
Abstract
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder that involves functional and structural defects in selective central nervous system (CNS) regions, harming the individual capability to process and respond to external stimuli, including impaired verbal and non-verbal communications. Etiological causes of ASD have not been fully clarified; however, prenatal activation of the innate immune system by external stimuli might infiltrate peripheral immune cells into the fetal CNS and activate cytokine secretion by microglia and astrocytes. For instance, genomic and postmortem histological analysis has identified proinflammatory gene signatures, microglia-related expressed genes, and neuroinflammatory markers in the brain during ASD diagnosis. Active neuroinflammation might also occur during the developmental stage, promoting the establishment of a defective brain connectome and increasing susceptibility to ASD after birth. While still under investigation, we tested the hypothesis whether the monocyte chemoattractant protein-1 (MCP-1) signaling is prenatally programmed to favor peripheral immune cell infiltration and activate microglia into the fetal CNS, setting susceptibility to autism-like behavior. In this review, we will comprehensively provide the current understanding of the prenatal activation of MCP-1 signaling by external stimuli during the developmental stage as a new selective node to promote neuroinflammation, brain structural alterations, and behavioral defects associated to ASD diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autism spectrum disorders (ASD) are neurodevelopmental disorders that harm the central nervous system (CNS) and hinder an individual’s capability to process and respond to external stimuli, including impaired verbal and non-verbal communications [1]. People diagnosed with ASD also show repetitive or stereotypical behaviors ranging from mild to severe, as described by the Diagnostic and Statistical Manual of Mental Disorders, 5th edition [1]. Based on this categorization, autism is currently considered a spectrum of deficits, where behavioral and cognitive impairments are commonly and selectively observed [2]. According to the Autism and Developmental Disabilities Monitoring (ADDM) Network 2020 of the USA, autism shows a prevalence of 23.1 per 1000 children aged 8 years in Maryland to 44.9 per 1000 in California [3]. In fact, in 2020, one in 36 children aged 8 years (approximately 4% of boys and 1% of girls) were diagnosed with ASD [3].
Etiological causes of ASD have not been fully clarified; however, environmental factors, genetics, epigenetics, exposure to teratogenic agents, xenobiotics, and, importantly, immune activation might modulate ASD susceptibility [4, 5]. Notably, dysregulation of innate and adaptive immunity contributes to ASD susceptibility. See [6]. Accordingly, in a recent breakthrough report, authors confirmed the contribution of immune-related genomic signature on ASD susceptibility. This study was one of the large-scale exome-sequencing experimental protocols in autism, identifying up to 102 genes strongly associated with ASD and with neurodevelopmental delay neuronal abnormalities [7]. Notably, this study pointed out that a proinflammatory gene signature and microglia-related expressed genes were enriched in people with ASD [7]. It is believed that peripheral inflammatory profiles might infiltrate the CNS and increase microglia activation, exacerbating brain inflammation during ASD occurrence. For instance, an increase in the mRNA expression for IL-1β, IL-4, and IFN-γ was found elevated in peripheral blood mononucleate cells (PBMC) of ASD individuals [8]. Also, accumulation of IL-1β and IL-6 cytokines and tumor necrosis factor alpha (TNF-α) was detected in the plasma or postmortem brain samples of people with ASD [9, 10]. Notably, proinflammatory accumulation in people with ASD might occur very early in development. A recent report identified that newborn children diagnosed with ASD showed increased levels of IL-6, IL-8, eotaxin-1, interferon-γ, and IL-12p70 [11]. This suggest that newborns with elevated levels of proinflammatory cytokines might be at risk to be subsequently diagnosed with ASD.
While still under investigation, several reports have confirmed that systemic proinflammatory profiles might infiltrate the CNS during early stages of development, such as the embryonic stage. If this is true, ASD susceptibility might be programed during prenatal stages. Prenatal programming occurs when the fetus is exposed to external positive or negative stimuli, setting normal or abnormal physiological outcomes in the newborn [12]. This hypothesis was originally raised from epidemiological data suggesting a concept referred to as the “developmental origins of health and disease,” confirming that selective stimuli in utero during critical developmental periods can disrupt selective physiological pathways in the fetus that persist throughout adulthood [13]. Accordingly, during development, peripheral immune cells might infiltrate the fetal CNS and activate cytokine secretion by microglia and astrocytes, turning them into a proinflammatory state, releasing cytokines and triggering a positive feedback signal for a proper neural growth and development [14]. By itself, under physiological conditions, microglia regulate neurogenesis, synaptic plasticity, and synaptic striping, in addition to being the mayor antigen-presenting cells (APC) in the CNS [15]. However, microglia overactivation might lead to neuroinflammation during developmental stages and increase the ASD occurrence [16]. We and others have reported that immune activation during developmental stage in animal models sets neuronal defects that lead to behavioral anomalies in the newborn such as sociability defects [17], as well as depression-like [18, 19] addiction-like [20,21,22], and overfeeding-like behaviors [23]. This evidence supports the notion that accumulation of proinflammatory cytokines and microglia activation during prenatal stages contributes to the onset and progression of psychiatric disorders including ASD.
In this review, we will comprehensively provide a conceptual outline of our current understanding of the monocyte chemoattractant protein-1 (MCP-1), a selective chemokine that promotes neuroinflammation in the brain during ASD occurrence. We will also provide evidence of the activation of MCP-1 signaling by external stimuli during the developmental stage as a new selective node to prime neuroinflammation, brain structural alterations, and behavioral defects associated to ASD diagnosis in the newborn.
The Monocyte Chemoattractant Protein-1 (MCP-1)/CC Chemokine Ligand-2 (CCL2) Chemokine Signaling
The chemokines are small molecular–weight proteins of 8–14 kDa that are secreted in response to proinflammatory microenvironment cytokines and displaying chemotactic signaling for cellular migration to the site of inflammation. The structure of all the chemokines includes three main chemical domains: an α-helix that projects and two antiparallel β-pleated sheets linked to a third β-pleated sheet. The α-helix and the antiparallel β-pleated sheets are stabilized by cysteine links from the N-terminal region [24]. According to the chemical structure of cysteine residues, chemokines are divided into four subfamilies: CC (C–C motif chemokine ligands—CCL), CXC (C-X-C motif chemokine ligands—CXCL), CX3C (C-X3-C motif chemokine ligands—CX3CL), and C (C motif ligands—XCL) chemokines [25]. Chemokines bind to chemokine G protein‐coupled heptahelical receptors, normally expressed in the cell surface of white blood immune cells such as monocytes, neutrophils, and lymphocytes [24]. As expected, G protein-coupled receptors are transmembrane proteins composed of a short N-terminal extracellular domain linked to seven hydrophobic transmembrane domains and by three intracellular and extracellular loops and a serine/threonine-rich C-terminal intracellular region. The latter couples to a heterotrimeric G-protein complex modulating intracellular signaling. Chemokine receptors are divided into two families of heptahelical surface molecules that bind to chemokines: conventional chemokine receptors (cCKRs) and atypical chemokine receptors (ACKRs) all of which are sensitive to chemokine gradient concentrations [26].
The MCP-1/CC chemokine ligand-2 (CCL2) belongs to the CC subfamily that integrates cysteine residues just closely adjoined to the N-terminus [24]. Also, other MCPs found in humans include the MCP-2 (CCL8), MCP-3 (CCL7), and MCP-4 (CCL13) which share about ∼ 60% homology each other [24]. During a physiological scenario, MCP-1 is produced by many cell types in the peripheral and central systems, including fibroblasts, endothelial, smooth muscle, and monocytes and also astrocytes and microglia [17, 27,28,29]. Mechanistically, MCP-1 mainly binds to the protein G-coupled–receptor CCR2, which according to its terminal carboxyl domain is divided into CCR2A and CCR2B displaying target cell selectivity. For instance, CCR2B is abundantly expressed at the cell surface whereas the CCR2A is detected predominantly in the cytoplasm,in fact, the CCR2B is the predominant isoform of the CCR2 able to be activated by MCP-1. As expected, CCR2A and CCR2B display selective signaling downstream pathways followed MCP-1 binding [30, 31]. CCR2 signaling integrates significant redundancy and promiscuity when activated. Accordingly, CCR2 might be activated by different ligands, such as MCP-2, MCP-3, MCP-4, MCP-5, or C–C motif chemokine ligand 16,however: MCP-1 shows significant higher activity for CCR2 [32]. In addition, MCP-1 also binds to the atypical chemokine receptor that lacks G protein binding motif (ACKR1 and ACKR2).
Activation of CCR2 receptors favors chemotactic activity whereas ACKR1 and ACKR2 activation associates to phagocytic activity [33]. MCP-1 binding to various G protein‐mediated signaling cascades on the p38 mitogen‐activated protein kinase (MAPK) and Janus kinase (JAK)/STAT3, MAPK and on the phosphatidylinositol 3‐kinase (PI3K)/AKT cell migration-sensitive site [34]. Several reports have confirmed that after CCL2R activation, MCP-1 signaling regulates the migration and infiltration of monocytes, T lymphocytes, and natural killer (NK) cells to specific inflammatory response sites and plays a vital role in antiapoptosis, angiogenesis, and cell migration [34].
MCP-1 Disrupts Brain Functionality in ASD
Preclinical evidence has confirmed the role of MCP-1 on brain functionality by modulating neuronal survival and differentiation (Fig. 1). In the brain, MCP-1 is expressed in astrocytes, neurons, or oligodendrocyte-precursor cells in various structures, such as the cerebral cortex, basal ganglia, hippocampus, hypothalamus, substantia nigra, pons, cerebellum, and in the spinal cord [35], and mainly expressed in the developing mouse midbrain [36,37,38]. Accordingly, MCP-1 exposure to rat embryonic cells promoted differentiation of neurons toward the dopaminergic lineage [36] and favored an increase in neuritogenesis [36]. Similarly, CCR2-MCP1 signaling activates the migration of neuronal progenitor cells to the site of inflammatory microdomains [39].

