
Abstract
The purpose of this review is to summarize the current knowledge regarding the reciprocal associations between brain-derived neurotrophic factor (BDNF) and immune-inflammatory pathways and how these links may explain the involvement of this neurotrophin in the immune pathophysiology of mood disorders and schizophrenia. Toward this end, we delineated the protein–protein interaction (PPI) network centered around BDNF and searched PubMed, Scopus, Google Scholar, and Science Direct for papers dealing with the involvement of BDNF in the major psychosis, neurodevelopment, neuronal functions, and immune-inflammatory and related pathways. The PPI network was built based on the significant interactions of BDNF with neurotrophic (NTRK2, NTF4, and NGFR), immune (cytokines, STAT3, TRAF6), and cell–cell junction (CTNNB, CDH1) DEPs (differentially expressed proteins). Enrichment analysis shows that the most significant terms associated with this PPI network are the tyrosine kinase receptor (TRKR) and Src homology region two domain-containing phosphatase-2 (SHP2) pathways, tyrosine kinase receptor signaling pathways, positive regulation of kinase and transferase activity, cytokine signaling, and negative regulation of the immune response. The participation of BDNF in the immune response and its interactions with neuroprotective and cell–cell adhesion DEPs is probably a conserved regulatory process which protects against the many detrimental effects of immune activation and hyperinflammation including neurotoxicity. Lowered BDNF levels in mood disorders and schizophrenia (a) are associated with disruptions in neurotrophic signaling and activated immune-inflammatory pathways leading to neurotoxicity and (b) may interact with the reduced expression of other DEPs (CTNNB1, CDH1, or DISC1) leading to multiple aberrations in synapse and axonal functions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maes and Smith [1, 2] developed novel theories in 1995 based on pioneering findings that the major psychoses, including major depressive disorder (MDD), bipolar disorder (BD), and schizophrenia, are characterized by activated immune-inflammatory pathways. Thus, the monocyte-T-lymphocyte theory of major depression postulated that increased M1 macrophage activity associated with increased complement factors and acute phase proteins induced by pro-inflammatory cytokines such as interleukin (IL)-1 and IL-6, as well as increased T-helper 1 (Th-1) activity associated with IL-2 and interferon (IFN), underpins the pathogenesis of this condition [1]. Smith and Maes [2] also pioneered the macrophage – Th-1 lymphocyte theory of schizophrenia, hypothesizing that prenatal bacterial and viral infections may cause neurodevelopmental disorders, increasing postnatal susceptibility to immune-inflammatory stimuli via the neurotoxic effects of the aforementioned cytokines, the associated nitro-oxidative pathways, and induction of the tryptophan catabolite (TRYCAT) pathway with increased levels of neurotoxic TRYCATs.
There is now evidence that immune-inflammatory pathways play a critical role in the pathophysiology of major psychosis. Recent conceptualizations of both affective disorders and schizophrenia indicate that the major psychoses are associated with activation of the immune-inflammatory response system (IRS) and concurrent activation of the compensatory immune regulatory system (CIRS), which tends to downregulate the immune response and protects against an excessive immune response and hyperinflammation [3, 4]. Both MDD and BD are associated with IRS activation markers such as M1, Th-1, and Th-17 activation, as well as CIRS activation markers such as negative immune-regulatory cytokines including Th-2 (e.g., IL-4) and T regulatory (Treg) cytokines (e.g., IL-10), increased levels of soluble cytokine receptors which may inhibit cytokine signaling, e.g., the soluble tumor necrosis factor (TNF) receptor (sTNFR) and sIL-2R and sIL-1R antagonist (sIL-1RA), and some positive acute phase proteins including haptoglobin (Hp) [3]. Similarly, schizophrenia is associated with the same markers of IRS activation, such as M1, Th-1, and Th-17 activation, as well as CIRS activation, such as increased Treg and Th-2 shifts [4]. In both affective disorders (MDD and BD) and schizophrenia, acute episodes are associated with a shift in the homeostatic IRS/CIRS setpoint, as shown by an increased IRS/CIRS ratio, indicating a net immune-inflammatory response [3, 4].
According to the aforementioned theories, numerous different pathways including some cytokines (e.g., IL-1β, IL-6, TNF-α) and chemokines such as IL-8 (CXCL-8) and exotoxin (CCL-11), TRYCATS, and nitro-oxidative stress increase neurotoxicity in vulnerable brain regions, resulting in neuro-affective toxicity in affective disorders and more widespread neurotoxicity in schizophrenia [3, 4]. Additionally, enrichment and annotation analysis of differentially expressed proteins (DEPs) in first-episode psychosis (FEP) and new findings in schizophrenia indicate that activation of nuclear factor-κB, Toll-like receptor (TLR)-4, and Janus kinases/signal transducer and activator of transcription (JAK-STAT) pathways are involved in FEP and play a role in the subsequent transition from FEP to first episode schizophrenia (FES) [5]. Notably, both affective disorders and FES are associated with increased Gram-negative bacteria translocation via increased gut permeability, and it is believed that these biotic stimuli contribute to affective disorders and schizophrenia by activating the TLR2/4 complex, eliciting neuro-immune and nitro-oxidative pathways in the major psychosis, autoimmune responses in affective disorders, and intracellular pathways in FEP/FES [5, 6].
However, decreased neuroprotection because of reduced levels of neurotrophic factors also contributes to affective disorders and schizophrenia [5, 7]. Among these neurotrophic factors is brain-derived neurotrophic factor (BDNF), a multifunctional neurotrophin with numerous functions in the brain [8,9,10]. The evidence for the participation of BDNF and BDNF/TRK tropomyosin receptor kinase (encoded by the NTRK1 gene and binds BDNF) signaling in a wide range of neurophysiological processes continues to mount, including synaptic plasticity, connections, structure and formation, synaptogenesis, axonal growth, sprouting and guidance, dendritic arborization, neuronal differentiation and survival, neurogenesis, and modulation of neurotransmitter release [5, 11]. BDNF is found in nearly every brain region and participates in all stages of brain development and aging [12, 13]. It is regarded as an instructive mediator of structural and functional plasticity in the central nervous system [12]. Not only the major psychoses, but also other neuropsychiatric illnesses such as Alzheimer’s, Huntington’s, and Parkinson’s disease, as well as anxiety disorders, have been associated with decreased or altered BDNF signaling [14, 15]. While BDNF is not a biomarker for neurodegenerative or neuropsychiatric diseases due to its nonspecific deregulation in a variety of pathological conditions, it is believed to be specifically associated with the occurrence and/or progression of mnemonic symptoms associated with a variety of conditions characterized by cognitive deficits [12].
The purpose of this review is to summarize the current knowledge regarding the associations between BDNF and immune-inflammatory pathways in general and in the major psychoses more specifically.
Methods
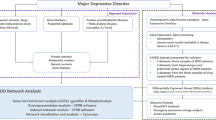
In this narrative review, we examine (a) the role of BDNF in the major psychoses with respect to its role in neurodevelopment and neuronal plasticity and (b) the reciprocal associations between BDNF and immune-inflammatory pathways. Toward this end, we delineated the protein–protein interaction (PPI) network in which BDNF participates and searched PubMed, Scopus, Google Scholar, and Science Direct for papers dealing with involvement of BDNF in the major psychosis, neurodevelopment, neuronal functions, and immune-inflammatory and related pathways.
BDNF and immune pathways play a role in complex mood and psychotic disorders, involving interactions between genetic factors, immune and BDNF pathways, and their intracellular signaling networks, metabolic pathways, and protein/enzyme alterations. Until recently, the majority of genetic, pathway, and metabolic studies on BDNF in psychiatry have focused on identifying and characterizing individual gene variants, as well as BDNF mRNA and protein levels in a variety of neuropsychiatric diseases. Nonetheless, BDNF and immune genes/proteins have a small to moderate effect on susceptibility and outcome in complex diseases, such as schizophrenia [5]. Enrichment and annotation analyses may shed light on how BDNF and immune genes and proteins interact to affect disease susceptibility. Additionally, network analysis of PPIs may reveal novel drug targets for modulating BDNF, immune pathways, and transcription factors. The objectives of this section are to define (a) the BDNF interactome and its interactions with the neuroimmune network; (b) the biological functions, pathways, molecular patterns, and cellular components that define the BDNF networks; and (c) the possible trigger factors for changes in the BDNF-neuroimmune interactome.
The enrichment and annotation analyses were conducted as described previously [5]. We constructed three types of expanded networks: (1) a first-order PPI network based on BDNF as only seed protein and 10 interactions in the first shell and none in the second (with a minimum required interaction score of 0.900 and set organism: homo sapiens), (2) a first order–enlarged PPI network with BDNF as only seed protein and 50 interactions in the first shell and none in the second shell (again with a minimum required interaction score of 0.900 and set organism: homo sapiens), and (3) giant networks based on the network established in or the combination of the latter with the major M1, Th-1, Th-2, Th-17, and Treg genes common to affective disorders and schizophrenia [3, 4]. The networks were constructed using STRING version 11.0 (https://string-db.org), a predictive database, and the IntAct Molecular Interaction Database (https://www.ebi.ac.uk/intact/), a primary database based on peer-reviewed publications. STRING and the Cytoscape plugin Network Analyzer were used to compute the network characteristics. The network backbone was defined as a collection of top hubs (nodes with the highest degree) and top non-hub bottlenecks (nodes with the highest betweenness centrality). STRING, Gene Ontology (GO) net (https://tools.dice-database.org/Gonet/), OmicsNet (OmicsNet), and Metascape (https://metascape.org) were used to visualize the physical interactions between the proteins/genes. The different PPI networks were analyzed for their enrichment scores and annotated terms using the following tools: (a) STRING to establish GO biological processes and molecular functions, as well as KEGG (https://genome.jp/kegg/) and REACTOME (the European Bioinformatics Institute Pathway Database; https://reactome.org) pathways, (b) OmicsNet (using InAct, mirNET, TTRUST, and KEGG metabolic reactions) to establish REACTOME and PANTHER (PANTHER—Gene List Analysis (pantherdb.org) biological processes) and the OmicsNet regulation explorer to establish TTRUST transcriptional regulatory relationships (www.grnpedia.org/trrust); and (c) Metascape to establish and visualize the over-represented REACTOME, KEGG, PANTHER, and Wiki (WikiPathways—WikiPathways) pathways. The latter are based on accumulative hypergeometric p-values that are used for filtering, after which the remaining terms are hierarchically clustered into a tree and cast into term clusters using a 0.3 kappa score threshold. As such, Metascape was also combined with Molecular Complex Detection (MCODE) to delineate and visualize enriched ontology clusters.
