD Rats
A sample of recorded activities in the BMT is shown in Fig. 4a. Two-way RM ANOVA showed a significant difference in distance moved to reach the target box over the 3 days of training in terms of the time (F (2, 21) = 157.0, P < 0.001), treatment (F (4, 84) = 9.71, P < 0.001), and time × treatment interaction effect (F (8, 84) = 4.48, P < 0.001). Subsequent Tukey’s post hoc test revealed a significant increase in the AD group compared to the control group on the second (P < 0.01) and third (P < 0.001) days. Oral administration of SEL resulted in a significant decrease in the AS group compared to the AD group on the second (P < 0.05) and third (P < 0.01) days of learning (Fig. 4b). However, the injection of PBS in the rats of the PBS group and administration of selegiline in the SEL group had no significant effect on the distance moved to reach the target box over the 3 days of training compared to the control group (P > 0.05 for each comparison).

The effects of selegiline on the BMT. Samples of recorded activities (a), distance traveled (b), escape latency (c), search errors of the learning days (d), time spent in the target zone (e), and search errors of the probe day (f) in the Barnes maze test. Data is presented as means ± SD of 8 animals per group (two- and one-way ANOVA, Tukey’s post hoc test). ns, no significance; *P < 0.05, **P < 0.01, ***P < 0.001
Similarly, two-way RM ANOVA showed a significant difference in the time taken to reach the target box over the 3 days of training in terms of the time (F (2, 21) = 134.7, P < 0.001) and treatment (F (4, 84) = 10.37, P < 0.001). However, there was no significant time × treatment interaction effect (F (8, 84) = 0.939, P = 0.488). Tukey’s post hoc test revealed a significant increase in the AD group compared to the control group on the second (P < 0.05) and third (P < 0.01) days. Oral administration of SEL resulted in a significant decrease in the AS group compared to the AD group on the second (P < 0.05) and third (P < 0.05) days of learning (Fig. 4c). However, the injection of PBS in the rats of the PBS group and administration of selegiline in the SEL group had no significant effect on the time taken to reach the target box over the 3 days of training compared to the control group (P > 0.05 for each comparison).
Additionally, two-way RM ANOVA showed a significant difference in the number of errors in searching for the hidden box over the 3 days of training in terms of the time (F (2, 21) = 453.5, P < 0.001) and treatment (F (4, 84) = 8.30, P < 0.001). However, there was no significant time × treatment interaction effect (F (8, 84) = 0.931, P = 0.495). Tukey’s post hoc test revealed a significant increase in the AD group compared to the control group on the second (P < 0.01) and third (P < 0.05) days. Oral administration of SEL resulted in a significant decrease in the AS group compared to the AD group on the second (P < 0.05) and third (P < 0.05) days of learning (Fig. 4d). However, the injection of PBS in the rats of the PBS group and administration of selegiline in the SEL group had no significant effect on the number of errors in searching for the hidden box over the 3 days of training compared to the control group (P > 0.05 for each comparison).
Furthermore, one-way ANOVA showed a significant difference in the time spent in the target zone on the probe day among the groups (F (4, 35) = 7.11, P < 0.001). Tukey’s post hoc test revealed a significant decrease in the AD group compared to the control group (P < 0.01), and a significant increase in the AS group compared to the AD group (P < 0.05, Fig. 4e).
Additionally, one-way ANOVA showed a significant difference in the number of errors in searching for the target hole on the probe day (F (4, 35) = 5.67, P = 0.001). Therefore, the AD group had a significant increase compared to the control group (P < 0.01), and the AS group showed a significant decrease compared to the AD group (P < 0.05, Fig. 4f).
The Effects of SEL on the fEPSP Slopes and PS Amplitudes of DG Granular Cells Layer in AD Rats
Field potentials were recorded from the hippocampal DG after stimulation of the hippocampal PP. According to Fig. 5e, a sample of the evoked field potential in the DG was recorded before HFS delivery (stable baseline response) and after tetanus.