MCP-1 signaling modulates brain function. A MCP-1 signaling is expressed in astrocytes, neurons, microglia, and oligodendrocytes. B MCP-1 signaling modulates neuritogenesis and neuronal migration to several microdomains and also incentivizes the dopaminergic lineage. C MCP-1 signaling is physiologically expressed in the cortex, basal ganglia, hippocampus, hypothalamus, substantia nigra, pons, cerebellum, and spinal cord. Created by Biorender
Pathological MCP-1 overexpression disrupts brain function and favors ASD susceptibility (Fig. 2). Clinical studies reported that people with ASD exhibited an increase in MCP-1 immunostaining in the anterior cingulate gyrus, cerebellum, and brain tissue homogenates, as well as up to 12-fold MCP-1 increase in the CSF [40] and in the amniotic fluid [41] when compared with controls. A recent transcriptomic and modeling study closely identified the MCP-1-CCR2 pathway as one of the innate immune responses found in ASD individuals [42]. Biological assessment of behavior outcomes reported that accumulation of MCP-1 in plasma was correlated with more impaired behavior in people with ASD such as visual reception, fine motor skills, expressive language, and daily living skills [9] according to the Adaptive Behavior Scale in Autism [43] or to the Autism Diagnostic Observation Schedule-second edition (ADOS-2) [44], while a recent clinical report showed significant decrease of MCP-1 levels in the CSF of adult patients with ASD [45]. In any case, it seems that MCP-1 signaling regulates brain function in humans and when uncontrolled is found in ASD individuals.

MCP-1 signaling potentially affects brain function in ASD subjects. MCP-1 is found accumulated in anterior cingulate cortex, cerebellum, and cerebrospinal fluid (CSF) of ASD individuals. MCP-1 binds to a G protein‐mediated signaling cascade on the CCR2 to activate the p38 mitogen‐activated protein kinase (MAPK) and Janus kinase (JAK)/STAT3. Preclinical evidence documented that MCP-1 signaling is associated to an increase of excitatory synaptic transmission, neuroinflammation, disruption of blood–brain barrier, and immune cell recruitment into the brain of autism-like related animal models. Created by Biorender
Several preclinical studies have provided some clues of the role of MCP-1 signaling which affects brain function and prime defective behavior in ASD individuals by assisting three molecular mechanisms: (1) disruption of neuronal activity and synaptic plasticity, (2) disruption of integrity of the blood–brain barrier (BBB), and (3) microglia activation. Accordingly, experimental studies have demonstrated that exposure to elevated levels of MCP-1 leads to increased excitatory synaptic transmission, creating an imbalance between inhibitory and excitatory signals within the brain [46, 47]. This disruption in synaptic homeostasis can impair the normal functioning of neural circuits involved in social cognition, language processing, and sensory integration [46], [47], which are known to be potentially affected in individuals with ASD.
Furthermore, MCP-1 can also disrupt the integrity of the blood–brain barrier (BBB) and impact the susceptibility to ASD occurrence. The BBB is a highly specialized barrier that regulates the exchange of molecules between the bloodstream and the brain. Disruption of the BBB allows immune cells and inflammatory mediators to infiltrate the brain, leading to neuroinflammation and subsequent neuronal damage. MCP-1 has been shown to increase the permeability of the BBB by promoting the adhesion and transmigration of monocytes and other immune cells across the barrier [48, 49]. This breach in the BBB further exacerbates the inflammatory response within the brain, contributing to the neurodevelopmental abnormalities seen in ASD [50].
A third molecular mechanism supporting the role of MCP-1 signaling on defective behavior in ASD individuals is by microglia activation. Initial research has highlighted the interplay between genetic in microglial dysfunction and MCP-1 dysregulation in ASD. For instance, genetic variations in genes associated with microglial function and immune response have also been identified in individuals with ASD [51]. Preclinical studies confirmed that MCP-1 modulates the function of microglia, the resident immune cells in the central nervous system. Microglia play a crucial role in brain development, immune surveillance, and synaptic remodeling. They constantly survey the brain environment, ensuring its proper functioning [52]. While preclinical or clinical studies have not totally defined the role MCP-1 on microglia signaling, some preclinical evidence has reported that MCP-1 is primarily secreted by activated microglia and astrocytes, acting as a chemoattractant, recruiting immune cells to sites of inflammation. In addition, it seems that might induce MCP-1 expression suggesting a crosstalk interplay between microglia and MCP-1. For instance, microglia rapidly induce MCP-1 expression in response to harmful stresses via NF-κB and p38 MAPK signaling pathways which by themselves are also involved in microglial activation [53]. In ASD, elevated levels of MCP-1 have been observed in the brains of affected individuals, suggesting its potential involvement in neuroinflammatory processes [54]. In fact, microglia in individuals with ASD exhibit an aberrant phenotype characterized by increased activation and proinflammatory responses [55]. This dysregulated immune response can contribute to the neuroinflammation observed in ASD, which has been implicated in the pathophysiology of the disorder [40]. It is expected that an increased activation in microglia can lead to the release of proinflammatory cytokines, chemokines, and reactive oxygen species, resulting in synaptic dysfunction and impaired neural circuitry [56]. These changes in neural connectivity and communication have been associated with the core symptoms of ASD, including social communication deficits and repetitive behaviors [57].
In conclusion, MCP-1, a chemokine involved in the inflammatory response, appears to disrupt brain functionality in individuals with ASD. Elevated levels of MCP-1 in the autistic brain can directly impact neuronal activity, modulate microglial function, and compromise the integrity of the blood–brain barrier. These mechanisms might contribute to the synaptic deficits, neuronal dysfunction, and neurodevelopmental abnormalities observed in ASD. Further research is needed to fully understand the intricate interactions between MCP-1 and molecular factors involved in ASD pathogenesis, which could potentially open new avenues for therapeutic interventions targeting neuroinflammation in this complex disorder.
Obesity or Maternal Exposure to High-Energy Diets Activates MCP-1-Dependent Neuroinflammation in ASD
As commented, ASD susceptibility is modulated by several stimuli including environmental factors, genetics, epigenetics, exposure to teratogenic agents, xenobiotics, infections, and diet [4, 5]. Clinical studies have confirmed crosstalk between maternal obesity or maternal exposure to high-energy diets on immune system activation and ASD susceptibility [4, 5]. It is believed that a chronic low-grade proinflammatory profile is in part promoted by the interaction of fatty acids released from adipose tissue expandability in obese mothers or from mothers exposed to high-energy diets with toll-like receptors, a pathological process known as metabolic inflammation [58].
While several authors have proposed metabolic inflammation as a major trigger of ASD, some reports have confirmed that some ASD individuals display a genetic susceptibility for physiological immune activation. Recent genome-wide association studies confirmed that people with ASD integrated common genomic variations affecting immune pathway activation and responses [59, 60] and confirming enriched proinflammatory and microglia-related genes [6, 7]. For instance, mRNA expression for IL-1β, IL-4, and IFN-γ was found elevated in peripheral blood mononucleate cells (PBMC) of ASD individuals [8]. Also, accumulation of IL-1β and IL-6 cytokines and tumor necrosis factor alpha (TNF-α) was detected in the plasma or postmortem brain samples of people with ASD [9, 10]. Also, active microglia have been found in several brain-associated areas of the mesocorticolimbic circuit, including cerebellum, PFC [55, 61], ACG, and OFC [61].
Mechanistically, it is believed that activation of the proinflammatory profile is linked to the toll-like receptor 4 (TLR4)/IKK/NF-κB pathway in microglia [62]. In fact, we reported that obese murine models show upregulation of the TBK1-related IKK marker [63, 64], a downstream target of TLR4 activation [65]. A potential scenario sets that the TLR4/IKK/NF-κB pathway allows IL-1β secretion, astrocyte activation in the choroid plexus, integrating peripheral B and T cells and a macrophage response by secreting chemoattractant molecules such as MCP-1 [5, 17]. In fact, selective depletion of TLR2 and TLR4 reestablishes social interaction in a mouse model of repeated social defeat stress by decreasing microglial activation in the PFC [66].
While this evidence supports the notion that active microglia leads to proinflammatory cytokine release, recent reports suggest that it is unclear whether individual microglia states or combinations of states of activation are protective or detrimental (or both) in the context of disease progression [52, 67]. Accordingly, a decrease in microglia number disrupts circuit establishment in humans [68]. Authors reported that a homozygous mutation in the colony-stimulating-factor 1 receptor promotes a major decrease in microglia and leukoencephalopathy [68]. Individuals with leukoencephalopathy displayed an absence of myelin-rich tracts in the corpus callosum and cingulate sulcus and dystrophic basal ganglia, thalamus, and hippocampus [68], suggesting a defective myelin turnover associated to colony-stimulating-factor 1 signaling. Accordingly, microglia actively modulates proper neurodevelopment by regulating neurogenesis, synaptic plasticity, and synaptic striping and also myelin conformation [56, 69, 70], suggesting that the colony-stimulating factor 1 receptor signaling coordinates microglial maturation during embryonic development [71]. In fact, a recent preclinical report confirmed that prenatal microglia are less responsive to immune challenges by secreting proinflammatory cytokines compared to the adult microglia [72]. Authors did not analyze the contribution of colony-stimulating factor 1 receptor signaling on behavioral traits of individuals, in part because individuals carrying the colony-stimulating factor 1 receptor mutation died at 10 months of age from streptococcal bacteremia. Based on the inaccessibility of living human brains for molecular characterization and ethical and technological limitations to test microglial function in human brains, it is still unknown whether microglial transition states during prenatal stages set behavioral traits found in people with ASD. Also, it is still unknown whether ASD behavioral outcomes are related to MCP-1 released from microglia in obesity or in response to high-energy diets.