Human BDNF
The human BDNF gene is composed of 11 exons and nine alternative promoters, with the coding sequence located in exon IX and present in all BDNF messenger RNA isoforms. The presence of many gene controlling regions suggests for complex BDNF expression in response to a wide variety of stimuli [16]. Alternative splicing enables the formation of transcripts that are tissue- and stimulus-specific [17]. BDNF is released as a mixture of pro- and mature forms with opposing effects and involvement of distinct cellular pathways. BDNF is translated as a proneurotrophin (pro-BDNF) and is cleaved in the cytoplasm by endoproteases or in the extracellular matrix by plasmin or matrix metalloproteinases to produce the mature BDNF [11].
Both mature BDNF and pro-BDNF can be secreted and bind to the neurotrophin receptor p75 NTR and induce apoptosis [18]. Only the mature BDNF binds to tyrosine kinase receptors (TrkB) and activates a variety of signaling cascades [19]. By creating cleavage-resistant pro-BDNF knock-in mouse, Yang et al. [20] demonstrated that pro-BDNF negatively regulates hippocampal dendritic complexity and spine density through p75(NTR). Low synaptic transmission and enhanced long-term depression were detected suggesting that pro-BDNF has distinct functions from those of mature BDNF, which acts as a biologically active factor that regulates hippocampal structure, synaptic transmission, and plasticity [20]. When activated, the phosphatidylinositol-3-kinase (PI3K), the Ras-mitogen-activated protein kinase (MAPK), and the phospholipase C (PLC-) pathways result in the phosphorylation of transcription factors and the de novo transcription of the BDNF gene [19], ultimately promoting cell survival [21]. On the other hand, truncated TrkB receptors act as dominant negative inhibitors of BDNF signaling, whereas proBDNF helps regulate BDNF in physiological conditions [22].
Apart from the two isoforms, a single nucleotide polymorphism at position 66 within the BDNF gene’s pro-domain encoding region influences neurotrophin’s functionality [23]. The Val66Met substitution is shown to result into a condensed protein explaining neuronal growth cone retraction [24]. The Met66 prodomain variant is also proposed to function as a ligand with independent significance for BDNF communication [25]. BDNF isoforms appear to play a role in the regulation of processes throughout the brain’s development, as well as in diverse cellular populations and body systems.
BDNF PPI Networks
Figure 1 shows a first-order PPI network constructed using STRING with BDNF as seed protein and 10 interactions in the first shell (none in the second shell; minimum required interaction score > 0.900). This network consists of 11 nodes, and the number of edges (n = 26) exceeds the expected number of edges (n = 11), with p-enrichment value of 9.54e-05, and average node degree = 4.73 and average local clustering coefficient = 0.811. Table 1 shows the descriptions and functions of the ten added genes of this PPI network.

A first-order protein–protein interaction network constructed using STRING with brain-derived neurotrophic factor (BDNF) as seed protein and 10 interactions in the first shell (none in the second shell; minimum required interaction score > 0.900)
Figure 2 shows a PPI network constructed using STRING with BDNF as seed protein and 50 interactions in the first shell (none in the second shell; minimum required interaction score > 0.900). This network consists of 51 nodes, and the number of edges (n = 380) exceeds the expected number of edges (n = 115), with p-enrichment value of < 1e-16 and average node degree = 14.9 and average local clustering coefficient = 0.709. Degree and betweenness centrality analysis showed that BDNF (degree = 50), NGF (35), NTRK2 (27), NTRK1 (27), HRAS (25), and GRB2 (25) were the top five hubs. BDNF (betweenness centrality = 0.3987), NGF (0.0767), NTRK2 (0.0245), NTRK1 (0.0239), and NGFR (0.0214) were the top bottlenecks in this network. As such the backbone of this enlarged network consists of BDNF (seed gene), NGF, NTRK2, NTRK1, HRAS, GRB2, and NGFR. Table 1 shows the names and descriptions of NGF, HRAS, and GRB2.

A protein–protein interaction network constructed using STRING with brain-derived neurotrophic factor (BDNF) as seed protein and 50 interactions in the first shell (none in the second shell; minimum required interaction score > 0.900)
Table 2 shows the top twenty biological GO-enriched terms in this PPI network, indicating that the five most important over-represented terms are the neurotrophin signaling pathway, transmembrane receptor protein tyrosine kinase signaling pathway, cell surface receptor signaling pathway, and positive regulation of cell communication and signaling. Figure 3 displays the top 10 KEGG terms that were over-expressed in the BDNF-enlarged network with the neurotrophin, RAS and MAPK, and ErbB signaling pathways as top pathways. Figure 4 displays the top 10 performing cellular GO terms that are accumulated in the BDNF PPI network indicating that neuron projection, axon, and dendrites are the top terms followed at a distance by focal adhesion, cell junctions, phosphatidylinositol 3-kinase complex, Sin3 complex, and catenin complex. Figure 5 shows a giant PPI network constructed using OmicsNet and IntAct using BDNF and the 50 BDNF-interacting proteins shown in Fig. 2. This giant network consists of 294 nodes and 308 edges, and the hubs in this enlarged network are GRB2 (degree = 729), TRAF6 (373), TP53 (367), HDAC1 (234), CRK (227), PIK3R1 (181), CTBBB1 (153), CDC42 (129), and STAT3 (129). Table 3 shows the top REACTOME and PANTHER pathways enriched in this PPI network.

Top 10 KEGG terms that are over-expressed in the brain-derived neurotrophic factor (BDNF) enlarged network

The top 10 performing cellular GO terms that are accumulated in the brain-derived neurotrophic factor protein–protein interaction network

A giant protein–protein interaction network constructed using OmicsNet and IntAct using brain-derived neurotrophic factor (BDNF) and 50 BDNF-interacting proteins as determined using STRING
BDNF and Key Molecular Pathways
Transcriptional activation of BDNF occurs when PI3K (phosphatidylinositol 3-Phosphate) is activated, which results in mTOR-dependent translation of BDNF [26]. Apart from binding to low affinity p75NTR, cleaved, mature BDNF binds to its high-affinity receptor tyrosine kinase B (TrkB), activating several signaling cascades which induce an increase in Ca2 + intake, phosphorylation of transcription factors, and de novo expression of the BDNF gene [8]. BDNF-dependent phospholipase C-gamma (PLC-γ) can provoke short-term signaling by increasing Ca2 + neuronal response and inhibit inflammatory-dependent apoptosis cascade by suppression of glycogen synthase kinase 3-beta (GSK-3β) [10].
BDNF can also influence gene regulation via activating transcription factors such as NF-κB and CREB, which activate Akt and Erk, respectively, through downstream pathways [27]. By modifying these genes, BDNF may modulate neuronal survival, development, and long-term potency [28]. Furthermore, BDNF-independent transactivation of TrkB can also play a vital role in the neurotrophic pathway regulated by adenosine, zinc, epidermal growth factor (EGF), glucocorticoids, and pituitary adenylate-cyclase-activating polypeptide (PACAP), further enhancing TrkB signaling in the synapse [11].
Based on the analysis of PPI networks, BDNF seems to interact most widely with the neurotrophin receptors, catenin β1 (a downstream component of the canonical Wnt signaling pathway), the TNFR superfamily member 16 (a low affinity receptor binding to NGF, BDNF, NT-3, and NT-4), and sortilin (SORT1, a sorting receptor in the Golgi compartment). The interactions of BDNF with neurotrophin receptors NTRK1, NTRK2, and NTRK3 are important for the early stages of neurodevelopment. While NTRK3 is required for initial neurite outgrowth and regulation of surviving cells numbers, BDNF promotes survival and differentiation of motoneurons. Depletion of NT-3 or BDNF may affect the development of the peripheral or central nervous systems [29]. Moreover, synergistic interactions between BDNF and NTRK3 may be crucial for spiral ganglion neuron survival during the final stages of development [30]. By single-cell transcriptome analysis BDNF is identified as a STAT3 target gene in a unique population of regenerating type II alveolar pneumocytes, and hence the therapeutic importance of the STAT3-BDNF-TrkB axis in regulating alveolar epithelial regeneration after lung injury is recognized [31].
Sortilin promotes neuronal apoptosis by mediating endocytosis of the proapoptotic precursor forms of BDNF (proBDNF) and NGFB (proNGFB). The sortilin pathway has distinct roles in pro-neurotrophin-induced apoptotic signaling in pathological conditions, as well as in specific stages of neuronal development and aging [32].
BDNF, Embryonic Brain Development, and Neurodevelopment
Being the most abundant neurotrophin in the mammalian CNS, BDNF plays a key role in the development and physiology of the fetal brain [10, 33]. Its biological role is not only limited to neurogenesis but also affects the processes of placental angiogenesis and fetal development [34,35,36]. Functionally, BDNF acts through the TrkB and p75 NGFR receptors, located in the central and peripheral nervous system of the fetus in the midpregnancy [37, 38]. The binding of proBDNF to 75NTR receptors determines synaptic pruning in the prenatal brain and initiates the proBDNF / p75NTR / sortiline signaling cascade [39]. This results in activation of c-Jun amino-terminal kinase (JNK) that is involved in neuronal apoptosis [40, 41]. On the other hand, the binding of the mature domain of BDNF to the p75NTR receptors, RIP2 (serine / threonine protein kinase 2) / TRAF6 (tumor necrosis factor 6 receptor factor), initiates a pathway to ensure the survival and maintenance of neurons during embryogenesis [40, 42].
During the early brain development, BDNF coordinates the processes of glio-, neuro-, and synaptogenesis on one hand and the elimination of improperly formed connections on the other, in order to form the functional and morphological integrity of the nervous system [10, 43,44,45]. During the fourth month of pregnancy, the number of BDNF-positive neurons increases which is associated with its involvement in the development of the frontal lobe of the fetal brain [46].