The effects of selegiline (0.5 mg/kg/day for 30 consecutive days) on the evoked field potential in the hippocampal DG after HFS in AD rats. a, b Time course diagrams showing the changes in fEPSP slope and PS amplitude, respectively, prior to HFS and 5, 30, and 60 min after HFS of the PP (two-way RM ANOVA, Tukey’s post hoc test). ##P < 0.01, ###P < 0.001 compared between the control group and AD group; &P < 0.05, &&P < 0.01 compared between AD group and AS group. c, d The percentage changes of fEPSP slope and PS amplitude during LTP induction, respectively (one-way ANOVA, Tukey’s post hoc test). Data are expressed as means ± SD % of baseline of 8 animals per group. ***P < 0.001. e Evoked field potentials in the DG of the experimental groups were measured before and 60 min after HFS
fEPSP slope was significantly affected by both time points (F (3, 28) = 123.5, P < 0.001) and treatment (F (4, 112) = 15.37, P < 0.001) in a two-way RM ANOVA (Fig. 5a). However, there was no significant time × treatment interaction effect (F (12, 112) = 1.795, P = 0.057). Tukey’s post hoc analysis indicated a significant decrease at different times of 5 (P < 0.001), 30 (P < 0.001), and 60 min (P < 0.001) after HFS in the AD group than in the control group. Oral administration of SEL prevented the decremental effect of Aβ on the slope of fEPSP in the AS group at different times of 5 (P < 0.01), 30 (P < 0.05), and 60 min (P < 0.01); so that their magnitudes were similar to animals in the control group. However, the injection of PBS in the rats of the PBS group and administration of selegiline in the SEL group had no significant effect at different times of 5,30, and 60 min after HFS on fEPSP slope compared to the control group (P > 0.05 for each comparison).
The mean percent fEPSP slope change during 60 min after HFS (F (4, 35) = 20.34, P < 0.001) was significantly smaller in the AD group in comparison to the control group (P < 0.001, Fig. 5c); This suggests that HFS did not considerably change the fEPSP slope and LTP induction was impaired in AD group.
According to the two-way ANOVA, the PS amplitudes of the granular cells layer are significantly influenced by time points (F (3, 28) = 67.83, P < 0.001), treatments (F (4, 112) = 18.04, P < 0.001), and time × treatment interaction effect (F (12, 112) = 2.11, P = 0.021). Tukey’s post hoc analysis indicated a significant decrease at different times of 5 (P < 0.01), 30 (P < 0.001), and 60 min (P < 0.001) after HFS in the AD group than in the control group. Oral administration of SEL prevented these changes, so there was a significant difference in PS amplitudes in the AS group at different times of 5 (P < 0.01), 30 (P < 0.01), and 60 min (P < 0.01) compared to the AD group (Fig. 5b). However, the injection of PBS in the rats of the PBS group and administration of selegiline in the SEL group had no significant effect at different times of 5,30, and 60 min after HFS on PS amplitude compared to the control group (P > 0.05 for each comparison).
The mean percent PS amplitude change during 60 min after HFS (F (4, 35) = 8.94, P < 0.001) was significantly smaller in the AD group in comparison to the control group (P < 0.001, Fig. 5d); this suggests that HFS did not considerably change the PS amplitude and LTP induction was impaired in AD group.
Effect of SEL and Aβ on TTG and MDA in AD Rats
One-way analysis of variance showed a significant difference in the plasma levels of MDA among the groups (F (4, 35) = 11.4, P < 0.001). Therefore, the AD group showed a significant increase compared to the control group (P < 0.001). Additionally, the AS group had a significant decrease compared to the AD group (P < 0.05, Fig. 6a).

The effects of selegiline (0.5 mg/kg/day, for 30 consecutive days) on the plasma parameters of malondialdehyde (MDA) (a) and total thiol group (TTG) (b) of AD rats using assay kits. Data is presented as means ± SD of 8 animals per group (one-way ANOVA, Tukey’s post hoc test). *P < 0.05 and ***P < 0.001
Similarly, one-way ANOVA showed a significant difference in the plasma levels of TTG among the groups (F (4, 35) = 11.13, P < 0.001). The AD group showed a significant decrease compared to the control group (P < 0.001). Additionally, the AS group significantly increased compared to the AD group (P < 0.05, Fig. 6b).