We next described major findings supporting the role of prenatal programming on microglia phenotypes and microglia-MCP-1 release associated to ASD susceptibility after birth.
Prenatal Programming by External Stimuli Regulates MCP-1-Dependent Inflammation and Autism-Related Behavior
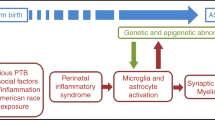
Several epidemiological reports have confirmed that prenatal programming favors a proinflammatory profile program in people with ASD, which might occur very early in development (Fig. 3). For instance, prenatal programming potentially might explain genomic signatures of active immune and inflammatory responses in postmortem brains of people with ASD [73, 74]. Maternal inflammation leads to elevated proinflammatory cytokines such as IFN gamma, and IL-4, IL-5, and IL-6 might affect the fetal brain development and subsequent cognitive disease outcomes [75]. Accordingly, authors identified that the intellectual disability in ASD was associated with an increase in GM-CSF, IFN-γ, IL-1α, and IL-6 plasma levels and lower plasma levels of IL-8 and MCP-1 during mid-gestation of mothers [76]. Under this scenario, it is expected that plasma proinflammatory profile infiltrates the fetal brain allowing microglia activation and neuroinflammation during prenatal stages. While still under investigation, initial reports identified accumulation of IL-6 and C-X-C motif chemokine ligand 10 (CXCL-10), also known as interferon-γ-inducible protein 10 (IP-10) in the amniotic fluid of mothers during the mid-trimester of pregnancy [77]. Notably, authors documented that IL-6 and CXCL-10 accumulation is associated to preterm delivery before or after 32 weeks of gestation, respectively [77]. While the authors did not determine the association of IL-6 and CXCL-10 on ASD susceptibility, this report confirmed the deleterious role of prenatal inflammation on newborn health. A recent report also identified that newborn children diagnosed with ASD showed increased levels of IL-6, IL-8, eotaxin-1, interferon-γ, and IL-12p70 [11]. This suggest that elevated levels of proinflammatory cytokines in the newborn might be a risk factor in the development of some behavioral traits found in ASD individuals.

Prenatal programming by external stimuli primes MCP-1 signaling in ASD subjects. Several external stimuli during prenatal stages prime the MCP-1 signaling in the brain of animal models diagnosed with autism-like behavior. Prenatal exposure to stress, high-energy diet, and pollutants increases IL-6 in plasma. Prenatal exposure to tobacco, obesity, or infections accumulated MCP-1 in plasma or kidney, respectively. Also, prenatal exposure to high-energy diets and pollutants affected cortical development and promoted neuroinflammation and microglia activation. Prenatal exposure to pollutants also decreases dopaminergic neurotransmission. Created by Biorender
We propose that MCP-1 signaling orchestrates brain neuroinflammation during prenatal stages, modulating behavioral traits in ASD after birth [17]. For major experimental evidence of immune dysregulation in ASD, see [6]. Here, we tested whether MCP-1 signaling is prenatally programmed in the newborn, favoring inflammatory profiles, microglia activity, and susceptibility to autism-like behavior. We focus our hypothesis on the role of prenatal programming of MCP-1 signaling by infections, maternal obesity, diet, stress, smoking, and pollution.
Prenatal Programing of MCP-1 Signaling by Infections
Preclinical reports have confirmed that fetal exposure to infections during pregnancy modulates the susceptibility to mental illnesses in the newborn. Authors reported that prenatal programming by maternal immune activation after rubella virus exposure [78,79,80] was associated to ASD susceptibility in the offspring. Accordingly, maternal immune activation appearing in the first 3 months of embryonic development has been involved in the disruption of neurodevelopment and potentially affects behaviors in the newborn [81]. Prenatal programming of inflammation and ASD behavior in the offspring was initially reported to be primed by pharmacologic activation of the toll-like-receptor 3 signaling [82]. In an elegant preclinical report, Kim et al. demonstrated that prenatal exposure to Poly I:C in mice favors IL-17A increase and alteration in cortical microstructures, which correlates with autism-like behavior in the mouse offspring [82], as well as in non-human and human primates [83]. Notably, defects in sociability correlates with an increase of proinflammatory cytokines INF-γ, IL-6, IL-17a, and TNF-α in plasma [82, 83] and high expression of IL-6, tool-like receptor 4 (TLR4), and MCP-1 on the fetal brain [84]. Also, the toll-like receptor signaling integrates a potential signaling pathway associated to MCP-1 secretion. For instance, pharmacologic activation of the toll-like-receptor 7/8 during maternal programming increased plasma MCP-1 in maternal and fetal brains, which correlates with microglia activation [85].
Experimental data also confirmed that a microglia activation shows a time-dependent response to a proinflammatory profile in the brain after a maternal immune activation by Poly(I:C) inoculation in mice [86]. Authors reported that maternal immune activation promoted changes in microglia motility in the brain as early as E18 and are sustained through to adolescence in mice (postnatal day 42) [86]. Notably, maternal immune activation induced earlier (at E12) caused sustained alterations in the patterns of microglial process motility and asocial behavior in the offspring, which is associated to increased IL-6 expression in prenatal microglia [86].
Prenatal Programing of MCP-1 Signaling by Obesity
As commented, several reports confirmed that maternal obesity predisposes ASD appearance in the offspring. Preclinical data reported that obese mice showed increased microglial activity that correlates with phagocytosis and diminished dendritic spine density in the PFC and hippocampus [87,88,89]. As expected, obesity disrupts offspring connectome found in the dopaminergic circuitry of the substantia nigra and the nigrostriatal tract [90]. Also, in a recent study, authors confirmed that maternal weight gain during pregnancy increases the risk of ASD after birth [91]. In fact, weight gain during pregnancy was suggested as an important risk factor for ASD compared to pre-pregnancy obesity [92]. Accordingly, insufficient rates of weight gain during the second trimester and excessive rates of weight gain during the third trimester were associated with a higher risk of neurodevelopmental disorders in offspring including ASD [93, 94]. For instance, maternal obesity is linked to up to 1.39% and 1.59% of ASD cases and a greater likelihood of having a child with ASD compared with their leaner counterparts [95,96,97,98]. Physiologically, adipose tissue accumulation and expansion has been associated to MCP-1 release, favoring macrophage infiltration into the adipose tissue and setting a proinflammatory cytokine profile [99]. Humans showing failure in adipose tissue expansion experienced a major increase in the proinflammatory profile in plasma including accumulation of MCP-1 [100]. This evidence supports the notion that adipose tissue expansion during maternal obesity promotes MCP-1 accumulation and immune cell infiltration, exacerbating the proinflammatory profile. Although the association of MCP-1 signaling in obesity programming ASD susceptibility has not been firmly established, this data provides evidence of the deleterious effect linked to maternal/fetal adipose tissue accumulation.
Prenatal Programming of MCP-1 Signaling by Diet
Maternal exposure to high-energy diets favors defective social novelty [101] and suppresses social interactions in the newborn [5, 102]. Experimental studies by our group and others have reported that prenatal exposure to high-energy diets sets a systemic and central proinflammatory profile in the male offspring diagnosed with asocial behavior [17]. Maternal exposure to high-energy diets during fetal development substantially increases the MCP-1 plasma levels [17, 103, 104] and also favors microglia activation in the hypothalamus [105] and gliosis in the hippocampus of the offspring [19]. As commented, active microglia have been found in several brain-associated areas of the mesocorticolimbic circuit, including cerebellum, PFC [55, 61], ACG, and OFC [61]. It is expected that active microglia and astrocytes underlie neuroinflammation and synaptic degradation in ASD [106]. Accordingly, exposure to high-energy diets activates microglia [107] and promotes excessive synaptic stripping, affecting hippocampal plasticity [108]. We reported that maternal exposure to high-energy diets during pregnancy promoted structural brain abnormalities showing a decrease in hippocampal and nucleus accumbens volume in the offspring [19]. Also, maternal exposure to high-energy diets decreased the total volume of the brain as well as the volume of the medial amygdala and basal forebrain of the offspring [109]. In fact, a decrease of the synaptophysin marker in the hippocampus was found in mice exposed to high-energy diets during programming [19]. Also, a decrease in dendritic spines and synaptic maturation were found in the primary somatosensory cortex of offspring exposed to high-energy diets [110]. We and others reported a priming phenotype of microglia after prenatal exposure to high-energy diets, demonstrated by exacerbated IL-6 upon an LPS-induced immune challenge [103, 105]. While the authors did not analyze MCP-1 release after prenatal exposure to high-energy diets, we reported that administration of MCP-1 antibody in mice decreased plasma IL-6 levels, reduced microglia complexity, and improved social behavior [17]. These studies suggest that prenatal exposure to high-energy diets primes MCP-1 signaling pathways modulating microglia plasticity and cytokine release that assist social behavior in the offspring.