TrkB receptors for the neuroprotein are expressed at the beginning of neurulation in the neural tube [29]. An in situ hybridization study of mouse and chicken embryos showed that TrkB is detected in the hindbrain in the area of the neuraxis, which generates motor neurons [47,48,49]. Likewise, BDNF acts as a mitogen for motoneurons and guides neuronal differentiation, and blocking BDNF/TrkB receptors with trkB-Ig reduces the number of neurons [29, 48]. Therefore, BDNF deficiency leads to fewer motor neurons, which affects the development of the central or peripheral nervous system. Evidence of this are animals with knocked out BDNF and TrkB, which have severe neuronal deficits, difficulty in coordination, and short life [50,51,52]. The lowest BDNF mRNA expression was detected in the brain of early rat embryos, and it gradually increased [53] between embryonic days 11–13, and the time corresponded to the beginning of neurogenesis [54, 55] These findings can be explained by dynamically developing neuronal populations during the early stages of ontogenesis.
In addition, BDNF supplied by the mother during pregnancy supports the development of the fetal nervous system. BDNF concentrations in the amniotic fluid influence its levels in the fetus [56,57,58,59,60,61]. Thus, fetal brain development is a strictly regulated process, as evidenced by pathological changes occurring with the rapid increase of BDNF in the fetal brain [62, 63]. For example, injection of the protein into the brain ventricles of the mouse embryo in vitro leads to premature neurogenesis and abnormal cell proliferation [62].
BDNF has also a key role in some pregnancy-associated disorders, whereby high BDNF levels in the amniotic fluid are associated with reduced fetal growth. Thus, macrosomic and small for gestational age fetuses show higher BDNF amnion fluid levels [34]. According to the authors, these changes are a compensatory mechanism associated with the accelerated development and maturation of the fetal brain, which limits its growth [34]. Roth and Sweatt (2011) emphasize that adverse environmental stressors during pregnancy may modulate cortical BDNF gene expression. This epigenetic mechanism leads to methylation [64,65,66,67] and changes in the genes regulated by BDNF, significantly reducing the expression of BDNF in the hippocampus [68, 69], blood, and brain [66, 70]. This reflects the role of epigenetic factors and the influence of stress in the early life of the individual.
Various animal models reveal that elevated levels of hippocampal BDNF are associated with higher levels of maternal care, and the result is a high propensity to the social interaction of the individual in adulthood [71]. On the other hand, the isolation from the mother is associated with reduced levels of both mRNA and protein in the prefrontal cortex, amygdala, and hippocampus [72]. This again shows the key role of BDNF in brain development and the regulation of complex behavioral responses.
BDNF as Master Regulator of Neurogenesis and Synaptic Plasticity
The term neuroplasticity describes the ability of the brain to reorganize its neural circuits in response to experience, which is of fundamental importance for learning and memory [73]. In turn, neurogenesis allows the incorporation of newly generated neurons into the existing neural networks [74]. As a member of the neurotrophin family, BDNF controls neuronal development and promotes neuronal function and plasticity through various mechanisms.
The initial hypothesis that BDNF may contribute to synaptic plasticity arose from the discovery that high frequency stimulation (HFS) can promote overexpression of BDNF in the hippocampus [75, 76]. Direct evidence of such contribution was provided by Figurov et al. [77], who described that BDNF treatment promotes the induction of long-term potentiation (LTP) in the hippocampus. Furthermore, the observed effects were reversed following the application of TrkB-IgG, which scavenges endogenous BDNF, thus decreasing the strength of LTP. In addition, in BDNF knockout mice, hippocampal LTP was significantly affected [78, 79]. In line with these findings, application of exogenous BDNF can trigger LTP in a protein synthesis-dependent manner and can modulate translation in dendrites [80].
In general, BDNF-mediated regulation of synaptic plasticity depends on the two main types of BDNF receptors. These include the Trk family and the p75 neurotrophin receptor (p75NTR), part of the TNF receptor superfamily [81]. By interacting with these receptors, BDNF triggers various pathways of intracellular signaling, especially the MAPK-, PI3K-, and PLCγ-dependent signaling cascades [19], to regulate neuronal differentiation and survival, axonal growth, and dendritic pruning [82].
The TrkB is highly selective and interacts with great affinity with BDNF and NT-4, but not with the other two neurotrophins, NGF and NT-3. Conversely, all four neurotrophins recognize p75NTR with equal affinity [83]. Among the signal transducers involved in MAPK and PI3K pathways, MAPK/ERK and PIK3CA are closely activated following BDNF-TrkB binding. Behavioral studies revealed the role of BDNF-MAPK pathway in mood control and anxiety [84]. BDNF activates MAPK signaling to upregulate Bcl-2 gene expression in neural stem cells (NSCs), which inhibits apoptosis and promotes cell survival. Furthermore, siRNA-mediated suppression of BDNF synthesis or U0126 inhibition of the MAPK signal transduction significantly decreases Bcl-2 levels, which confirms that the neuroprotective effect of BDNF is MAPK-dependent [85].
It has been suggested that the interaction between BDNF and TrkB initiates MAPK/ERK signal transduction to increase the dendritic spine density in hippocampal neurons. The activated MAPK pathway also promotes enhanced spine maturation. Inhibition of TrkB with k-252a disrupts the effects of BDNF on spine density and morphology [83].
In the past two decades, emerging evidence has suggested the involvement of MMP-9 (matrix metalloproteinase 9) in neuronal plasticity [86]. MMP-9 is an endopeptidase that takes part in the remodeling of the extracellular matrix. The enzyme has been associated with LTP and memory, as well as with dendritic spine reorganization. In line with this association is the fact that elevated MMP-9 expression can result from KCl-induced neuronal depolarization and chemically evoked seizures. Researchers have demonstrated that MMP-9 transcription is modulated by synaptic activity and depends on TrkB signaling [86]. The latter experimental data suggest that bicuculline administration in rat primary cortical neurons enhances synaptic activity and upregulates the expression of BDNF. The upregulated BDNF then regulates the MMP-9 levels in a mechanism that requires MAPK/ERK-signaling and c-Fos expression. Apparently, the observed changes in both BDNF and MMP-9 expression levels resulted from the elevated synaptic activity, which is a crucial trigger of neuronal plasticity [86].
The complex interplay between local protein synthesis and remodeling of the actin cytoskeleton plays a key role in the growth cones of axons and may also participate in LTP [87, 88]. The BDNF receptor, TrkB, appears pivotal for the signal network that regulates the local production of many factors involved in cytoskeletal dynamics including RhoA and LIMK1 [89, 90]. Stabile LTP in gyrus dentatus requires persistent Arc synthesis for the phosphorylation of cofilin and the steady accumulation of synaptic F-actin content [91]. In this context, application of exogenous BDNF significantly elevates the Arc-mediated LTP [91], while LTP in gyrus dentatus strongly associates with increased TrkB activation and enhanced release of BDNF [92, 93].
Numerous studies on cultured hippocampal neurons have demonstrated that BDNF regulates synaptic transmission and enhances N-methyl-D-aspartic acid receptor (NMDAR) function [94]. BDNF also stimulates the maturation of neuromuscular synapses and increases their activity in vitro [95]. In addition to the excitatory transmission, BDNF also regulates inhibitory neuronal signaling [96]. It promotes neuronal potentiation while affecting both pre- and postsynaptic processes. In the presynaptic compartment, BDNF improves glutamate secretion in cortical neurons in vitro by activating the PLC-γ (phospholipase C-γ)/Ca2+ pathway, thus increasing the frequency of the miniature excitatory postsynaptic currents [97]. Conversely, the elevated neuronal activity resulting in LTP was shown to cause significant hippocampal BDNF overexpression, suggesting the presence of regulatory feedback mechanisms [76]. On the postsynaptic side, BDNF increases the sensitivity of the NMDAR and modulates its opening probability [98]. NMDAR-mediated Ca2+ influx is essential for neuronal differentiation, neuronal migration, synaptogenesis, synaptic remodeling, long-lasting changes in synaptic efficacy, such as LTP and LTD (long-term depression), and cognitive functions such as learning and memory [99].
NMDAR activation results in increased Ca2+ influx that couples synaptic activation and the intracellular signaling pathways, which regulate neuronal differentiation, synaptic remodeling, and plasticity [99]. It has also been reported that BDNF increases the expression of voltage-dependent Ca2+ and voltage-dependent Na+ channels on the surface of the neurons. As for the role of BDNF in the inhibitory neural circuits, BDNF regulates the formation of GABAergic synapses in the hippocampus [100]. Moreover, artificially applied BDNF decreases the inhibitory synaptic GABAergic currents in the hippocampus via TrkB receptor activation [101].
Apart from its well-established role in remodeling the existing neuronal circuits, BDNF has an important function in neural maturation and neurogenesis. It is involved in the proliferation of NPCs and the prolonged maintenance of newly emerging neurons [102,103,104]. Direct application of BDNF in the hippocampus and its administration in peripheral blood have been correlated with strong stimulation of hippocampal neurogenesis [103]. According to Kuipers et al. [105], BDNF-induced neurogenesis in gyrus dentatus is associated with LTP, while inhibition of Arc translation restricts the induction of LTP and its positive effects on neurogenesis [104, 106].
BDNF and the Wnt/Catenin Pathway
Using confocal microscopy, Bamji et al. [107] demonstrated that BDNF supports neuronal plasticity by disrupting the association between cadherin and β-catenin (CTNNB1) in neuronal synapses. This results in enhanced mobility of synaptic vesicles, as well as in long-term increase in synapse number. BDNF hampers cadherin–β-catenin interactions by increasing β-catenin phosphorylation [107]. This phosphorylation occurs on tyrosine 654 and is catalyzed by Src kinases. At the same time, BDNF stimulates the formation of new synapses, which is blocked by the artificial preservation of cadherin–β-catenin interactions.