The Effect of SEL and Aβ on the Histological Changes in the Hippocampus
Hematoxylin & Eosin staining was conducted to confirm the histological changes in the hippocampus’s DG and CA1 regions in the brains of the rats. As illustrated in Fig. 7a, a significant drop was found in the number of intact neurons of the hippocampal CA1 (F (2, 9) = 77.43; P < 0.001) and DG (F (2, 9) = 84.95; P < 0.001) regions of the AD group in comparison to the control group. Interestingly, the administration of SEL prevented neuronal death in the AS group. There was a significant difference in the number of intact neurons in the hippocampal CA1 (P < 0.001) and DG (P < 0.001) regions between the AS group and the AD group (Fig. 7b).

Effects of selegiline and Aβ on histological changes in the hippocampal CA1 and DG regions of rats. a The figure illustrates intact neurons (identified by black arrows) and dark neurons (identified by blue arrows, H & E stain, × 40 magnification). b The quantitative data of the number of intact neurons. Data is presented as means ± SD of 4 animals per group (one-way ANOVA, Bonferroni post hoc test). ***P < 0.001
Discussion
This study’s key findings are as follows: (1) SEL alleviated Aβ-induced spatial cognitive impairment in the rats. (2) SEL improved hippocampal synaptic plasticity deficit caused by Aβ in the rats. (3) SEL modulated the oxidant/antioxidant status in the Aβ-infused rats. (4) SEL ameliorated neuronal loss in the rat hippocampus. (5) Aβ and treatment with the SEL did not affect body weight.
The BMT used in the current work is a widely employed model in behavioral neuroscience to investigate spatial learning and memory abilities [49]. In the present study, the findings of BMT showed that Aβ injection leads to spatial learning impairment, as the time and distance traveled to reach the target box and the number of search errors increased in the rats during 3 days of learning. Moreover, the target zone occupancy was markedly alleviated in the probe trial, indicating impaired spatial memory in the Aβ-induced AD rats. These findings were consistent with previous studies [50, 51]. However, SEL treatment reduced the distance traveled, the latency to reach the target box, and the number of search errors. On the other hand, treatment with SEL prolonged the target zone occupancy in the Aβ-induced AD rats. These results suggest that oral SEL treatment was able to improve spatial learning and memory impairments in the Aβ-infused rats. The findings of this study are consistent with previous studies, demonstrating that SEL could improve spatial learning and memory deficits in animal models of cerebral ischemia [17] and scopolamine-induced brain damage [21]. However, in an AD animal model, this is the first study to report that SEL is beneficial for spatial cognitive decline caused by Aβ.
LTP induction in the hippocampus is used to study the cellular basis of learning and memory [52]. LTP can be induced by activating N-methyl-D-aspartate (NMDA) glutamate receptors, which typically occur during the simultaneous activation of pre-synaptic and postsynaptic neurons [53]. In the present study, Aβ injection affected synaptic performance and inhibited LTP induction in the DG region of the hippocampus. The effects of Aβ injection on hippocampal synaptic plasticity were evident in the reduction of fEPSP slope and PS amplitude. There is considerable electrophysiological evidence from rodent models showing that Aβ injection can interfere with synaptic homeostasis and lead to suppression of LTP in the DG region of the hippocampus and cognitive impairments [54,55,56]. Strong evidence suggests that Aβ increases pre-synaptic calcium and alters glutamate levels in hippocampal synapses, leading to excitotoxicity [57]. The synaptic performance of the hippocampus can be compromised as a result of these detrimental changes caused by Aβ, leading to impairments in learning and memory mediated by the hippocampus. In this regard, it has been shown that spatial cognitive decline resulting from Aβ administration as an AD model is closely related to the suppression of hippocampal DG LTP [56]. However, treatment with SEL improved LTP impairment in the hippocampus of AD rats by increasing fEPSP slopes and PS amplitude. The possible mechanisms underlying SEL’s protective action against hippocampal LTP impairment in AD rats can be attributed, at least in part, to the following mechanisms: (1) modulation of hippocampal dopaminergic transmission [28]; (2) regulation of Ca2+/calmodulin-dependent protein kinase IIα (CaMKIIα) phosphorylation [58]; (3) reduction of tonic gamma-aminobutyric acid (GABA) inhibition [59]; (4) enhancement of exiatability of the different hippocampus subfields by augmenting basal firing rate, sodium/potassium ATPase (Na+/K+-ATPase), and protein kinase C activities [60]; and (5) stimulation of nitric oxide (NO) production [31]. However, the mechanisms mentioned above were not investigated in this study. Therefore, it is strongly recommended to investigate them in future research. While previous studies have demonstrated SEL’s protective effects against hippocampal LTP impairment under both in vitro [59] and in vivo [28] conditions, our current study is the first to highlight SEL’s neuroplastic properties in an AD model. Based on the present study, SEL may attenuate Aβ-induced LTP impairment in DG granule cells, potentially explaining its beneficial effects on spatial learning and memory impairments caused by Aβ.