Prenatal Programing of MCP-1 Signaling by Stress
Stress is frequently referred to experiences in life that might be beneficial or negative and even traumatic, but all of them affect our daily lives [111]. Stress can cause an imbalance of neural circuitry affecting cognitive and motor behavior and, also, the systemic physiology via neuroendocrine, autonomic, metabolic, and immune disruptions [111]. Also, maternal exposure to high-energy diet or maternal obesity, maternal infection, or maternal smoking/pollutants might be also considered as prenatal stressors. Accordingly, authors reported that prenatal stress caused fetal MCP-1 secretion in mice, promoting brain inflammation leading to defective sociability [84]. Notably, prenatal stress increases fetal brain IL-6 in a CCL2-dependent manner [84]. In fact, MCP-1 expression was detected in placenta and fetal brain [84], suggesting that MCP-1 signaling integrates prenatal stimuli, coding for IL-6 expression in the intrauterine environment, potentially leading to defective behavior after birth [112], [92]. This resembles what we found in prenatal programing of MCP-1 signaling by diet: prenatal programming by stress promotes MCP-1 plasma accumulation leading to IL-6 secretion and defective behavior in the newborn. If this is true, MCP-1-IL-6 signaling might provide a context-dependent pathway that defines a proinflammatory profile and sets defective behavioral outcomes in the newborn.
Several reports have documented the role of prenatal IL-6 signaling on defective behavior in the newborn. Clinical evidence reported that maternal IL-6 penetrates into the fetal compartment [113, 114]. Accordingly, recent studies in humans have confirmed the role of maternal IL-6 concentrations during pregnancy and defective offspring behavior [115,116,117,118]. These studies demonstrated that IL-6 accumulation during maternal immune activation is associated to defective cognitive performance coded by the cortex, amygdala, and frontolimbic circuit in the newborn. ASD shows functional and structural defects in CNS regions, such as the prefrontal cortex, the amygdala, the hippocampus, and the cerebellum [4], so the fact that IL-6–dependent maternal immune activation affects brain circuits suggests that MCP-1 signaling might be an initial contributor of the immune response. Finally, IL-6 itself might amplify leukocyte accumulation at sites of inflammation by activating STAT3 through increased local production of MCP-1 and ICAM-1 [119]. We recently proposed that physiological or pathological outcomes of IL-6 signaling are related to its pleiotropic effects and levels in the brain by microglia, astrocytes, neurons, and endothelial cells and also by peripheral infiltrating macrophages or T lymphocytes [120]. In this context, MCP-1-IL-6 signaling might mutually interplay during prenatal programming in assisting brain circuit establishment during pregnancy, coding for ASD-related behaviors after birth.
Prenatal Programing of MCP-1 Signaling by Smoking
Epidemiological studies documented that smoking during pregnancy programs brain function and behavior after birth. Women who smoked during pregnancy prime food preferences in the offspring: infants born from mothers who smoked consumed more carbohydrates than protein [121]. Preclinical data also confirmed that exposure to tobacco smoke during pregnancy incentivizes consumption of palatable foods in the offspring [122]. For many years, nicotine has been considered the main psychoactive substance present in tobacco smoke, which promotes higher levels of anxiety [123]. Previous findings identified elevated MCP-1 levels in middle-aged smokers compared to age-matched non-smokers [124], which seem to increase with a longer smoking history (~ 15 years) [125].
Prenatal exposure to nicotine was also associated to a proinflammatory profile in the offspring. A preclinical model of maternal cigarette smoke exposure in mice documented MCP-1 accumulation in the kidney of offspring [126]. Also, humans exposed to environmental tobacco smoke developed significant elevation of inflammatory markers in plasma, including the MCP-1 [127].
Evidence on the association between maternal prenatal smoking and the likelihood for autism has been contradictory and, in some cases, conflicting, based on some studies supporting association effects and others failing to prove a real risk. A recent clinical report integrating 72 cohorts in the Environmental Influences on Child Health Outcomes consortium reported that maternal prenatal tobacco smoking is consistently associated with an increase in autism-related symptoms in the general population and modestly associated with elevated risk for a diagnosis of ASD when looking at a combined analysis from multiple studies that each included both pre- and full-term births [128]. Conversely, in a Finnish national birth cohort, authors reported that prenatal maternal levels of serum cotinine, a biomarker for tobacco exposure, were not associated with the odds of autism [129], which was also confirmed by other authors [130], [131]. In any case, despite experimental evidence supporting MCP-1 accumulation in the offspring of mothers exposed to tobacco smoke, no causality between MCP-1 signaling on neuroinflammation and autism susceptibility in the offspring has been confirmed.
Prenatal Programing of MCP-1 Signaling by Pollutants
Air pollution is known to affect neurological function and to have effects on the fetus in utero US-EPA [132]Several recent studies have reported associations between perinatal exposure to air pollution and ASD susceptibility in children, including agricultural pesticides [133] [134]. Air pollutant exposure is becoming one of the most consistent environmental risk factors for neurodevelopmental disorders. In particular, using a prospective cohort of 116,430 US female nurses recruited in 1989, authors reported that higher maternal exposure to particulate matter (< 2.5 μm) during pregnancy, particularly the third trimester, was associated with greater odds of a child having ASD [135].
Preclinical models documented that exposure to particulate matter < 2.5 μm in mice increased the infiltration of inflammatory cells TNF-α and IL-6, as well as MCP-1 gene and protein expression levels in the liver, kidney, spleen, and thymus in a dose-dependent manner [136] and during vascular dysfunction in the brain of rodents [137]. Also, prenatal exposure to a high dose of particulate matter < 2.5 μm impaired the development of the cerebral cortex in mice [138] and promoted defective dopamine neurotransmission associated to hyperactive responses in mice [139] and in rabbits [140]. Experimental evidence confirmed that air pollutants activate neuroinflammation [141]. While the molecular pathways regarding the exposure to particulate matter < 2.5 μm during exposure to air pollutants on MCP-1 levels and neuroinflammation have not been totally established, the MCP-1 chemokine actively participates in the innate immune activation, which is expected to be activated during exposure to air pollutants [141].
A recent report documented that prenatal exposure to air pollutants is exacerbated by stress. Authors designed a murine model co-exposing pregnant dams to an environmental pollutant and limited-resource stress and found that both robustly activate the maternal immune system [142]. Notably, male offspring, but not females, developed defective social interaction that correlates with diminish microglial function within the anterior cingulate cortex [142]. In fact, prenatal exposure to particulate matter < 2.5 μm affects brain structure and promotes defective socialization in the newborn [143]. This suggest that an early postnatal impairment of microglial phagocytic function is sufficient to induce social behavior impairments in male offspring.
Potential Molecular Determinates of MCP-1 Signaling in Autism-Related Behavior
As commented, MCP-1 is expressed in astrocytes, neurons, or oligodendrocyte precursor cells in the brain [35] and mainly expressed in the developing mouse midbrain [36,37,38]. MCP-1 signaling promotes neuronal differentiation [36] and neuritogenesis [36] and incentivizes migration of neuronal progenitor cells to the site of inflammation [39]. One hypothesis states that MCP-1 signaling recruits immune cell types into the brain during neurodevelopment promoting neuroinflammation and a defective establishment of the brain connectome.
Several reports have provided experimental data supporting the role of MCP-1 signaling during neurodevelopment on brain function and ASD susceptibility. Initially, sex-dependent neonatal immune signatures were identified in subjects diagnosed with ASD [144]. Authors reported that male newborns diagnosed with ASD have higher levels of inflammatory 6CKINE, MPIF-1, and also MCP-1 than female newborns [144]. While still under investigation, no data is available to precisely identify the cellular source of MCP-1 in people with ASD,however, it seems to depend on the selective diagnosis of the pathological brain. Reports have traced MCP-1 release from glia, astrocytes, and microglia. For instance, Müller cell-derived MCP-1 might diffuse throughout the neural retina and photoreceptors at the onset of degeneration [145]. As expected, retinal MCP-1-CCR2 signaling recruits peripheral monocytes into the site of neurodegeneration [145]. A recent report also confirmed the production of MCP-1 from astrocytes of mice after MPTP exposure in a murine model of Parkinson disease [146]. Also, during chronic traumatic encephalopathy in humans, authors reported MCP-1 expression and microglia accumulation into the dorsolateral frontal cortex [147]. In fact, microglia response during cerebral hemorrhage seems to contribute to MCP-1 release [148]. In fact, activating the expression of the MCP-1 receptor, CCR2, in bone marrow cells improved memory capacities and decreased soluble Aβ accumulation in mice [149], confirming that the MCP-1-CCR2 signaling incentivizes cell migration toward sites of inflammation to preserve brain homeostasis [39]. This evidence confirms that MCP-1 secretion from neurons, astrocytes, and microglia supports peripheral immune cell recruitment into the brain, modulating neurodegeneration.
The contribution of MCP-1 signaling on cellular recruitment into the brain has also been reported in prenatal stages. During development, tissue-resident macrophage populations first arrive in the CNS from embryonic precursors starting from E8.5 [150]. Microglia are derived from erythromyeloid progenitors from yolk sac cells, which infiltrate the brain during early prenatal day 9 (E9) embryogenesis in mice and in the first trimester in humans [150, 151]. While still under investigation, very early in development, the initial seeding of the pre-macrophages partially depends on the chemokine receptor CX3CR1 signaling at E9.5 and E10.5 [152]. MCP-1-CCR2 signaling contributes to microglia recruitment in embryonic development [153]. In fact, intracerebral stimulation by low doses of MCP-1 efficiently enhanced fetal cell mobilization into the mouse brain [154]. Central immune cell infiltration in prenatal stages might also be promoted in response to inflammatory stimulus [155]. Authors reported that during prenatal inflammation the choroid plexus secretes MCP-1, recruiting peripheral macrophages into the brain by the embryonic choroid plexus-blood-cerebrospinal fluid interface [155]. In fact, postnatal (3 to 11 days) exposure to pharmacologic inflammatory stimulus in mice followed by toll-like receptors 2 agonist increased cerebral microglia density and MCP-1 levels [156]. Conversely, we reported that inhibiting the MCP-1-CCR2 signaling by systemic administration of AbMCP-1 in mice offspring decreased neuroinflammation and microglia activation [17]. This evidence supports the hypothesis that systemic and central MCP-1 signaling during prenatal stages promotes myeloid cell recruitment and microglia seeding in the brain.