β-catenin is a protein with versatile functions, which participates in the cell-cell adhesion and in the canonical Wnt signaling pathway [108]. It forms a complex with the cytoplasmic domain of cadherin and binds to the actin fibers of the cytoskeleton via α-catenin [109]. The cadherin–catenin complex engages in intercellular adhesion and plays a pivotal role in synaptogenesis and synaptic remodeling, affecting both synaptic size and strength [110]. Destabilization and reassociation of the cadherin–catenin complex contribute to morphological changes in neurons that result in new synapse formation. From another perspective, β-catenin functions as a transducer in the Wnt signaling pathway. In the absence of Wnt activation, β-catenin is phosphorylated by glycogen synthase kinase-3β (GSK-3β) and readily degraded by the proteasome [111]. Conversely, Wnt signaling stabilizes β-catenin by inhibiting GSK-3β, which allows the translocation of β-catenin into the nucleus. There β-catenin interacts with transcription factors of the TCF/LEF family to regulate the expression of Wnt-targeted genes [112, 113]. Accordingly, experimental evidence suggests that Wnt signaling is implicated in the regulation of synaptic plasticity in hippocampal slices.
Maguschak et al. [114] have confirmed the role of β-catenin in the amygdala-dependent fear memory. The application of LiCl, which unspecifically inhibits β-catenin degradation, results in increased fear retention. Consolidation of fear memory involves changes in both the expression and phosphorylation levels of β-catenin. The observed changes are accompanied by dynamic regulation of the β-catenin/cadherin complex. Genetic analyses involving region-specific deletion of loxP-flanked CTNB1 further confirmed the importance of β-catenin for long-term memory consolidation [114].
Alterations in β-catenin phosphorylation and distribution are involved not only in long-term memory but also in trauma response [115]. Heat acclimation provides robust antecedent neuroprotection against traumatic brain injury via a mechanism involving tyrosine phosphorylation of β-catenin and weakening of the β-catenin/cadherin complex. According to Umschweif et al. [115], high basal levels of BDNF and N-cadherin stimulate a rapid increase in the Akt phosphorylation upon a cerebral injury, leading to the inhibition of both JNK (c-Jun N-terminal kinase) and GSK-3β. The inhibition of GSK-3β alters ser33/37thr41 phosphorylation of β-catenin, while elevated levels of BDNF result in induced tyrosine 654 phosphorylation of β-catenin. The new phosphorylation state allows β-catenin to escape degradation or nuclear translocation, which is crucial for the reorganization of β-catenin/cadherin complexes. This complex dynamic of β-catenin/cadherin interactions promotes the reestablishment of intercellular adhesion and plays a key role in trauma recovery.
Numerous studies have shown that BDNF significantly contributes to the longevity, proliferation, and differentiation of neural stem cells (NSCs) through the Wnt/β-catenin signaling pathway [116, 117]. Transfection of human embryonic spinal cord NSCs (hESC-NSCs) with a plasmid vector expressing BDNF stimulates cell growth in vitro. The plasmid-mediated overexpression of BDNF upregulates the expression of Wnt, Frizzled, and Dsh, while decreasing the levels of GSK-3β. The opposite effects are evoked following siRNA silencing of BDNF.
BDNF increases neuronal growth in terms of perikaryon size and neurite length through the Wnt/β-catenin signaling pathway. Experimental data from Western blot and qRT-PCR analyses have demonstrated that BDNF overexpression upregulates key Wnt signaling factors, Wnt, Frizzled, and Dsh, and their downstream target β-catenin, while downregulates GSK-3β. These expression changes can be reverted with the application of BDNF siRNA, which also decreases neuronal growth. Furthermore, BDNF signaling factors, Wnt pathway components, and β-catenin were all downregulated, whereas GSK-3β was upregulated. Further treatment of the neurons with the GSK-3β inhibitor 6-bromoindirubin-3′-oxime (BIO) suppresses the effects of BDNF on neuronal growth and reduces the BDNF-dependent activation of the Wnt signaling pathway. These findings confirm that BDNF stimulates neuronal growth by modulating the Wnt/β-catenin signaling pathway [118].
In addition, BDNF can markedly stimulate the differentiation of induced pluripotent stem cells (iPSCs) into NSCs through the Wnt/β-catenin and MAPK/ERK signaling cascades. Zhang et al. demonstrated that BDNF increases the expression of β-catenin and ERK 5 in BDNF-treated iPSCs in comparison with untreated ones [119]. The authors have also suggested that the inhibition of ERK5 affects the activity of the Wnt/β-catenin signaling pathway. This finding implies a functional overlapping between Wnt/β-catenin and MAPK signaling in the context of BDNF-induced iPSCs differentiation.
Altogether these vast experimental data confirm the role of BDNF as an important regulator of the Wnt/β-catenin signaling pathway. However, Wnt/β-catenin signaling is not only regulated by BDNF but also participates in the activity-dependent control of BDNF expression [120]. It has been widely acknowledged that BDNF expression is regulated by neuronal activity which elevates BDNF transcription via an increased Ca2+ influx through ligand- and voltage-gated calcium channels. The Ca2+ entry triggers the activation of the cAMP response element-binding protein (CREB). CREB serves as a transcription factor that binds to the cAMP/Ca2+-response elements within the BDNF promoter, thus increasing the transcription of the BDNF gene. The activation of CREB can occur through multiple Ca2+-dependent signal transduction pathways such as the cAMP/PKA, Ras/MAPK, and calmodulin/calmodulin kinase pathways. Despite that CREB activation constitutes the primary mechanism for activity-regulated BDNF transcription, the activity-dependent expression of BDNF also relies on the Wnt/β-catenin signaling. Synaptic activity and NMDAR stimulation modulate the Wnt/β-catenin pathway and the transcription of Wnt/β-catenin target genes. The activation of the Wnt/β-catenin pathway is mediated by Frizzled receptors together with LRP5 and LRP6 co-receptors. This leads to the translocation of β-catenin into the nucleus where it supports the transcription of its target genes including BDNF and Axin2 [120].
BDNF and the JAK/STAT Pathway
Apart from the ERK1/2, PI3K, and PLC pathways, BDNF can induce JAK/STAT signaling to stimulate neurite growth [121,122,123,124]. In the rat major pelvic ganglion (MPG), BDNF increases the phosphorylation of JAK2, STAT1, and STAT3, thus promoting neurite elongation [124]. The neurite-stimulating effect of exogenous BDNF on cultured MPGs is dosage-dependent with an optimal range between 25 and 50 ng/ml. In the presence of BDNF, this effect can be significantly reduced by the JAK/STAT inhibitor AG490, which confirms the predominant role of the JAK/STAT pathway in the BDNF-related neurite stimulation.
After nerve injury, BDNF activates the JAK/STAT pathway in Schwann cells to facilitate nerve regeneration [125], and BDNF stimulates the phosphorylation of STAT1 and STAT3, which in Schwann cells occurs in a delayed manner. Following an initial peak 1 h after BDNF treatment, the levels of STAT1/2 decrease to baseline levels and peak again 24 h after treatment. As a result of BDNF administration, Schwann cells produce cytokines, especially OSM-M and IL-6 that promote nerve regeneration.
Using a full transcriptome sequencing approach on BDNF-treated neurons with and without JAK/STAT inhibitors, Hixson et al. [126] have determined the complete pool of genes that undergo BDNF-dependent JAK/STAT-mediated regulation in cultured cortical neurons. Their analyses revealed 2869 differentially regulated genes whose expression changes after BDNF application. One thousand five hundred fifty-nine of these 2659 genes appeared to be most strongly associated with JAK/STAT signaling in neurons. Among the targets of BDNF-induced JAK/STAT signaling, the authors identified genes for ion channels, neurotransmitter receptors, and several factors of synaptic plasticity and neurogenesis. This work has also revealed that the BDNF-induced JAK/STAT pathway regulates many epilepsy-associated genes. The presumed mechanism by which BDNF triggers the JAK/STAT activation in neurons is non-canonical and potentially involves an interaction between STAT3 and Heterochromatin Protein 1 alpha (HP1α).
A major BDNF downstream mediator, STAT3 (Signal transducer and activator of transcription 3), has been associated with the prenatal development of the neocortex [127, 128] and the neuronal fitness in response to insulin-like growth factor [129]. STAT3 regulates axonal outgrowth [130, 131] and participates in the formation of hypothalamic neural circuits [132]. Tang et al. [133] have described significant upregulation of STAT3 at both mRNA and protein levels in rat brain samples following movement training. STAT3 binds to the promoters of brain-related genes, including BDNF, thus mediating their transcriptional control [133].
Oncogenic Properties of BDNF
Alternatively, BDNF is well-known to exhibit oncogenic properties, which are closely related to its neurogenetic activity [134, 135]. Researchers have suggested that glial malignancies may arise from neural stem cells that undergo an aberrant differentiation. It has been also shown that the interaction between BDNF and TrkB stimulates Ras activation, which intensifies the cell cycle progression [134]. Accordingly, overexpression of BDNF and TrkB significantly contributes to oncogenesis of neuroblastoma. In neuroblastoma cells, BDNF reduces the upregulated expression of p53 and promotes the survival of tumor cells following etoposide treatment [136]. BDNF may stimulate cell proliferation in glioblastoma multiforme and in retinoblastoma. Intriguingly, the oncogenic activity of BDNF can be markedly suppressed by p53 which upregulates the long non-coding antisense RNA of the BDNF gene, encoded by BDNF-AS. Using a ChIP analysis, Lv et al. [137] demonstrated that the p53 transcription factor readily binds to the promoter region of the BDNF-AS gene. This discovery was further validated by a luciferase reporter assay. The authors have also shown that p53 transfection dramatically increases the expression of BDNF-AS.
Upregulation of BDNF and TrkB has been demonstrated in a vast set of cancer types, including bladder cancer [138], breast cancer [139, 140], ovarian cancer [141], hepatocellular carcinoma [142, 143], gastric cancer [144], and colorectal cancer [145, 146]. When triggered by BDNF, TrkB promotes the activation of numerous downstream factors, such as Akt, Src, ERK, and MAPK, resulting in abnormal cell proliferation, ineffective apoptosis, invasion, metastasis, and chemotherapy resistance [147,148,149]. Cervical cancer cells manifest higher levels of TrkB and BDNF than normal cells. Notably, the siRNA-mediated knockdown of TrkB reduces the invasiveness of cancer cells in a way involving the suppression of N-cadherin, vimentin, MMP-2, and MMP-9. Conversely, the silencing of TrkB results in overexpression of the epithelial cadherin (E-cadherin) and the tissue inhibitor of metalloproteinases 2 (TIMP2). This newly established pattern of gene expression suppresses cell division, migration, and invasion [150].