Oxidative stress is a key pathological condition in neurological diseases and remains a major therapeutic target for AD [61]. Studies have shown that antioxidant factors improve hippocampal LTP induction and cognitive impairments resulting from AD [52, 62]. Consistent with previous studies [51, 63], in the present study, Aβ administration resulted in an imbalance of oxidative-antioxidant status in the plasma of rats, as evidenced by a decrease in TTG concentration (a non-enzymatic antioxidant) and an increase in MDA (a marker of lipid peroxidation) levels. Among all the antioxidants in the body, TTG constitutes a major part of the body’s total antioxidants, helping protect cellular structures and functions against ROS [64, 65]. In lipid peroxidation, ROS attacks unsaturated lipids in the cell membrane, making the brain, which has high levels of unsaturated lipids, highly vulnerable to oxidative stress [62, 66]. Studies have shown that Aβ plaques are always co-localized with oxidized lipids in brain tissue samples affected by AD [67]. MDA is an important product of lipid peroxidation and is involved in the pathological cascade in AD [68]. Interestingly, treatment with SEL improved the oxidative/antioxidant balance in the plasma by increasing TTG concentration and suppressing the increase in MDA levels, indicating its antioxidant capacity. Consistent with these results, the antioxidant property of SEL has been previously reported [69, 70]. Therefore, it is speculated that SEL’s ability to eliminate free radicals and prevent oxidative damage contributes to its protective effect against Aβ-induced impairments of hippocampal LTP and spatial learning and memory.
Neuronal loss is the outcome of various neurological diseases such as AD [71]. Some studies have shown that neuronal loss primarily occurs in regions with Aβ plaques, indicating a link between Aβ deposition and neuronal loss [72]. Furthermore, Aβ-associated neuronal loss can lead to behavioral impairments in AD [73]. Additionally, localized oxidative stress around the plaques has been shown to contribute to long-term toxicity and selective neuronal death in AD [71]. Consistent with previous studies [43, 51], Aβ injection in rats resulted in increased neuronal death in the hippocampal DG and CA1 regions. Interestingly, neuronal death was successfully prevented with SEL treatment in the hippocampus of Aβ-injected rats, which is in congruence with previous studies [17, 74]. The available evidence suggests that SEL’s ability to inhibit Aβ plaque formation and neuronal death may be part of the explanation for its protective effect against Aβ-induced deficits in LTP and spatial learning and memory.
Conclusions
The present study demonstrates, for the first time, that oral SEL treatment ameliorates spatial learning and memory deficits in AD rats, potentially through the modulation of oxidative status, reduction of neuronal death, and improvement of hippocampal LTP impairment. This study indicates that SEL may be a promising agent against AD-related cognitive decline. However, further research is needed to evaluate the mechanisms underlying SEL’s protective effects against AD-related cognitive decline in detail, especially the mechanisms involved in its antioxidant properties.
Data Availability
No datasets were generated or analysed during the current study.