Brain microglia initially originated from the yolk sac during embryogenesis have an estimated median life span of > 15 months in the mouse cortex [157,158,159] and of 4 years in humans, with some reports identifying a life span of 20 years [160]. This selective long-lived identity benefits microglia by preserving its embryogenic phagocytic, immune, and proinflammatory profiles,however, they also might be compromised, favoring negative outcomes in adulthood if they were negatively primed during prenatal programming such as by exposure to infections, diet, obesity, stress, smoking, or pollution. For instance, authors reported that exposure to hypercaloric diets primes an immunity phenotype by reprograming the innate cells toward an enhanced immune response at later stages [161], a molecular mechanism termed “trained immunity” [162]. Notably, the adaptive innate phenotype has also been reported in microglia [163]. Hence, if we assume that MCP-1–responsive microglia regulate a proper activity and experience-dependent refinement of brain structure, setting a mature mesocorticolimbic connectome during neurodevelopment [15], it is conceivable that, in individuals with ASD, prenatal stimuli of MCP-1 might prime microglial cells to be recruited and overresponsive, affecting the neuronal connectome and setting abnormal behavioral traits at earlier years of life. Accordingly, we reported that inhibiting the MCP-1-CCR2 signaling by systemic administration of AbMCP-1 in mouse offspring improved sociability, which associates with structural changes into the somatosensorial cortex [17]. In fact, intracerebral stimulation by low doses of MCP-1 efficiently enhanced fetal cell mobilization into the brain to reduce microglia activation and brain excitotoxic damage in postpartum mice [154]. Also, central accumulation of inflammatory monocytes in mice exhibiting anxiety-like behavior was associated with microglial priming [164]. This evidence supports the notion of the existence of MPC-1 signaling on myeloid cell recruitment, microglia responsive and microglial training, and defective behavior in the offspring.
Despite the epidemiological and preclinical evidence, one of the major challenges is still to understand the contribution of inflammation during prenatal stages on ASD susceptibility after birth. We conceive that MCP-1 signaling activates peripheral immune cell recruitment into the brain during prenatal stages, incentivizing neuroinflammation and defective brain development. Also, MCP-1 signaling might drive local cytokine synthesis by resident glia, amplifying central immune response to external challenges. If this hypothesis is true, MCP-1 signaling might participate in two main detrimental pathways, promoting the immune cell recruitment and exacerbation of the immune response to external stimuli.
Conclusions
Individuals diagnosed with ASD have found a seminal relationship between immunity and ASD susceptibility. Prenatal programming by several stimuli, such as exposure to infections, diet, stress, smoking, pollution, or even obesity, seems to be a potential environmental trigger which sets MCP-1 signaling, allowing myeloid cell recruitment and active microglia in the newborn’s brain. Primed microglia might contribute to micro and macro structural defects on several brain regions, affecting connectome establishment and favoring ASD susceptibility. A current limitation is the lack of causality of prenatal MCP-1 signaling on the myeloid cell recruitment and primed microglia in ASD individuals. A combination of postmortem tissue analysis coupled to spatial and temporal resolution neuroimaging studies, such as the recent positron emission tomography analysis of CNS-infiltrating myeloid cells in a mouse model [165], might be useful to confirm the role of MCP-1 molecule as trigger signaling to favor microglial priming and behavioral traits in people with ASD.
Data Availability
Not applicable.
References
Christensen D, Zubler J (2020) CE: from the CDC: understanding autism spectrum disorder. Am J Nurs 120(10):30–37
Dizitzer Y, Meiri G, Flusser H, Michaelovski A, Dinstein I, Menashe I (2020) Comorbidity and health services’ usage in children with autism spectrum disorder: a nested case-control study. Epidemiol Psychiatr Sci 28(29):e95
Maenner MJ, Warren Z, Williams A, Amoakohene E, Bakian AV, Bilder DA, Durkin MS et al (2023) Prevalence and characteristics of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveill Summ 72(2):1–14
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J (2018) Autism spectrum disorder. Lancet 392(10146):508–520
Maldonado-Ruiz R, Garza-Ocañas L, Camacho A (2019) Inflammatory domains modulate autism spectrum disorder susceptibility during maternal nutritional programming. Neurochem Int 126:109–117
Erbescu A, Papuc SM, Budisteanu M, Arghir A, Neagu M (2022) Re-emerging concepts of immune dysregulation in autism spectrum disorders. Front Psychiatry 19(13):1006612
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, de Rubeis S, An JY, Peng M et al (2020) Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180(3):568–584.e23
Ahmad SF, Nadeem A, Ansari MA, Bakheet SA, Al-Ayadhi LY, Attia SM (2017) Upregulation of IL-9 and JAK-STAT signaling pathway in children with autism. Prog Neuro-Psychopharmacology Biol Psychiatry 79(Pt B):472–480
Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J (2011) Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 25(1):40–45
Sciara AN, Beasley B, Crawford JD, Anderson EP, Carrasco T, Zheng S, Ordway GA, Chandley MJ (2020) Neuroinflammatory gene expression alterations in anterior cingulate cortical white and gray matter of males with autism spectrum disorder. Autism Res 13(6):870–884
Heuer LS, Croen LA, Jones KL, Yoshida CK, Hansen RL, Yolken R, Zerbo O et al (2019) An exploratory examination of neonatal cytokines and chemokines as predictors of autism risk: the early markers for autism study. Biol Psychiatry 86(4):255–264
Doi M, Usui N, Shimada S (2022) Prenatal environment and neurodevelopmental disorders. Front Endocrinol 15(13):860110
De BHA, Harding JE (2006) The developmental origins of adult disease (Barker) hypothesis. Aust N Z J Obstet Gynaecol 46(1):4–14
Buehler MR (2011) A proposed mechanism for autism: an aberrant neuroimmune response manifested as a psychiatric disorder. Med Hypotheses 76(6):863–870
Schafer DP, Stevens B (2015) Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol 7(10):a020545
Kim HJ, Cho MH, Shim WH, Kim JK, Jeon EY, Kim DH, Yoon SY (2017) Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol Psychiatry 22(11):1576–1584
Maldonado-Ruiz R, Trujillo-Villarreal LA, Montalvo-Martínez L, Mercado-Gómez OF, Arriaga-Ávila V, Garza-Ocañas L, Ortiz-López R, Garza-Villarreal EA et al (2022) MCP-1 signaling disrupts social behavior by modulating brain volumetric changes and microglia morphology. Mol Neurobiol 59(2):932–949
de la Garza AL, Garza-Cuellar MA, Silva-Hernandez IA, Cardenas-Perez RE, Reyes-Castro LA, Zambrano E, Gonzalez-Hernandez B, Garza-Ocañas L et al (2019) Maternal flavonoids intake reverts depression-like behaviour in rat female offspring. Nutrients 11(3):572
Trujillo-Villarreal LA, Romero-Díaz VJ, Marino-Martínez IA, Fuentes-Mera L, Ponce-Camacho MA, Devenyi GA, Mallar Chakravarty M, Camacho-Morales A et al (2021) Maternal cafeteria diet exposure primes depression-like behavior in the offspring evoking lower brain volume related to changes in synaptic terminals and gliosis. Transl Psychiatry 11(1):53
Camacho A, Montalvo-Martinez L, Cardenas-Perez RE, Fuentes-Mera L, Garza-Ocañas L (2017) Obesogenic diet intake during pregnancy programs aberrant synaptic plasticity and addiction-like behavior to a palatable food in offspring. Behav Brain Res 14(330):46–55
Montalvo-Martínez L, Cruz-Carrillo G, Maldonado-Ruiz R, Cárdenas-Tueme M, Bernal-Vega S, Garza-Ocañas L, Ortiz-López R, Reséndez-Pérez D et al (2022a) Maternal high-dense diet programs interferon type I signaling and microglia complexity in the nucleus accumbens shell of rats showing food addiction-like behavior. Neuroreport 33(12):495–503
Montalvo-Martínez L, Cruz-Carrillo G, Maldonado-Ruiz R, Trujillo-Villarreal LA, Cardenas-Tueme M, Viveros-Contreras R, Ortiz-López R, Camacho-Morales A (2022b) Transgenerational susceptibility to food addiction-like behavior in rats associates to a decrease of the anti-inflammatory IL-10 in plasma. Neurochem Res 47(10):3093–3103
Camacho-Morales A, Caballero-Benitez A, Vázquez-Cruz E, Maldonado-Ruiz R, Cárdenas-Tueme M, Rojas-Martinez A, Caballero-Hernández D (2022) Maternal programming by high-energy diets primes ghrelin sensitivity in the offspring of rats exposed to chronic immobilization stress. Nutr Res 107:37–47
Singh S, Anshita D, Ravichandiran V (2021) MCP-1: function, regulation, and involvement in disease. Int Immunopharmacol 101(Pt B):107598
Laing KJ, Secombes CJ (2004) Trout CC chemokines: comparison of their sequences and expression patterns. Mol Immunol. 41(8):793–808
Hughes CE, Nibbs RJB (2018) A guide to chemokines and their receptors. FEBS J 285(16):2944–2971