Except from cervical cancer, upregulation of TrkB and BDNF along with downregulation of E-cadherin has been demonstrated in salivary adenoid cystic carcinoma (SACC). Moreover, the levels of TrkB correlate negatively with the expression of E-cadherin in SACC. These alterations in gene expression have been associated with higher invasive potential of SACC cells and with poor prognosis of affected patients. In agreement with this, the exogenous addition of BDNF markedly activates TrkB and induces the progression of SACC cells, while TrkB inhibition by k252a significantly reduces SACC proliferation [151].
BDNF and Immunity
PPI Network of BDNF and Immune Molecules
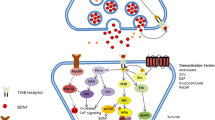
Recently, Maes et al. [5] reported the PPI network of first-episode psychosis (FEP) and first-episode schizophrenia (FES) which include BDNF and immune differentially expressed proteins (DEPs), and they constructed a dense PPI network with 92 nodes and 1063 edges (n = 1063). Immune genes (including IL-6, IL-2, IL10, and TNF-α) were top hubs, while BDNF, CTNNB1, and CDH1 were the most important non-hub bottlenecks. As such the backbone of this network consisted of CTNNB1, IL6, TNF, CDH1, IL4, IL10, and BDNF [5]. MCL cluster analysis performed at an inflation parameter of 2.5 showed two PPI clusters, namely an immune cluster, which comprised cytokines, chemokines and immune DEPs, and a cell–cell junction cluster, which comprised CTNNB1 and CDH1. Importantly, BDNF was allocated to the immune cluster and functioned as a switch between both clusters with many relevant interactions with DEPs in the cell–cell junction (e.g., with CTNNB1, NTRK2, CTNNA1, and CTNND1) and immune (e.g., STAT3, TRAF6, NTF4, NGFR, IL6, TNF, IL4, IL10, IFNG) clusters.
Based on this PPI, we here constructed a new PPI centered around BDNF only and added all Fig. 1 DEPs as well key Fig. 2 DEPs (HRAS, PIK3R1) to make a new immune, neurotrophic, and cell–cell junction PPI network, whereby all DEPs show relevant interactions with BDNF. Figure 6 shows this new neurotrophic, immune-cell–cell junction PPI network centered around BDNF. This network consists of 39 nodes, and the number of edges (n = 362) exceeds the expected number of edges (n = 123), with p-enrichment value of 1.0e-016 and average node degree = 18.6 and average local clustering coefficient = 0.739. Metascape enrichment analysis shows that the most significant terms associated with this network were the SHP2 (log10 p = − 26.2) and TRKR (log10 p = − 25.7) pathway and signaling by receptor tyrosine kinases (log10 p = − 24.7).

A protein–protein interaction network centered around brain-derived factor (BDNF) and comprising major neurotrophic, immune-inflammatory, and cell–cell junction genes
Table 4 and Fig. 7 show the results of MCODE analysis with GO enrichment analysis applied to the three molecular components to extract “biological meanings”. The top terms retained in the first MCODE component revolve around signaling by receptor tyrosine kinases and NTRKs, positive regulation of kinase and transferase activity in the second MCODE component, and cytokine signaling in the immune system and negative regulation of the immune response in the third component. This indicates that changes in BDNF may affect a wide variety of neurotrophic, immune, and cell–cell junction functions and their associated signaling pathways. As such, a wide spectrum of processes is controlled by BDNF, and therefore, BDNF is a crucially involved in different intercellular communications and intracellular functions and is implicated in the bidirectional interactions between the immune system and neuronal functions.

Results of molecular complex detection (MCODE) analysis with enrichment analysis applied to the three molecular components
Brunelli et al. [152] showed that human PBMCs produce and secrete BDNF isoforms as part of the physiological stress response showing that immune cells and BDNF are interconnected. In a rat model of optic nerve injury, mRNA expression of BDNF in T cells is demonstrated together with mRNAs of other neurotrophins [153]. BDNF and NGF levels are regulated by TNF-α and its receptors even under immunologically unchallenged conditions [154]. Additionally, BDNF may have anti-inflammatory and anti-apoptotic properties via its regulation of the MyD88/NFB and PI3K/AKT signaling pathways [155]. Pretreatment with BDNF may increase the expression of IL-10 and Trk and inhibit the production of IL-6, TNF-α, and IL-1 and NF-κB following bacterial infection.
BDNF is implicated in neuroinflammation and shows neuroprotective effects. For example, in a mouse model of aging, the critical role of BDNF-TrkB signaling in regulating microglia inflammation was shown. Decreases of BDNF-TrkB signaling in microglia during aging are associated with their activation in the substantia nigra, while systemic delivery of BDNF reversed aging-related microglial activation [13]. The authors also used a cultured microglial cell line, and BDNF blocked LPS-induced microglial activation. Activation of the TrkB/Erk/CREB pathway is necessary for the BDNF-induced antimicroglial activation response. BDNF overexpression in the hippocampus diminishes synaptic impairments and improves neuroinflammation induced by hyperglycemia, which may be mediated by inhibiting the HMGB1/RAGE/NF-κB signaling pathway [156]. A study on Alzheimer’s dementia revealed associations between BDNF and proBDNF with immune markers, such as VEGF, EGF, and CD95 + CD3 + ratio [157].
In multiple sclerosis, BDNF secreted by immune cells is associated with sites of higher inflammatory activity and may represent a crucial factor associated with the white matter volume [158]. An animal model of MS mice deficient for BDNF in immune cells displays an attenuated immune response in the acute phase of the disease but progressive disability with enhanced axonal loss in the chronic phase. In mice deficient for CNS-derived BDNF, a more severe course and an overall increased axonal loss was observed [159]. This indicated again the protective role of BDNF in autoimmune demyelination by mediating axon protection. Another study comparing BDNF to Tau proteins revealed that BDNF is a good biomarker for the diagnosis of MS but not for severity or progression of the disease [160]. High proBDNF expression is found in circulating lymphocytes and infiltrated inflammatory cells at the lesion sites of the brain and spinal cord of patients [161]. The latter authors showed that the systemic administration of anti-proBDNF blocking antibodies attenuated clinical severity scores. Yoshimura et al. [162] reported that IFN-β caused a significant increase in serum BDNF levels produced by T cells as well as in TrkB expression levels in peripheral blood mononuclear cells of MS patients.
After brain damage or inflammation, BDNF shows a neuroprotective effect resulting from its binding to the TrkB receptor and activation of a signaling cascade [163]. This leads to anti-inflammatory and anti-apoptotic effects by modulating MyD88 / NF-κB and PI3K / AKT signaling pathways [155, 164]. Activation of the MyD88 pathway triggers NF-κB and MAPK, which increase the expression of cytokines and proinflammatory mediators [165, 166]. Therefore, BDNF is a part of an endogenous defense pathway during bacterial and viral inflammatory processes and helps to eliminate the negative effect of the inflammatory response [165]. BDNF is involved in the modulation of the inflammatory response and in the regulation of its detrimental effects [163, 165, 167]. By blocking the activation of caspase-3 [168,169,170], BDNF reduces the translocation of apoptosis-inducing factor (AIF) [171] and the excitotoxicity of glutamate. Moreover, BDNF augments the activity of antioxidant enzymes [172] and improves the binding capacity of intracellular calcium increasing cell survival [168, 173].
BDNF is implicated not only in neuroinflammation but also in inflammatory processes in visceral organs. Qiao et al. [174] showed that both BDNF and pro-BDNF levels were increased in inflamed urinary bladder. The BDNF high affinity receptor TrkB and general receptor p75 expression levels were elevated, with an increased level of TrkB tyrosine phosphorylation/activity. These results suggest that in vivo BDNF release is an important event in neurogenic inflammatory states and implies a possible pro-proliferative effect of BDNF in the inflamed bladder. The significant interplay between BDNF and immune mediators is also indicated by increased BDNF serum levels and increased NGFR expression on T-cells in patients with rheumatoid arthritis [175]. Furthermore, mRNA for TrkA, TrkB, and p75 receptors were expressed in the injured nerve, suggesting that these specific receptors can mediate the effects of the T-cell-derived neurotrophins [153].
Therefore, it is safe to posit that the participation of BDNF in the immune cluster and its interactions with neuroprotective and cell–cell adhesion DEPs is a conserved regulatory process which protects against the many detrimental effects of immune activation and hyperinflammation [5]. Overall, the results of the PPI network in Fig. 6 and the above review on the interactions between BDNF and the immune system indicate that (a) BDNF belongs to the immune response PPI network, (b) BDNF regulates many immune functions and signaling and may downregulate the immune-inflammatory response, and (c) lowered BDNF levels during chronic stress are associated with increased inflammatory responses.
BDNF and Immune Cell Interactions and Inflammatory Pathways in the Major Psychoses
Major Depression
Depression is regarded as a life-threatening disease as well as a severe cause of disability [176]. The growing body of evidence in the literature supports the central role of a moderate chronic immune-inflammatory process as a driver for the onset and progression of depression, as evidenced by over 10,000 publications published on the subject [3]. Glial and endothelial cells in the brain produce cytokines and chemokines [177] that function as neuromodulators in brain development and to maintain a healthy brain homeostasis in general [178, 179]. Nonetheless, major depressive individuals have heightened immune-inflammatory responses, including increased levels of acute phase reactants as well as elevated proinflammatory cytokines and receptors, chemokines, and soluble adhesion molecules in the CSF, post-mortem tissue, and peripheral blood [3].