References
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM (2021) Alzheimer’s disease. Lancet 397(10284):1577–1590
Armstrong RA (2019) Risk factors for Alzheimer’s disease. Folia Neuropathol 57(2):87–105
Breijyeh Z, Karaman R (2020) Comprehensive review on Alzheimer’s disease: causes and treatment. Molecules 25(24):5789
Armstrong RA (2009) The molecular biology of senile plaques and neurofibrillary tangles in Alzheimer’s disease. Folia Neuropathol 47(4):289–299
Tamagno E, Guglielmotto M, Vasciaveo V, Tabaton M (2021) Oxidative stress and beta amyloid in Alzheimer’s disease Which comes first: the chicken or the egg? Antioxidants 10(9):1479
Emilsson L, Saetre P, Balciuniene J, Castensson A, Cairns N, Jazin EE (2002) Increased monoamine oxidase messenger RNA expression levels in frontal cortex of Alzheimer’s disease patients. Neurosci Lett 326(1):56–60
Gulyás B, Pavlova E, Kása P, Gulya K, Bakota L, Várszegi S, Keller E, Horváth MC et al (2011) Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int 58(1):60–68
Rahman K (2007) Studies on free radicals, antioxidants, and co-factors. Clin Interv Aging 2(2):219–236
Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, Nordberg A (2012) Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med 53(1):37–46
Behl T, Kaur D, Sehgal A, Singh S, Sharma N, Zengin G, Andronie-Cioara FL, Toma MM et al (2021) Role of monoamine oxidase activity in Alzheimer’s disease: an insight into the therapeutic potential of inhibitors. Molecules 26(12):3724
Waters CH, Sethi KD, Hauser RA, Molho E, Bertoni JM (2004) Zydis selegiline reduces off time in Parkinson’s disease patients with motor fluctuations: a 3-month, randomized, placebo-controlled study. Mov Disord 19(4):426–432
Pu Y, Qian F, Guo J, Sha Y, Qian Y (2022) Selegiline protects against lipopolysaccharide (LPS)–induced impairment of the blood–brain barrier through regulating the NF-κB/MLCK/p-MLC signaling pathway. Neurotox Res 40(1):267–275
Magyar K, Szende B (2004) (−)-Deprenyl, a selective MAO-B inhibitor, with apoptotic and anti-apoptotic properties. Neurotoxicology 25(1–2):233–242
Wu Y, Kazumura K, Maruyama W, Osawa T, Naoi M (2015) Rasagiline and selegiline suppress calcium efflux from mitochondria by PK11195-induced opening of mitochondrial permeability transition pore: a novel anti-apoptotic function for neuroprotection. J Neural Transm 122:1399–1407
Abdanipour A, Mirzaei M, Anarkooli IJ, Mohammadi P (2023) Effect of selegiline as a monomine oxidase B inhibitor on the expression of neurotrophin mRNA levels in a contusion rat model of spinal cord injury. Neurol Res 45(3):241–247
Gyárfás T, Knuuttila J, Lindholm P, Rantamäki T, Castrén E (2010) Regulation of brain-derived neurotrophic factor (BDNF) and cerebral dopamine neurotrophic factor (CDNF) by anti-parkinsonian drug therapy in vivo. Cell Mol Neurobiol 30:361–368
Amini-Khoei H, Saghaei E, Mobini G-R, Sabzevary-Ghahfarokhi M, Ahmadi R, Bagheri N, Mokhtari T (2019) Possible involvement of PI3K/AKT/mTOR signaling pathway in the protective effect of selegiline (deprenyl) against memory impairment following ischemia reperfusion in rat. Neuropeptides 77:101942
Knoll J (1989) The pharmacology of selegiline ((−) deprenyl) New aspects. Acta Neurologica Scandinavica 80:83–91
Soliman GF, Hashem G, Fawzy MG, Ibrahim W (2019) Neuroprotective effects of metformin versus selegiline on Parkinson’s disease model by reserpine through the interrelation of α synuclein and antioxidants on behavioral changes in rats. Egypt J Basic Clin Pharmacol 9
Maia F, Pitombeira B, Araujo D, Cunha G, Viana G (2004) l-Deprenyl prevents lipid peroxidation and memory deficits produced by cerebral ischemia in rats. Cell Mol Neurobiol 24:87–100
Goverdhan P, Sravanthi A, Mamatha T (2012) Neuroprotective effects of meloxicam and selegiline in scopolamine-induced cognitive impairment and oxidative stress. Int J Alzheimer’s Disease 2012:974013