Barna BP, Pettay J, Barnett GH, Zhou P, Iwasaki K, Estes ML (1994) Regulation of monocyte chemoattractant protein-1 expression in adult human non-neoplastic astrocytes is sensitive to tumor necrosis factor (TNF) or antibody to the 55-kDa TNF receptor. J Neuroimmunol 50(1):101–107
Cushing SD, Berliner JA, Valente AJ, Territo MC, Navab M, Parhami F, Gerrity R, Schwartz CJ, Fogelman AM (1990) Minimally modified low density lipoprotein induces monocyte chemotactic protein 1 in human endothelial cells and smooth muscle cells. Proc Natl Acad Sci U S A 87(13):5134–5138
Standiford TJ, Kunkel SL, Phan SH, Rollins BJ, Strieter RM (1991) Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J Biol Chem 266(15):9912–9918
Apel AK, Cheng RKY, Tautermann CS, Brauchle M, Huang CY, Pautsch A, Hennig M, Nar H et al (2019) Crystal structure of CC chemokine receptor 2A in complex with an orthosteric antagonist provides insights for the design of selective antagonists. Structure 27(3):427–438.e5
Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, Coughlin SR (1994) Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc Natl Acad Sci U S A 91(7):2752–2756
White GE, Iqbal AJ, Greaves DR (2013) CC chemokine receptors and chronic inflammation-therapeutic opportunities and pharmacological challenges. Pharmacol Rev 65(1):47–89
Lokeshwar BL, Kallifatidis G, Hoy JJ (2020) Atypical chemokine receptors in tumor cell growth and metastasis. Adv Cancer Res 145:1–27
Xu M, Wang Y, Xia R, Wei Y, Wei X (2021) Role of the CCL2-CCR2 signalling axis in cancer: mechanisms and therapeutic targeting. Cell Prolif 54(10):e13115
Banisadr G, Gosselin RD, Mechighel P, Kitabgi P, Rostène W, Parsadaniantz SM (2005) Highly regionalized neuronal expression of monocyte chemoattractant protein-1 (MCP-1/CCL2) in rat brain: evidence for its colocalization with neurotransmitters and neuropeptides. J Comp Neurol 489(3):275–292
Edman LC, Mira H, Arenas E (2008) The β-chemokines CCL2 and CCL7 are two novel differentiation factors for midbrain dopaminergic precursors and neurons. Exp Cell Res 314(10):2123–2130
Gaupp S, Arezzo J, Dutta DJ, John GR, Raine CS (2011) On the occurrence of hypomyelination in a transgenic mouse model: a consequence of the myelin basic protein promoter? J Neuropathol Exp Neurol 70(12):1138–1150
Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Väremo L, Giacomello S et al (2018) Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev Cell 46(4):504–517.e7
Belmadani A, Tran PB, Ren D, Miller RJ (2006) Chemokines regulate the migration of neural progenitors to sites of neuroinflammation. J Neurosci 26(12):3182–3191
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57(1):67–81. https://doi.org/10.1002/ana.20315
Abdallah MW, Larsen N, Grove J, Nørgaard-Pedersen B, Thorsen P, Mortensen EL, Hougaard DM (2012) Amniotic fluid chemokines and autism spectrum disorders: an exploratory study utilizing a Danish Historic Birth Cohort. Brain Behav Immun 26(1):170–176. https://doi.org/10.1016/j.bbi.2011.09.003
Alshammery S, Patel S, Jones HF, Han VX, Gloss BS, Gold WA, Dale RC (2022) Common targetable inflammatory pathways in brain transcriptome of autism spectrum disorders and Tourette syndrome. Front Neurosci 15(16):999346. https://doi.org/10.3389/fnins.2022.999346
Napolioni V, Ober-Reynolds B, Szelinger S, Corneveaux JJ, Pawlowski T, Ober-Reynolds S, Kirwan J, Persico AM, Melmed RD, Craig DW, Smith CJ, Huentelman MJ (2013) Plasma cytokine profiling in sibling pairs discordant for autism spectrum disorder. J Neuroinflammation 14(10):38. https://doi.org/10.1186/1742-2094-10-38
Peng G, Peng X, Tong T, Zhang X, Xu M, Peng X (2021) Correlation analysis of expression of CC and CXC chemokines in children with autism spectrum disorder. Medicine (Baltimore) 100(24):e26391. https://doi.org/10.1097/MD.0000000000026391
Runge K, Fiebich BL, Kuzior H, Rausch J, Maier SJ, Dersch R, Nickel K, Domschke K, van Elst LT, Endres D (2023) Altered cytokine levels in the cerebrospinal fluid of adult patients with autism spectrum disorder. J Psychiatr Res 158:134–142. https://doi.org/10.1016/j.jpsychires
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL et al (2009) JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 29(13):4096–4108
Jung H, Toth PT, White FA, Miller RJ (2008) Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem 104(1):254–263
Banks WA, Kastin AJ, Broadwell RD (1995) Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 2(4):241–248
Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van RN, Andjelkovic AV (2005) Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab 25(5):593–606
de Los Angeles Robinson-Agramonte M, García EN, Guerra JF, Hurtado YV, Antonucci N, Semprún-Hernández N, Schultz S, Siniscalco D (2022) Immune dysregulation in autism spectrum disorder: what do we know about it? Int J Mol Sci 23(6):3033
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H et al (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459(7246):569–573
Camacho-Morales A (2022) Glycolytic metabolism supports microglia training during age-related neurodegeneration. Pharmacol Rep 4(5):818–831
Kim JE, Park H, Lee JE, Kang TC (2020) CDDO-Me inhibits microglial activation and monocyte infiltration by abrogating NFκB- and p38 MAPK-mediated signaling pathways following status epilepticus. Cells 9(5):1123. https://doi.org/10.3390/cells9051123
Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, Ji L, Brown T, Malik M (2009) Elevated immune response in the brain of autistic patients. J. Neuroimmunol 207(1-2):111–116
Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP (2010) Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol. Psychiatry 68(4):368–376
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 80 333(6048):1456–1458
Tang G, Gudsnuk K, Kuo SH, Cotrina ML, Rosoklija G, Sosunov A, Sonders MS et al (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83(5):1131–1143
Cai D, Khor S (2019) “Hypothalamic microinflammation” paradigm in aging and metabolic diseases. Cell Metab 30(1):19–35
Arenella M, Cadby G, De WW, Jones RM, Whitehouse AJO, Moses EK, Fornito A et al (2022) Potential role for immune-related genes in autism spectrum disorders: evidence from genome-wide association meta-analysis of autistic traits. Autism 26(2):361–372
Golovina E, Fadason T, Lints TJ, Walker C, Vickers MH, O’Sullivan JM (2021) Understanding the impact of SNPs associated with autism spectrum disorder on biological pathways in the human fetal and adult cortex. Sci Rep 11(1):15867
Tetreault NA, Hakeem AY, Jiang S, Williams BA, Allman E, Wold BJ, Allman JM (2012) Microglia in the cerebral cortex in autism. J Autism Dev Disord 42(12):2569–2584
Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DML et al (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. Neurosci 29(2):359–370
Delint-Ramirez I, Maldonado Ruiz R, Torre-Villalvazo I, Fuentes-Mera L, Garza Ocañas L, Tovar A, Camacho A (2015) Genetic obesity alters recruitment of TANK-binding kinase 1 and AKT into hypothalamic lipid rafts domains. Neurochem Int 80:23–32
Diaz B, Fuentes-Mera L, Tovar A, Montiel T, Massieu L, Martínez-Rodríguez HG, Camacho A (2015) Saturated lipids decrease mitofusin 2 leading to endoplasmic reticulum stress activation and insulin resistance in hypothalamic cells. Brain Res 1627:80–89
Leulier F, Parquet C, Pili-Floury S, Ryu J. H, Caroff M, Lee WJ, Mengin-Lecreulx D, Lemaitre B. (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4(5):491–496
Nie X, Kitaoka S, Tanaka K, Segi-Nishida E, Imoto Y, Ogawa A, Nakano F et al (2018) The innate immune receptors TLR2/4 mediate repeated social defeat stress-induced social avoidance through prefrontal microglial activation. Neuron 99(3):464–479.e7
Yvanka de Soysa T, Therrien M, Walker AC, Stevens B (2022) Redefining microglia states: lessons and limits of human and mouse models to study microglia states in neurodegenerative diseases. Semin Immunol 60:101651
Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, Young E et al (2019) Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet 104(5):936–947
Cárdenas-Tueme M, Montalvo-Martínez L, Maldonado-Ruiz R, Camacho-Morales A, Reséndez-Pérez D (2020) Neurodegenerative susceptibility during maternal nutritional programing: are central and peripheral innate immune training relevant? Front Neurosci 4(14):13
Lloyd AF, Miron VE (2019) The pro-remyelination properties of microglia in the central nervous system. Nat Rev Neurol 15(8):447–458
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (6005):330, 841–335
Yaqubi M, Groh A, Dorion M-F, Afanasiev E, Luo J, Hashemi H, Sinha S et al (2023) Analysis of the microglia transcriptome across the human lifespan using single cell RNA sequencing. J Neuroinflammation 20(1):132
Gupta S, Ellis SE, Ashar FN, Moes A, Bader JS, Zhan J, West AB, Arking DE (2014) Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 5:5748
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474(7351):380–384
Goines PE, Croen LA, Braunschweig D, Yoshida CK, Grether J, Hansen R, Kharrazi M, Ashwood P et al (2011) Increased midgestational IFN-γ, IL-4 and IL-5 in women bearing a child with autism: a case-control study. Mol Autism 2(2):13