The literature shows that neurotrophic pathways, as well as activated immune-inflammatory pathways, have a role in the etiology of MDD. Thus, neurotrophic and inflammatory processes may interact in depression, with BDNF playing a key role due to its impact on brain plasticity and neuronal functioning [180,181,182]. There is evidence that BDNF levels are reduced in MDD/MDE patients, but that levels in remitted patients are comparable to those in healthy people [183]. In a cohort study, Hsieh et al. [184] compared the methylation patterns of BDNF exon IX promoter as well as the serum BDNF protein and mRNA levels in 51 patients with MDD and 62 healthy controls. The authors’ results suggest that in peripheral blood, the expression of BDNF protein and mRNA is significantly lower in MDD as compared with healthy controls [184]. These observations are concordant with the findings of Schroter et al. [185], who also described lower serum BDNF levels in patients with MDD. According to Chiou and Huang [186], drug-naïve patients with first-episode MDD exhibit lower BDNF protein levels than healthy individuals. Moreover, a ROC (receiver operating characteristic) analysis conducted by the same authors [187] showed that BDNF expression can discriminate male patients with MDD from controls with moderate accuracy. Additionally, alterations in the methylation patterns of histone H3 at lysine 9 within the BDNF exon IV have been observed in patients with schizophrenia [188]. BDNF levels that are decreased can also be utilized to externally validate the diagnosis of MDD. Chen and coauthors were able to design an algorithm based on serum BDNF, cortisol, and IFN-γ levels with an accurate confusion matrix (AUC of 0.884, 86.7 percent sensitivity, and 83.3 percent specificity). The Val66Met polymorphism (rs6265) of BDNF is linked to reduced BDNF levels as well as higher inflammation in depressed patients [189,190,191,192].
Inflammation generated by lipopolysaccharides (LPS) may alter BDNF/TrkB signaling in the prefrontal cortex, hippocampus, and nucleus accumbens, which is associated with the onset of depressive behaviors [120]. This is critical because mood symptoms are related with increased bacterial translocation, which results in elevated levels of IgA/IgM directed against Gram-negative bacteria’s LPS, while the latter are associated with enhanced immunological and autoimmune responses [6]. Inflammatory chemokines and cytokines, such as CXCL1/2, activate the GSK3b pathway in mice, generating depression-like behaviors [193]. LPS administration can induce depressive-like behaviors in mice models of inflammation by modulating the N-methyl-D-aspartate (NMDA) receptor functions [194], whereas LPS-treated mice lacking the NMDA receptor GluN2A subunit do not develop depressive-like behaviors and have increased levels of proBDNF and BDNF in the prefrontal cortex (PFC) and/or hippocampus [195]. This shows that BDNF-mediated alterations in the GluN2A subunit may contribute to depression associated with neuroinflammation.
Additionally, chronic stress-induced depression-like behaviors are related with increased chemokine production (e.g., CXCL1/CXCL2) and GSK3b pathway regulation, which disrupts the CREB-BDNF pathway. Similarly, chronic stress-induced depression-like behaviors may be reversed by inhibiting CXCR2, which regulates GSK3b, apoptosis, and the CREB-BDNF signaling pathway [193]. All of this evidence suggests that activation of immune-inflammatory pathways plays a critical role in the development of major depression or depressive-like behaviors and that targeting these pathways may be a viable therapeutic strategy [196, 197].
Because BDNF levels are decreased in critical cortical and limbic brain regions in depressed patients, novel therapeutic approaches may focus on increasing BDNF levels [9, 192, 198,199,200]. Another strategy involves the use of PPARg agonists, which have been shown to raise BDNF levels [9, 201,202,203]. In the rodent, deep brain stimulation (DBS) of the medial forebrain bundle (MFB), a novel treatment for treatment-resistant depression [204, 205], ameliorates anhedonia-like behaviors induced by chronic unpredictable mild stress in association with normalization of the plasma, CSF, and hippocampus BDNF levels.
Heterozygous (BDNF + /) individuals exhibit anhedonia- and anxiety-like behaviors in association with changes in neurogulin-1, glucocorticoid receptor, and disrupted-in-schizophrenia (DISC) 1 gene expression in the prefrontal cortex, all of which may play a role in depression [206]. These findings imply that a decrease in BDNF levels alters the expression of genes related with affective disorders, potentially precipitating the onset of depressive-like symptoms. The BDNF exon IV promoter is involved in BDNF transcription and depressive-like behavior in humans and rodents. Patients with MDD who had hypomethylation of the CpG-87 site in the promoter IV region have a worse response to antidepressant therapy [207, 208]. Additional research established a relationship between stress exposure and a particular methylation pattern in the BDNF promoter IV, which results in depression [207, 209]. Promoter IV mutant mice (BDNF-KIV) have severe abnormalities in prefrontal GABAergic interneurons and decreased GABAergic activity in conjunction with depressive-like behaviors [210]. Additionally, antidepressant therapy boosts BDNF mRNA and protein levels in the cerebral cortex and hippocampus in rodent models due to histone acetylation in the promoter regions [211, 212].
Schizophrenia
As described in the “Introduction” [1], the early monocyte-T-lymphocyte theory of schizophrenia postulated that prenatal bacterial or viral infections could result in neurodevelopmental disorders, which, when combined with subsequent immune hits, could result in the onset of schizophrenia via increased neurotoxicity [2]. A recent meta-analysis showed that schizophrenia has been linked to decreased BDNF levels (Hedges g = 0.458, p 0.004) [213]. BDNF levels are adversely related with a greater immune-inflammatory response in antipsychotic naïve first-episode psychosis (FEP), which may progress to deficit schizophrenia [214]. Additionally, an interaction between polymorphisms in the BDNF and NTRK2 genes increase susceptibility to paranoid schizophrenia [215]. The BDNF Met and DISC1 Cys mutations are associated with schizophrenia, and this BDNF variant is connected with a biomarker associated with schizophrenia, namely dysfunctions in exploratory eye movement [216]. Another study established a mutational profile for the BDNF (rs6265) and DISC1 (rs821597) variants in schizophrenia patients compared to healthy controls, demonstrating that the BDNF variant was associated with global cognitive impairment, working memory impairment, and attention deficits [217].
As discussed before in the section titled “PPI network of BDNF and immune molecules,” decreased BDNF levels in FEP are associated with the immunological cluster (which contains M1, Th1, Th2, and Treg cytokines and pro-inflammatory chemokines and reflects the immune-inflammatory response in FEP). Not only is BDNF connected with immunological (cytokines, STAT3, TRAF6) and neurotrophic (NTRK2, NTF4, and NGFR) DEPs in this FEP PPI network, but also with cell–cell junction (CTNNB1, CTNNA1, CTNND1, CDH1) DEPs [5]. BDNF not only acts as a neurotrophic and immunological regulating factor within this intricate yet tightly interconnected network, but also connects the immune cluster to a cell–cell junction cluster that regulates the beta-catenin complex, adherence junction architecture, and Wnt/catenin signaling pathways [5]. Thus, it seems that the immune-inflammatory response in FEP/FES involves not only IRS and CIRS components, but also neurotrophic (BDNF, NTF4, and NGFR) and cell adhesion (CTNNB1 and CDH1) components [5]. As BDNF levels are decreased in FEP/FES, the neurotoxic effects of the IRS may not be adequately mitigated by the neuroprotective and CIRS capabilities of BDNF and its related cell–cell adhesion network. As a result, decreased BDNF levels contribute to FEP and its progression to deficit schizophrenia by impairing neuroprotective and immunological regulatory functions [5].
Additionally, unique combinations of the downregulated BDNF, DISC1, CHD1, and CTNNB1 DEPs are related with abnormalities in synaptic formation (CDH1 and BDNF), positive control of axonogenesis (BDNF and DISC1), and neuron projection development (BDNF, CTNNB1, and CDH1) [5]. The BDNF and CTNNB1 genes exhibit very significant database-annotated interactions (STRING), and the BDNF and CTNNB1 pathways are also functionally linked [5]. BDNF-mediated breakdown of cadherin–catenin complexes is associated with increased synaptic density in hippocampal neurons [107]. Polymorphisms in BDNF are associated with changes in the Wnt/-catenin pathway, which may in part govern BDNF synthesis [218, 219]. This may explain why individuals with impaired CIRS functions, such as deficiencies in natural IgM, protective cytokine receptors, neurotrophin/Trk, and Wnt/catenin signaling, decreased DISC1 expression, and interactions between diminished BDNF, CDH1, CTNNB, and DISC1 expression may be at an increased risk of developing FEP and FES following immune hits [5].
Conclusions
A growing body of research indicates the involvement of immune-inflammatory pathways in the development of neuropsychiatric disorders. In the present review, we have summarized the link between BDNF and immune-inflammatory pathways and underlined how those interactions delineate the neuro-immune pathophysiology of mood disorders and schizophrenia. The built PPI network showed significant interactions between BDNF and neurotrophic (NTRK2, NTF4, and NGFR), immune (cytokines, STAT3, TRAF6), and cell–cell junction (CTNNB, CDH1) DEPs in the major psychosis, neurodevelopment, neuronal functions, and immune-inflammatory and related pathways. TRKR and Src homology region SHP2 pathways, Trk signaling pathways, positive regulation of kinase and transferase activity, cytokine signaling, and negative regulation of the immune response were the most significant participants identified to belong to this PPI network. We conclude that decreased BDNF levels in mood disorders and schizophrenia most probably contribute to alterations in neurotrophic signaling and increase the vulnerability to activation of immune-inflammatory pathways, followed by neurotoxicity. Also, BDNF may cause alterations in the expression of other DEPs (CTNNB1, CDH1, or DISC1) leading to multiple aberrations in synaptic and axonal functions. The available data suggest that BDNF is an instructive mediator of structural and functional plasticity in the CNS and its interactions with the immune pathways are the key components in major psychiatric disorders. Consequently, future research which examines the expression of BDNF in psychiatric disorders should examine not only BDNF but also the expression (protein-mRNA) of its major interacting proteins as determined in this review. Moreover, such measurements should be added to the existing precision nomothetic models of affective disorders and schizophrenia which were constructed based on the neurotoxic effects of immune and nitro-oxidative pathways [6, 220,221,222]. As such, these neurotoxicity precision models may be enriched and improved by adding the neuroprotective BDNF-associated pathways.
Data Availability
Not applicable.