Wahdan SA, Tadros MG, Khalifa AE (2017) Antioxidant and antiapoptotic actions of selegiline protect against 3-NP-induced neurotoxicity in rats. Naunyn-Schmiedeberg’s Arch Pharmacol 390:905–917
Zhu J, Hamm R, Reeves T, Povlishock J, Phillips L (2000) Postinjury administration of L-deprenyl improves cognitive function and enhances neuroplasticity after traumatic brain injury. Exp Neurol 166(1):136–152
Ahmari M, Sharafi A, Mahmoudi J, Jafari-Anarkoli I, Gharbavi M, Hosseini M-J (2020) Selegiline (l-deprenyl) mitigated oxidative stress, cognitive abnormalities, and histopathological change in rats: alternative therapy in transient global ischemia. J Mol Neurosci 70:1639–1648
Amiri A, Noorbala AA, Nejatisafa AA, Ghoreishi A, Derakhshan MK, Khodaie-Ardakani MR, Hajiazim M et al (2008) Efficacy of selegiline add on therapy to risperidone in the treatment of the negative symptoms of schizophrenia: a double-blind randomized placebo-controlled study. Hum Psychopharmacol Clin Exp 23(2):79–86
Gerlach M, Youdim M, Riederer P (1996) Pharmacology of selegiline. Neurology 47 (6_suppl_3) 137S-145S
LI G (1964) Monoamine oxidase inhibitors Adverse reactions and possible mechanisms. JAMA 190:456–462
Ishikawa T, Okano M, Minami A, Tsunekawa H, Satoyoshi H, Tsukamoto Y, Takahata K et al (2019) Selegiline ameliorates depression-like behaviors in rodents and modulates hippocampal dopaminergic transmission and synaptic plasticity. Behav Brain Res 359:353–361
Pazini AM, Gomes GM, Villarinho JG, Da Cunha C, Pinheiro F, Ferreira AP, Mello CF, Ferreira J et al (2013) Selegiline reverses Aβ 25–35-induced cognitive deficit in male mice. Neurochem Res 38:2287–2294
Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J et al (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med 336(17):1216–1222
Thomas T (2000) Monoamine oxidase-B inhibitors in the treatment of Alzheimers disease. Neurobiol Aging 21(2):343–348
Tsunekawa H, Noda Y, Mouri A, Yoneda F, Nabeshima T (2008) Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25–35). Behav Brain Res 190(2):224–232
Wilcock G, Birks J, Whitehead A, Evans SJG (2002) The effect of selegiline in the treatment of people with Alzheimer’s disease: a meta-analysis of published trials. Int J Geriatr Psychiatry 17(2):175–183
Arib O, Rat P, Molimard R, Chait A, Faure P, de Beaurepaire R (2010) Electrophysiological characterization of harmane-induced activation of mesolimbic dopamine neurons. Eur J Pharmacol 629(1–3):47–52
Bickford P, Adams C, Boyson S, Curella P, Gerhardt G, Heron C, Ivy G, Lin A et al (1997) Long-term treatment of male F344 rats with deprenyl: assessment of effects on longevity, behavior, and brain function. Neurobiol Aging 18(3):309–318
Carrillo M, Kanai S, Sato Y, Nokubo M, Ivy G, Kitani K (1993) The optimal dosage of (−) deprenyl for increasing superoxide dismutase activities in several brain regions decreases with age in male Fischer 344 rats. Life Sci 52(24):1925–1934
Carrillo M, Kitani K, Kanai S, Sato Y, Ivy G, Miyasaka K (1996) Long term treatment with (−) deprenyl reduces the optimal dose as well as the effective dose range for increasing antioxidant enzyme activities in old mouse brain. Life Sci 59(13):1047–1057
Muralikrishnan D, Samantaray S, Mohanakumar KP (2003) D-deprenyl protects nigrostriatal neurons against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced dopaminergic neurotoxicity. Synapse 50(1):7–13
Zhang F, Lu J, Zhang J-g, Xie J-x (2015) Protective effects of a polysaccharide from Spirulina platensis on dopaminergic neurons in an MPTP-induced Parkinson’s disease model in C57BL/6J mice. Neural Regen Res 10(2):308–313
Arabi A, Karimi SA, Salehi I, Haddadi R, Komaki A (2023) Effects of sesamin on Aβ1-42-induced oxidative stress and LTP impairment in a rat model of Alzheimer’s disease. Metab Brain Dis 38(5):1503–1511
Basir HS, Mirazi N, Komaki A, Hosseini A (2024) Cacao consumption improves passive avoidance memory impairment in a rat model of Alzheimer’s disease: the role of hippocampal synaptic plasticity and oxidative stress. Front Pharmacol 15:1379264