Jones KL, Croen LA, Yoshida CK, Heuer L, Hansen R, Zerbo O, Delorenze GN et al (2017) Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol Psychiatry 22(2):273–279
Gervasi MT, Romero R, Bracalente G, Erez O, Dong Z, Hassan SS, Yeo L, Yoon BH et al (2012) Midtrimester amniotic fluid concentrations of interleukin-6 and interferon-gamma-inducible protein-10: evidence for heterogeneity of intra-amniotic inflammation and associations with spontaneous early (<32 weeks) and late (>32 weeks) preterm delivery. J Perinat Med 40(4):329–343
Atladóttir HÓ, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET (2010) Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 40(12):1423–1430
Chess S (1971) Autism in children with congenital rubella. J Autism Child Schizophr 1(1):33–47
Croen LA, Qian Y, Ashwood P, Zerbo O, Schendel D, Pinto-Martin J, Daniele Fallin M et al (2019) Infection and fever in pregnancy and autism spectrum disorders: findings from the study to explore early development. Autism Res 12(10):1551–1561
Courchesne E, Pramparo T, Gazestani V, Lombardo M, Pierce K, Lewis N (2018) The ASD living biology: from cell proliferation to clinical phenotype. Mol Psychiatry. 24(1):88–107
Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, Yeon Kim J et al (2017) Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549(7673):482–487
Careaga M, Murai T, Bauman MD (2017) Maternal immune activation and autism spectrum disorder: from rodents to nonhuman and human primates. Biol Psychiatry 81(5):391–401
Chen HJ, Antonson AM, Rajasekera TA, Patterson JM, Bailey MT, Gur TL (2020) Prenatal stress causes intrauterine inflammation and serotonergic dysfunction, and long-term behavioral deficits through microbe- and CCL2-dependent mechanisms. Transl Psychiatry 10(1):191
Kwon J, Suessmilch M, McColl A, Cavanagh J, Morris BJ (2021) Distinct trans-placental effects of maternal immune activation by TLR3 and TLR7 agonists: implications for schizophrenia risk. Sci Rep 11(1):23841
Ozaki K, Kato D, Ikegami A, Hashimoto A, Sugio S, Guo Z, Shibushita M et al (2020) Maternal immune activation induces sustained changes in fetal microglia motility. Sci Rep 10(1):21378
Bocarsly ME, Fasolino M, Kane GA, Lamarca EA, Kirschen GW, Karatsoreos IN, McEwen BS, Gould E (2015) Obesity diminishes synaptic markers, alters microglial morphology, and impairs cognitive function. Proc Natl Acad Sci U S A 112(51):15731–15736
Cope EC, Lamarca EA, Monari PK, Olson LB, Martinez S, Zych AD, Katchur NJ, Gould E (2018) Microglia play an active role in obesity-associated cognitive decline. J Neurosci 38(41):8889–8904
Tozuka Y, Kumon M, Wada E, Onodera M, Mochizuki H, Wada K (2010) Maternal obesity impairs hippocampal BDNF production and spatial learning performance in young mouse offspring. Neurochem Int 57(3):235–247
Lippert RN, Hess S, Klemm P, Burgeno LM, Jahans-Price T, Walton ME, Kloppenburg P, Brüning JC (2020) Maternal highfat diet during lactation reprograms the dopaminergic circuitry in mice. J Clin Invest 130(7):3761–3776
Su L, Chen C, Lu L, Xiang AH, Dodds L, He K (2020) Association between gestational weight gain and autism spectrum disorder in offspring: a meta-analysis. Obesity 28(11):2224–2231
Shen Y, Dong H, Lu X, Lian N, Xun G, Shi L, Xiao L, Zhao J, Ou J (2018) Associations among maternal pre-pregnancy body mass index, gestational weight gain and risk of autism in the Han Chinese population. BMC Psychiatry 18(1):11
Chen S, Fan M, Lee B, Dalman C, Karlsson H, Gardner R (2023) Rates of maternal weight gain over the course of pregnancy and offspring risk of neurodevelopmental disorders. BMC Med 21(1):108
Patti M, Croen L, Chen A, Fallin MD, Khoury J, Lyall K, Newschaffer C et al (2023) Prepregnancy BMI, gestational weight gain, and susceptibility to autism-related traits: the EARLI and HOME studies. Obesity (Silver Spring) 31(5):1415–1424
Andersen CH, Thomsen PH, Nohr EA, Lemcke S (2018) Maternal body mass index before pregnancy as a risk factor for ADHD and autism in children. Eur Child Adolesc Psychiatry 48(2):139–148
Gardner RM, Lee BK, Magnusson C, Rai D, Frisell T, Karlsson H, Idring S, Dalman C (2015) Maternal body mass index during early pregnancy, gestational weight gain, and risk of autism spectrum disorders: results from a Swedish total population and discordant sibling study. Int J Epidemiol 44(3):870–883
Li M, Fallin MD, Riley A, Landa R, Walker SO, Silverstein M, Caruso D et al (2016) The association of maternal obesity and diabetes with autism and other developmental disabilities. Pediatrics 137(2):e20152206
Reynolds LC, Inder TE, Neil JJ, Pineda RG, Rogers CE (2014) Maternal obesity and increased risk for autism and developmental delay among very preterm infants. J Perinatol 34(9):688–692
Dommel S, Blüher M (2021) Does C-C motif chemokine ligand 2 (CCL2) link obesity to a pro-inflammatory state? Int J Mol Sci . 22(3):1500
Nankam PAN, Cornely M, Klöting N, Blüher M (2022) Is subcutaneous adipose tissue expansion in people living with lipedema healthier and reflected by circulating parameters? Front Endocrinol (Lausanne). 13:1000094
Zilkha N, Kuperman Y, Kimchi T (2017) High-fat diet exacerbates cognitive rigidity and social deficiency in the BTBR mouse model of autism. Neuroscience 345:142–154
Veniaminova E, Cespuglio R, Cheung CW, Umriukhin A, Markova N, Shevtsova E, Lesch KP, Anthony DC, Strekalova T (2017) Autism-like behaviours and memory deficits result from a western diet in mice. Neural Plast 2017:9498247
Bordeleau M, Lacabanne C, Fernández De Cossío L, Vernoux N, Savage JC, González-Ibáñez F, Tremblay MÈ (2020) Microglial and peripheral immune priming is partially sexually dimorphic in adolescent mouse offspring exposed to maternal high-fat diet. J Neuroinflammation 17(1):264
Thompson MD, Derse A, Ferey JL, Reid M, Xie Y, Christ M, Chatterjee D et al (2019) Transgenerational impact of maternal obesogenic diet on offspring bile acid homeostasis and nonalcoholic fatty liver disease. Am J Physiol - Endocrinol Metab 316(4):E674–E686
Maldonado-Ruiz R, Cárdenas-Tueme M, Montalvo-Martínez L, Vidaltamayo R, Garza-Ocañas L, Reséndez-Perez D, Camacho A (2019) Priming of hypothalamic ghrelin signaling and microglia activation exacerbate feeding in rats’ offspring following maternal overnutrition. Nutrients 11(6):1241
Xiong Y, Chen J, Li Y (2023) Microglia and astrocytes underlie neuroinflammation and synaptic susceptibility in autism spectrum disorder. Front Neurosci. 17:1125428
Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I et al (2017) Microglial inflammatory signaling orchestrates the hypothalamic immune response to dietary excess and mediates obesity susceptibility. Cell Metab 26(1):185–197.e3
Hao S, Dey A, Yu X, Stranahan AM (2016) Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun 51:230–239
Fernandes DJ, Spring S, Roy AR, Qiu LR, Yee Y, Nieman BJ, Lerch JP, Palmert MR (2021) (2021) Exposure to maternal high-fat diet induces extensive changes in the brain of adult offspring. Transl Psychiatry 111(11):1–9
Hatanaka Y, Wada K, Kabuta T (2016) Maternal high-fat diet leads to persistent synaptic instability in mouse offspring via oxidative stress during lactation. Neurochem Int 97:99–108
McEwen BS (2017) Neurobiological and systemic effects of chronic stress. Chronic Stress (Thousand Oaks) 1:2470547017692328
Bronson SL, Bale TL (2014) Prenatal stress-induced increases in placental inflammation and offspring hyperactivity are male-specific and ameliorated by maternal antiinflammatory treatment. Endocrinology 155(7):2635–2646
Dahlgren J, Samuelsson AM, Jansson T, Holmäng A (2006) Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res 60(2):147–151
Zaretsky MV, Alexander JM, Byrd W, Bawdon RE (2004) Transfer of inflammatory cytokines across the placenta. Obstet Gynecol 103(3):546–550
Graham AM, Rasmussen JM, Rudolph MD, Heim CM, Gilmore JH, Styner M, Potkin SG et al (2018) Maternal systemic interleukin-6 during pregnancy is associated with newborn amygdala phenotypes and subsequent behavior at 2 years of age. Biol. Psychiatry 83(2):109–119
Rasmussen JM, Graham AM, Entringer S, Gilmore JH, Styner M, Fair DA, Wadhwa PD, Buss C (2019) Maternal interleukin-6 concentration during pregnancy is associated with variation in frontolimbic white matter and cognitive development in early life. Neuroimage 185:825–835
Rudolph MD, Graham AM, Feczko E, Miranda-Dominguez O, Rasmussen JM, Nardos R, Entringer S, Wadhwa PD, Buss C, Fair DA (2018) Maternal IL-6 during pregnancy can be estimated from newborn brain connectivity and predicts future working memory in offspring. Nat Neurosci 21(5):765–772
Spann MN, Monk C, Scheinost D, Peterson BS (2018) Maternal immune activation during the third trimester is associated with neonatal functional connectivity of the salience network and fetal to toddler behavior. J Neurosci 38(11):2877–2886
Romano M, Sironi M, Toniatti C, Polentarutti N, Fruscella P, Ghezzi P, Faggioni R et al (1997) Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 6(3):315–325
García-Juárez M, Camacho-Morales A (2022) Defining the role of anti- and pro-inflammatory outcomes of interleukin-6 in mental health. Neuroscience 1(492):32–46