Abbreviations
- BDNF:
-
Brain-derived neurotrophic factor
- BD:
-
Bipolar disorder
- CDH:
-
Cadherin gene/protein
- CDC:
-
Cell division cycle gene/protein
- ChIP analysis:
-
Chromatin immunoprecipitation analysis
- CIRS:
-
Compensatory immune regulatory system
- CPE:
-
Carboxypeptidase E
- CSF:
-
Cerebrospinal fluid
- CRK:
-
CRK proto-oncogene
- CREB:
-
CAMP response element-binding protein
- CTNNB:
-
Catenin beta protein
- CTBBB1:
-
Catenin beta 1 gene
- DBS:
-
Deep brain stimulation
- DISC1:
-
Disrupted in schizophrenia 1 protein
- DEPs:
-
Differentially expressed proteins
- FEP:
-
First-episode psychosis
- FES:
-
First-episode schizophrenia
- GABA:
-
Gamma aminobutyric acid
- GO:
-
Gene ontology
- GSK:
-
Glycogen synthase kinase
- GRB2:
-
Growth factor receptor-bound protein 2
- HDAC:
-
Histone deacetylase
- HMGB1:
-
High mobility group box 1 DNA-binding nuclear protein
- HRAS:
-
GTPase HRas
- Hp:
-
Haptoglobin
- hESC-NSCs:
-
Human embryonic spinal cord NSCs
- IFN:
-
Interferon
- IL:
-
Interleukin
- IRS:
-
Inflammatory response system
- JAK-STAT:
-
Janus kinases/signal transducer and activator of transcription
- JNK:
-
C-Jun N-terminal kinase
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- LIMK1:
-
LIM domain kinase 1
- LPS:
-
Lipopolysaccharide
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- MAPK:
-
Ras-mitogen-activated protein kinase
- MDD:
-
Major depressive disorder
- MECP2:
-
Methyl-CpG-binding protein 2
- MFB:
-
Medial forebrain bundle
- MMP:
-
Matrix metalloproteinase
- NMDAR:
-
N-Methyl-D-aspartate receptor
- NGF:
-
Beta-nerve growth factor
- NGFR:
-
Tumor necrosis factor receptor superfamily member 16
- NPC:
-
Intracellular cholesterol transporter
- NSC:
-
Neural stem cells
- NTF:
-
Neurotrophin
- NTRK1:
-
High affinity nerve growth factor receptor
- NTRK2:
-
BDNF/NT-3 growth factors receptor
- PBMC:
-
Peripheral blood mononuclear cells
- PFC:
-
Prefrontal cortex
- PI3K:
-
Phosphatidylinositol-3-kinase
- PIK3R:
-
Phosphatidylinositol-3-kinase receptor
- PLC:
-
Phospholipase C
- PPI:
-
Protein-protein interaction
- qRT-PCR:
-
Real-time quantitative reverse transcription
- RAGE:
-
Receptor for advanced glycation end products
- RIP2:
-
Serine/threonine protein kinase 2
- SHP2:
-
Two domain-containing phosphatase-2
- STAT3:
-
Signal transducer and activator of transcription 3
- STRING:
-
Significant database-annotated interactions
- SORT:
-
Sortilin
- Th:
-
T helper cells
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- TNFR:
-
Tumor necrosis factor receptor
- TP53:
-
Tumor protein p53
- TRAF6:
-
Tumor necrosis factor 6 receptor factor
- T reg:
-
T regulatory cells
- TRK:
-
Tropomyosin receptor kinase
- TRKR:
-
Tyrosine kinase receptor
- TRYCAT:
-
Tryptophan catabolite
References
Maes M, Smith R, Scharpe S (1995) The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology 20(2):111–116. https://doi.org/10.1016/0306-4530(94)00066-j
Smith RS, Maes M (1995) The macrophage-T-lymphocyte theory of schizophrenia: additional evidence. Med Hypotheses 45(2):135–141. https://doi.org/10.1016/0306-9877(95)90062-4
Maes M, Carvalho AF (2018) The compensatory immune-regulatory reflex system (CIRS) in depression and bipolar disorder. Mol Neurobiol 55(12):8885–8903. https://doi.org/10.1007/s12035-018-1016-x
Roomruangwong C, Noto C, Kanchanatawan B, Anderson G, Kubera M, Carvalho AF, Maes M (2020) The role of aberrations in the immune-inflammatory response system (IRS) and the compensatory immune-regulatory reflex system (CIRS) in different phenotypes of schizophrenia: the IRS-CIRS theory of schizophrenia. Mol Neurobiol 57(2):778–797. https://doi.org/10.1007/s12035-019-01737-z
Maes M, Vojdani A, Sirivichayakul S, Barbosa DS, Kanchanatawan B (2021) Inflammatory and oxidative pathways are new drug targets in multiple episode schizophrenia and leaky gut, Klebsiella pneumoniae, and c1q immune complexes are additional drug targets in first episode schizophrenia. Mol Neurobiol 58(7):3319–3334. https://doi.org/10.1007/s12035-021-02343-8
Simeonova D, Stoyanov D, Leunis JC, Murdjeva M, Maes M (2021) Construction of a nitro-oxidative stress-driven, mechanistic model of mood disorders: a nomothetic network approach. Nitric Oxide 106:45–54. https://doi.org/10.1016/j.niox.2020.11.001
Maes M, Berk M, Goehler L, Song C, Anderson G, Galecki P, Leonard B (2012) Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med 10:66. https://doi.org/10.1186/1741-7015-10-66
Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220:223–250. https://doi.org/10.1007/978-3-642-45106-5_9
Bjorkholm C, Monteggia LM (2016) BDNF - a key transducer of antidepressant effects. Neuropharmacology 102:72–79. https://doi.org/10.1016/j.neuropharm.2015.10.034
Kowianski P, Lietzau G, Czuba E, Waskow M, Steliga A, Morys J (2018) BDNF: a key factor with multipotent impact on brain signaling and synaptic plasticity. Cell Mol Neurobiol 38(3):579–593. https://doi.org/10.1007/s10571-017-0510-4
Lima Giacobbo B, Doorduin J, Klein HC, Dierckx R, Bromberg E, de Vries EFJ (2019) Brain-derived neurotrophic factor in brain disorders: focus on neuroinflammation. Mol Neurobiol 56(5):3295–3312. https://doi.org/10.1007/s12035-018-1283-6
Miranda M, Morici JF, Zanoni MB, Bekinschtein P (2019) Brain-derived neurotrophic factor: a key molecule for memory in the healthy and the pathological brain. Front Cell Neurosci 13:363. https://doi.org/10.3389/fncel.2019.00363
Wu SY, Pan BS, Tsai SF, Chiang YT, Huang BM, Mo FE, Kuo YM (2020) BDNF reverses aging-related microglial activation. J Neuroinflammation 17(1):210. https://doi.org/10.1186/s12974-020-01887-1
Colucci-D’Amato L, Cimaglia G (2020) Ruta graveolens as a potential source of neuroactive compounds to promote and restore neural functions. J Tradit Complement Med 10(3):309–314. https://doi.org/10.1016/j.jtcme.2020.05.002
Lin CC, Huang TL (2020) Brain-derived neurotrophic factor and mental disorders. Biomed J 43(2):134–142. https://doi.org/10.1016/j.bj.2020.01.001
Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90(3):397–406. https://doi.org/10.1016/j.ygeno.2007.05.004
Colucci-D'Amato L, Speranza L, Volpicelli F (2020) Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci 21 (20). doi:https://doi.org/10.3390/ijms21207777
Friedman WJ (2010) Proneurotrophins, seizures, and neuronal apoptosis. Neuroscientist 16(3):244–252. https://doi.org/10.1177/1073858409349903
Minichiello L (2009) TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 10(12):850–860. https://doi.org/10.1038/nrn2738
Yang J, Harte-Hargrove LC, Siao CJ, Marinic T, Clarke R, Ma Q, Jing D, Lafrancois JJ et al (2014) proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep 7(3):796–806. https://doi.org/10.1016/j.celrep.2014.03.040
Volosin M, Song W, Almeida RD, Kaplan DR, Hempstead BL, Friedman WJ (2006) Interaction of survival and death signaling in basal forebrain neurons: roles of neurotrophins and proneurotrophins. J Neurosci 26(29):7756–7766. https://doi.org/10.1523/JNEUROSCI.1560-06.2006
Haapasalo A, Sipola I, Larsson K, Akerman KE, Stoilov P, Stamm S, Wong G, Castren E (2002) Regulation of TRKB surface expression by brain-derived neurotrophic factor and truncated TRKB isoforms. J Biol Chem 277(45):43160–43167. https://doi.org/10.1074/jbc.M205202200
Chen ZY, Patel PD, Sant G, Meng CX, Teng KK, Hempstead BL, Lee FS (2004) Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J Neurosci 24(18):4401–4411. https://doi.org/10.1523/JNEUROSCI.0348-04.2004
Lohia R, Salari R, Brannigan G (2019) Sequence specificity despite intrinsic disorder: how a disease-associated Val/Met polymorphism rearranges tertiary interactions in a long disordered protein. PLoS Comput Biol 15(10):e1007390. https://doi.org/10.1371/journal.pcbi.1007390
Hempstead BL (2015) Brain-derived neurotrophic factor: three ligands, many actions. Trans Am Clin Climatol Assoc 126:9–19
Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H (2004) Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci 24(44):9760–9769. https://doi.org/10.1523/JNEUROSCI.1427-04.2004
Marini AM, Jiang X, Wu X, Tian F, Zhu D, Okagaki P, Lipsky RH (2004) Role of brain-derived neurotrophic factor and NF-kappaB in neuronal plasticity and survival: from genes to phenotype. Restor Neurol Neurosci 22(2):121–130
Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR (2002) Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci 22(17):7453–7461
Bernd P (2008) The role of neurotrophins during early development. Gene Expr 14(4):241–250. https://doi.org/10.3727/105221608786883799
Mou K, Hunsberger CL, Cleary JM, Davis RL (1997) Synergistic effects of BDNF and NT-3 on postnatal spiral ganglion neurons. J Comp Neurol 386(4):529–539
Paris AJ, Hayer KE, Oved JH, Avgousti DC, Toulmin SA, Zepp JA, Zacharias WJ, Katzen JB et al (2020) STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat Cell Biol 22(10):1197–1210. https://doi.org/10.1038/s41556-020-0569-x
Jansen P, Giehl K, Nyengaard JR, Teng K, Lioubinski O, Sjoegaard SS, Breiderhoff T, Gotthardt M et al (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat Neurosci 10(11):1449–1457. https://doi.org/10.1038/nn2000
Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T (2007) Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 85(3):525–535. https://doi.org/10.1002/jnr.21139