Halder T, Patel B, Acharya N (2022) Design and optimization of myricetin encapsulated nanostructured lipid carriers: in-vivo assessment against cognitive impairment in amyloid beta (1–42) intoxicated rats. Life Sci 297:120479
Puoyan-Majd S, Parnow A, Rashno M, Heidarimoghadam R, Komaki A (2024) The protective effects of high-intensity interval training combined with Q10 supplementation on learning and memory impairments in male rats with amyloid-β-induced Alzheimer’s disease. J Alzheimer’s Dis 99(s1):S67–S80
Gholipour P, Komaki A, Ramezani M, Parsa H (2022) Effects of the combination of high-intensity interval training and ecdysterone on learning and memory abilities, antioxidant enzyme activities, and neuronal population in an Amyloid-beta-induced rat model of Alzheimer’s disease. Physiol Behav 251:113817
Paxinos G, Watson C (2006) The rat brain in stereotaxic coordinates: hard cover. Elsevier
Harrison F, Hosseini A, McDonald M (2009) Endogenous anxiety and stress responses in water maze and Barnes maze spatial memory tasks. Behav Brain Res 198(1):247–251
Ghaderi S, Gholipour P, Komaki A, Shahidi S, Seif F, Bahrami-Tapehebur M, Salehi I, Zarei M et al (2024) Underlying mechanisms behind the neuroprotective effect of vanillic acid against diabetes-associated cognitive decline: an in vivo study in a rat model. Phytother Res 38(3):1262–77
Gholipour P, Ebrahimi Z, Mohammadkhani R, Ghahremani R, Salehi I, Sarihi A, Komaki A, Karimi SA (2024) Effects of (S)-3, 4-DCPG, an mGlu8 receptor agonist, on hippocampal long-term potentiation at perforant pathway-dentate gyrus synapses in prenatal valproic acid-induced rat model of autism. Sci Rep 14(1):13168
Gawel K, Gibula E, Marszalek-Grabska M, Filarowska J, Kotlinska JH (2019) Assessment of spatial learning and memory in the Barnes maze task in rodents—methodological consideration. Naunyn-Schmiedeberg’s Arch Pharmacol 392:1–18
Ahmadi N, Safari S, Mirazi N, Karimi SA, Komaki A (2021) Effects of vanillic acid on Aβ1-40-induced oxidative stress and learning and memory deficit in male rats. Brain Res Bull 170:264–273
Gholipour P, Komaki A, Parsa H, Ramezani M (2022) Therapeutic effects of high-intensity interval training exercise alone and its combination with ecdysterone against amyloid beta-induced rat model of Alzheimer’s disease: a behavioral, biochemical, and histological study. Neurochem Res 47:2090–108
Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361(6407):31–39
Lüscher C, Malenka RC (2012) NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol 4(6):a005710
Hu Z, Yu P, Zhang Y, Yang Y, Zhu M, Qin S, Xu J-T, Duan D et al (2022) Inhibition of the ISR abrogates mGluR5-dependent long-term depression and spatial memory deficits in a rat model of Alzheimer’s disease. Transl Psychiatry 12(1):1–11
Salimi M, Tabasi F, Abdolsamadi M, Dehghan S, Dehdar K, Nazari M, Javan M, Mirnajafi-Zadeh J et al (2022) Disrupted connectivity in the olfactory bulb-entorhinal cortex-dorsal hippocampus circuit is associated with recognition memory deficit in Alzheimer’s disease model. Sci Rep 12(1):1–13
Yang Y, Ji W-g, Zhang Y-j, Zhou L-p, Chen H, Yang N, Zhu Z-r (2021) Riluzole ameliorates soluble Aβ1–42-induced impairments in spatial memory by modulating the glutamatergic/GABAergic balance in the dentate gyrus. Prog Neuropsychopharmacol Biol Psychiatry 108:110077
Tofighi N, Asle-Rousta M, Rahnema M, Amini R (2021) Protective effect of alpha-linoleic acid on Aβ-induced oxidative stress, neuroinflammation, and memory impairment by alteration of α7 nAChR and NMDAR gene expression in the hippocampus of rats. Neurotoxicology 85:245–253
Okano M, Takahata K, Sugimoto J, Muraoka S (2019) Selegiline recovers synaptic plasticity in the medial prefrontal cortex and improves corresponding depression-like behavior in a mouse model of Parkinson’s disease. Front Behav Neurosci 13:176