Ayres C, Silveira PP, Barbieri MA, Portella AK, Bettiol H, Agranonik M, Silva AA, Goldani MZ (2011) Exposure to maternal smoking during fetal life affects food preferences in adulthood independent of the effects of intrauterine growth restriction. J Dev Orig Health Dis 2(3):162–167
Franke RM, Park M, Belluzzi JD, Leslie FM (2008) Prenatal nicotine exposure changes natural and drug-induced reinforcement in adolescent male rats. Eur J Neurosci 27(11):2952–2961
Conti AA, Tolomeo S, Steele JD, Baldacchino AM (2020) Severity of negative mood and anxiety symptoms occurring during acute abstinence from tobacco: a systematic review and meta-analysis. Neurosci Biobehav Rev 115:48–63
Kuschner WG, D’Alessandro A, Wong H, Blanc PD (1996) Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur Respir J 9(10):1989–1994
Kastelein TE, Duffield R, Marino FE (2015) Acute immune-inflammatory responses to a single bout of aerobic exercise in smokers; the effect of smoking history and status. Front Immunol 623(6):634
Al-Odat I, Chen H, Chan YL, Amgad S, Wong MG, Gill A, Pollock C, Saad S (2014) The impact of maternal cigarette smoke exposure in a rodent model on renal development in the offspring. PLoS One 9(7):e103443
Yu M, Zheng X, Peake J, Joad JP, Pinkerton KE (2008) Perinatal environmental tobacco smoke exposure alters the immune response and airway innervation in infant primates. J Allergy Clin Immunol 122(3):640–7.e1
Hertz-Picciotto I, Korrick SA, Ladd-Acosta C, Karagas MR, Lyall K, Schmidt RJ, Dunlop AL et al (2022) Maternal tobacco smoking and offspring autism spectrum disorder or traits in ECHO cohorts. Autism Res 15(3):551–569
Cheslack-Postava K, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Surcel HM, Brown AS (2021) A biomarkerbased study of prenatal smoking exposure and autism in a Finnish national birth cohort. Autism Res 14(11):2444–2453
Haglund NGS, Källén KBM (2011) Risk factors for autism and Asperger syndrome: perinatal factors and migration. Autism 15(2):163–183
Kalkbrenner AE, Meier SM, Madley-Dowd P, Ladd-Acosta C, Fallin MD, Parner E, Schendel D (2020) Familial confounding of the association between maternal smoking in pregnancy and autism spectrum disorder in offspring. Autism Res 13(1):134–144
US-EPA (2021) Health effects notebook for hazardous air pollutants
Dutheil F, Comptour A, Morlon R, Mermillod M, Pereira B, Baker JS, Charkhabi M, Clinchamps M et al (2021) Autismspectrum disorder and air pollution: a systematic review and meta-analysis. Environ Pollut 1(278):116856
Bertoletti ACC, Peres KK, Faccioli LS, Vacci MC, da Mata IR, Kuyven CJ, Dal Bosco SM (2023) Early exposure to agricultural pesticides and the occurrence of autism spectrum disorder: a systematic review. Rev Paul Pediatr 9(41):e2021360
Raz R, Roberts AL, Lyall K, Hart JE, Just AC, Laden F, Weisskopf MG (2015) Autism spectrum disorder and particulate matter air pollution before, during, and after pregnancy: a nested case–control analysis within the nurses’ health study II cohort. Environ Health Perspect 123(3):264–270
Wang J, Xue R, Li C, Hu L, Li Q, Sun Y, Chen Y et al (2023) Inhalation of subway fine particles induces murine extrapulmonary organs damage. Sci Total Environ 878:163181
Guan L, Geng X, Shen J, Yip J, Li F, Du H, Ji Z, Ding Y (2018) PM2.5 inhalation induces intracranial atherosclerosis which may be ameliorated by omega 3 fatty acids. Oncotarget 9(3):3765–3778
Zhang T, Zheng X, Wang X, Zhao H, Wang T, Zhang H, Li W (2018) Maternal exposure to PM2.5 during pregnancy induces impaired development of cerebral cortex in mice offspring. Int J Mol Sci. 19(1):257
Cui J, Fu Y, Lu R, Bi Y, Zhang L, Zhang C, Aschner M, Li X, Chen R (2019) Metabolomics analysis explores the rescue to neurobehavioral disorder induced by maternal PM 2.5 exposure in mice. Ecotoxicol Environ Saf 169:687–695
Bernal-Meléndez E, Lacroix MC, Bouillaud P, Callebert J, Olivier B, Persuy MA, Durieux D et al (2019) Repeated gestational exposure to diesel engine exhaust affects the fetal olfactory system and alters olfactory-based behavior in rabbit offspring. Part Fibre Toxicol 16(1):5
Brockmeyer S, D’Angiulli A (2016) How air pollution alters brain development: the role of neuroinflammation. Transl Neurosci 7(1):24–30
Block CL, Eroglu O, Mague SD, Smith CJ, Ceasrine AM, Sriworarat C, Blount C et al (2022) Prenatal environmental stressors impair postnatal microglia function and adult behavior in males. Cell Rep 40(5):111161
Peterson BS, Bansal R, Sawardekar S, Nati C, Elgabalawy ER, Hoepner LA, Garcia W et al (2022) Prenatal exposure to air pollution is associated with altered brain structure, function, and metabolism in childhood. J Child Psychol Psychiatry Allied Discip 63(11):1316–1331
Kim D, Iosif AM, Ramirez-Celis A, Ashwood P, Ames J, Lyall K, Berger K (2023) Neonatal immune signatures differ by sex regardless of neurodevelopmental disorder status: macrophage migration inhibitory factor (MIF) alone reveals a sex by diagnosis interaction effect. Brain Behav Immun. 111:328–333
Karlen SJ, Miller EB, Wang X, Levine ES, Zawadzki RJ, Burns ME (2018) Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J. Neuroinflammation 15(1):344
Qiao C, Niu G, Zhao W, Quan W, Zhou Y, Zhang M, Li T (2023) RIPK1-induced A1 reactive astrocytes in brain in MPTP-treated murine model of Parkinson’s disease. Brain Sci 13(5):733
Cherry JD, Meng G, Daley S, Xia W, Svirsky S, Alvarez VE, Nicks R et al (2020) CCL2 is associated with microglia and macrophage recruitment in chronic traumatic encephalopathy. J. Neuroinflammation 17(1):370
Xie ST, Chen AX, Song B, Fan J, Li W, Xing Z, Peng SY et al (2020) Suppression of microglial activation and monocyte infiltration ameliorates cerebellar hemorrhage induced-brain injury and ataxia. Brain Behav Immun 89:400–413
Naert G, Rivest S (2012) Hematopoietic CC-chemokine receptor 2 (CCR2) competent cells are protective for the cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Mol Med 18(1):297–313
Amann L, Masuda T, Prinz M (2023) Mechanisms of myeloid cell entry to the healthy and diseased central nervous system. Nat Immunol 24(3):393–407
Maldonado-Ruiz R, Fuentes-Mera L, Camacho A. (2017) Central modulation of neuroinflammation by neuropeptides and energy-sensing hormones during obesity. Biomed Res Int . 2017:7949582
Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, Jacome-Galarza CE et al (2016) Specification of tissue-resident macrophages during organogenesis. Science 80:353
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1-and Irf8-dependent pathways. Nat Neurosci 16(3):273–280
Sbeih M, Oulès B, Alkobtawi M, Schwendimann L, Ngô QT, Fontaine R, Teissier N, Gressens P, Aractingi S (2022) CCL2 recruits fetal microchimeric cells and dampens maternal brain damage in post-partum mice. Neurobiol Dis 174:105892
Cui J, Shipley FB, Shannon ML, Alturkistani O, Dani N, Webb MD, Sugden AU, Andermann ML, Lehtinen MK (2020) Inflammation of the embryonic choroid plexus barrier following maternal immune activation. Dev Cell 55(5):617–628.e6
Du X, Fleiss B, Li H, D’angelo B, Sun Y, Zhu C, Hagberg H, Levy O et al (2011) Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS One 6(5):e19583
Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T et al (2017) Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 18(2):391–405
Füger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermüller U, Wegenast-Braun BM et al (2017) Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci 20(10):1371–1376
Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar S, Datta M et al (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20(6):793–803
Réu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, Perl S et al (2017) The lifespan and turnover of microglia in the human brain. Cell Rep 20(4):779–784
Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ et al (2018) Western diet triggers NLRP3-dependent innate immune reprogramming. Cell 172(1-2):162:–175.e14
Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB et al (2020) Defining trained immunity and its role in health and disease. Nat Rev Immunol 20(6):375–388
Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J et al (2018) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556(7701):332–338
Weber MD, McKim DB, Niraula A, Witcher KG, Yin W, Sobol CG, Wang Y, Sawicki CM et al (2019) The influence of microglial elimination and repopulation on stress sensitization induced by repeated social defeat. Biol Psychiatry 85(8):667–678
Chaney A, Cropper H, Jain P, Wilson E, Simonetta F, Johnson E, Alam I et al (2023) PET imaging of TREM1 identifies CNS-infiltrating myeloid cells in a mouse model of multiple sclerosis. Sci Transl Med. 15(702):6267
Acknowledgements
We thank M.S. Alejandra Arreola-Triana for her support in editing this manuscript.
Author information
Authors and Affiliations
Contributions
MCT and ACM wrote the manuscript and design the figures.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Camacho-Morales, A., Cárdenas-Tueme, M. Prenatal Programming of Monocyte Chemotactic Protein-1 Signaling in Autism Susceptibility. Mol Neurobiol 61, 6119–6134 (2024). https://doi.org/10.1007/s12035-024-03940-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-024-03940-z