Srivastava I, Vazquez-Juarez E, Henning L, Gómez-Galán M, Lindskog M (2020) Blocking astrocytic GABA restores synaptic plasticity in prefrontal cortex of rat model of depression. Cells 9(7):1705
Singh R, Mishra M, Singh S, Sharma D (2012) Effect of L-deprenyl treatment on electrical activity, Na+, K+ ATPase, and protein kinase C activities in hippocampal subfields (CA1 and CA3) of aged rat brain. Indian J Exp Biol 50(2):101–109
Briyal S, Ranjan AK, Gulati A (2023) Oxidative stress: a target to treat Alzheimer’s disease and stroke. Neurochem Int 165:105509
Reed TT (2011) Lipid peroxidation and neurodegenerative disease. Free Radical Biol Med 51(7):1302–1319
Komaki H, Faraji N, Komaki A, Shahidi S, Etaee F, Raoufi S, Mirzaei F (2019) Investigation of protective effects of coenzyme Q10 on impaired synaptic plasticity in a male rat model of Alzheimer’s disease. Brain Res Bull 147:14–21
Chianeh YR, Prabhu K (2014) Protein thiols as an indicator of oxidative stress. Arch Med Rev J 23(3):443–456
Taysi S, Polat F, Gul M, Sari R, Bakan E (2002) Lipid peroxidation, some extracellular antioxidants, and antioxidant enzymes in serum of patients with rheumatoid arthritis. Rheumatol Int 21(5):200–204
Ayala A, Muñoz MF, Argüelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438
Benseny-Cases N, Klementieva O, Cotte M, Ferrer I, Cladera J (2014) Microspectroscopy (μFTIR) reveals co-localization of lipid oxidation and amyloid plaques in human Alzheimer disease brains. Anal Chem 86(24):12047–12054
Nazifi M, Oryan S, Esfahani DE, Ashrafpoor M (2021) The functional effects of piperine and piperine plus donepezil on hippocampal synaptic plasticity impairment in rat model of Alzheimer’s disease. Life Sci 265:118802
Tian Z, Wang X, Han T, Sun C (2023) Selegiline ameliorated dyslipidemia and hepatic steatosis in high-fat diet mice. Int Immunopharmacol 117:109901
Wahdan SA, Tadros MG, Khalifa AE (2023) Retraction note to: antioxidant and antiapoptotic actions of selegiline protect against 3-NP-induced neurotoxicity in rats. Springer
Xie H, Hou S, Jiang J, Sekutowicz M, Kelly J, Bacskai BJ (2013) Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc Natl Acad Sci 110(19):7904–7909
Zhang J, Wu N, Wang S, Yao Z, Xiao F, Lu J, Chen B (2021) Neuronal loss and microgliosis are restricted to the core of Aβ deposits in mouse models of Alzheimer’s disease. Aging Cell 20(6):e13380
Dunys J, Valverde A, Checler F (2018) Are N-and C-terminally truncated Aβ species key pathological triggers in Alzheimer’s disease? J Biol Chem 293(40):15419–15428
Kiray M, Bagriyanik H, Pekcetin C, Ergur B, Uysal N, Ozyurt D, Buldan Z (2006) Deprenyl and the relationship between its effects on spatial memory, oxidant stress and hippocampal neurons in aged male rats. Physiol Res 55(2):205
Acknowledgements
The current study was funded (Grant No. 99-371) by the Faculty of Basic Sciences, Bu-Ali Sina University, Hamadan, Iran. The authors are grateful to the staff of the Neurophysiology Research Center, Hamadan University of Medical Sciences for supporting this study.
Author information
Authors and Affiliations
Contributions
Hamid Shokati Basir, Naser Mirazi, Alireza Komaki, Behnam Mohamadpour, and Abdolkarim Hosseini contributed equally to this work. All experiments were performed by Hamid Shokati Basir, Naser Mirazi, and Alireza Komaki. Hamid Shokati Basir, Naser Mirazi, Alireza Komaki, Behnam Mohamadpour, and Abdolkarim Hosseini wrote the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Experimental methods and animal care were in accordance with the National Institutes of Health (NIH) and ARRIVE Guidelines and were approved by Bu Ali Sina University-Hamedan’s Ethics Committee (Ethic code: IR.BASU.REC.1400.001).
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Basir, H.S., Mirazi, N., Komaki, A. et al. Selegiline Improves Cognitive Impairment in the Rat Model of Alzheimer’s Disease. Mol Neurobiol (2024). https://doi.org/10.1007/s12035-024-04388-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12035-024-04388-x