Antonakopoulos N, Iliodromiti Z, Mastorakos G, Iavazzo C, Valsamakis G, Salakos N, Papageorghiou A, Margeli A et al (2018) Association between brain-derived neurotrophic factor (BDNF) levels in 2(nd) trimester amniotic fluid and fetal development. Mediators Inflamm 2018:8476217. https://doi.org/10.1155/2018/8476217
Kawamura K, Kawamura N, Sato W, Fukuda J, Kumagai J, Tanaka T (2009) Brain-derived neurotrophic factor promotes implantation and subsequent placental development by stimulating trophoblast cell growth and survival. Endocrinology 150(8):3774–3782. https://doi.org/10.1210/en.2009-0213
Kawamura K, Kawamura N, Fukuda J, Kumagai J, Hsueh AJ, Tanaka T (2007) Regulation of preimplantation embryo development by brain-derived neurotrophic factor. Dev Biol 311(1):147–158. https://doi.org/10.1016/j.ydbio.2007.08.026
Klein R, Parada LF, Coulier F, Barbacid M (1989) trkB, a novel tyrosine protein kinase receptor expressed during mouse neural development. EMBO J 8(12):3701–3709
Barbacid M (1994) The Trk family of neurotrophin receptors. J Neurobiol 25(11):1386–1403. https://doi.org/10.1002/neu.480251107
Deinhardt K, Chao MV (2014) Shaping neurons: long and short range effects of mature and proBDNF signalling upon neuronal structure. Neuropharmacology 76 Pt C:603–609. doi:https://doi.org/10.1016/j.neuropharm.2013.04.054
Anastasia A, Deinhardt K, Chao MV, Will NE, Irmady K, Lee FS, Hempstead BL, Bracken C (2013) Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat Commun 4:2490. https://doi.org/10.1038/ncomms3490
Teng HK, Teng KK, Lee R, Wright S, Tevar S, Almeida RD, Kermani P, Torkin R et al (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci 25(22):5455–5463. https://doi.org/10.1523/JNEUROSCI.5123-04.2005
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361(1473):1545–1564. https://doi.org/10.1098/rstb.2006.1894
Gonzalez A, Moya-Alvarado G, Gonzalez-Billaut C, Bronfman FC (2016) Cellular and molecular mechanisms regulating neuronal growth by brain-derived neurotrophic factor. Cytoskeleton (Hoboken) 73(10):612–628. https://doi.org/10.1002/cm.21312
Sasi M, Vignoli B, Canossa M, Blum R (2017) Neurobiology of local and intercellular BDNF signaling. Pflugers Arch 469(5–6):593–610. https://doi.org/10.1007/s00424-017-1964-4
Foltran RB, Diaz SL (2016) BDNF isoforms: a round trip ticket between neurogenesis and serotonin? J Neurochem 138(2):204–221. https://doi.org/10.1111/jnc.13658
Zheng LR, Zhu XQ, Huang XM, Gu Q, Xie DH (2013) Morphological study on early development of brain derived neurophic factor-positive neurons in the frontal lobe of human fetus. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 35(3):260–264. https://doi.org/10.3881/j.issn.1000-503X.2013.03.004
Yao L, Zhang D, Bernd P (1994) The onset of neurotrophin and trk mRNA expression in early embryonic tissues of the quail. Dev Biol 165(2):727–730. https://doi.org/10.1006/dbio.1994.1288
Jungbluth S, Koentges G, Lumsden A (1997) Coordination of early neural tube development by BDNF/trkB. Development 124(10):1877–1885
Zhang D, Yao L, Bernd P (1996) Expression of neurotrophin trk and p75 receptors in quail embryos undergoing gastrulation and neurulation. Dev Dyn 205(2):150–161. https://doi.org/10.1002/(SICI)1097-0177(199602)205:2%3c150::AID-AJA6%3e3.0.CO;2-H
Li H, Lin LY, Zhang Y, Lim Y, Rahman M, Beck A, Al-Hawwas M, Feng S et al (2020) Pro-BDNF knockout causes abnormal motor behaviours and early death in mice. Neuroscience 438:145–157. https://doi.org/10.1016/j.neuroscience.2020.05.007
Conover JC, Erickson JT, Katz DM, Bianchi LM, Poueymirou WT, McClain J, Pan L, Helgren M et al (1995) Neuronal deficits, not involving motor neurons, in mice lacking BDNF and/or NT4. Nature 375(6528):235–238. https://doi.org/10.1038/375235a0
Johnson EM, Craig ET, Yeh HH (2007) TrkB is necessary for pruning at the climbing fibre-Purkinje cell synapse in the developing murine cerebellum. J Physiol 582(Pt 2):629–646. https://doi.org/10.1113/jphysiol.2007.133561
Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD (1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5(4):501–509. https://doi.org/10.1016/0896-6273(90)90089-x
Almeida LE, Roby CD, Krueger BK (2014) Increased BDNF expression in fetal brain in the valproic acid model of autism. Mol Cell Neurosci 59:57–62. https://doi.org/10.1016/j.mcn.2014.01.007
Murray PS, Holmes PV (2011) An overview of brain-derived neurotrophic factor and implications for excitotoxic vulnerability in the hippocampus. Int J Pept 2011:654085. https://doi.org/10.1155/2011/654085
Deuschle M, Hendlmeier F, Witt S, Rietschel M, Gilles M, Sanchez-Guijo A, Fananas L, Hentze S et al (2018) Cortisol, cortisone, and BDNF in amniotic fluid in the second trimester of pregnancy: effect of early life and current maternal stress and socioeconomic status. Dev Psychopathol 30(3):971–980. https://doi.org/10.1017/S0954579418000147
Kodomari I, Wada E, Nakamura S, Wada K (2009) Maternal supply of BDNF to mouse fetal brain through the placenta. Neurochem Int 54(2):95–98. https://doi.org/10.1016/j.neuint.2008.11.005
DM Halepoto S Bashir L ALA 2014 Possible role of brain-derived neurotrophic factor (BDNF) in autism spectrum disorder: current status J Coll Physicians Surg Pak 24 4 274 278 04.2014/JCPSP.274278
Kimiwada T, Sakurai M, Ohashi H, Aoki S, Tominaga T, Wada K (2009) Clock genes regulate neurogenic transcription factors, including NeuroD1, and the neuronal differentiation of adult neural stem/progenitor cells. Neurochem Int 54(5–6):277–285. https://doi.org/10.1016/j.neuint.2008.12.005
Ohmiya M, Shudai T, Nitta A, Nomoto H, Furukawa Y, Furukawa S (2002) Brain-derived neurotrophic factor alters cell migration of particular progenitors in the developing mouse cerebral cortex. Neurosci Lett 317(1):21–24. https://doi.org/10.1016/s0304-3940(01)02412-0
Buchman VL, Davies AM (1993) Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development 118(3):989–1001
Fukumitsu H, Ohtsuka M, Murai R, Nakamura H, Itoh K, Furukawa S (2006) Brain-derived neurotrophic factor participates in determination of neuronal laminar fate in the developing mouse cerebral cortex. J Neurosci 26(51):13218–13230. https://doi.org/10.1523/JNEUROSCI.4251-06.2006
Bartkowska K, Paquin A, Gauthier AS, Kaplan DR, Miller FD (2007) Trk signaling regulates neural precursor cell proliferation and differentiation during cortical development. Development 134(24):4369–4380. https://doi.org/10.1242/dev.008227
Kertes DA, Bhatt SS, Kamin HS, Hughes DA, Rodney NC, Mulligan CJ (2017) BNDF methylation in mothers and newborns is associated with maternal exposure to war trauma. Clin Epigenetics 9:68. https://doi.org/10.1186/s13148-017-0367-x
Braithwaite EC, Kundakovic M, Ramchandani PG, Murphy SE, Champagne FA (2015) Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 10(5):408–417. https://doi.org/10.1080/15592294.2015.1039221
Gilmore JH, Jarskog LF, Vadlamudi S (2003) Maternal infection regulates BDNF and NGF expression in fetal and neonatal brain and maternal-fetal unit of the rat. J Neuroimmunol 138(1–2):49–55. https://doi.org/10.1016/s0165-5728(03)00095-x
Wang JM, Pei LX, Zhang YY, Cheng YX, Niu CL, Cui Y, Feng WS, Wang GF (2018) Ethanol extract of Rehmannia glutinosa exerts antidepressant-like effects on a rat chronic unpredictable mild stress model by involving monoamines and BDNF. Metab Brain Dis 33(3):885–892. https://doi.org/10.1007/s11011-018-0202-x
Smith MA, Makino S, Kvetnansky R, Post RM (1995) Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci 15(3 Pt 1):1768–1777
Buchmann AF, Hellweg R, Rietschel M, Treutlein J, Witt SH, Zimmermann US, Schmidt MH, Esser G et al (2013) BDNF Val 66 Met and 5-HTTLPR genotype moderate the impact of early psychosocial adversity on plasma brain-derived neurotrophic factor and depressive symptoms: a prospective study. Eur Neuropsychopharmacol 23(8):902–909. https://doi.org/10.1016/j.euroneuro.2012.09.003
Kundakovic M, Gudsnuk K, Herbstman JB, Tang D, Perera FP, Champagne FA (2015) DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci U S A 112(22):6807–6813. https://doi.org/10.1073/pnas.1408355111
Branchi I (2009) The mouse communal nest: investigating the epigenetic influences of the early social environment on brain and behavior development. Neurosci Biobehav Rev 33(4):551–559. https://doi.org/10.1016/j.neubiorev.2008.03.011
Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM (2007) Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci 25(10):3091–3098. https://doi.org/10.1111/j.1460-9568.2007.05522.x
Mateos-Aparicio P, Rodriguez-Moreno A (2019) The impact of studying brain plasticity. Front Cell Neurosci 13:66. https://doi.org/10.3389/fncel.2019.00066
Kee N, Teixeira CM, Wang AH, Frankland PW (2007) Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci 10(3):355–362. https://doi.org/10.1038/nn1847
Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ (1993) The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport 4(7):895–898. https://doi.org/10.1097/00001756-199307000-00014
Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M (1992) Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron 9(6):1081–1088. https://doi.org/10.1016/0896-6273(92)90067-n
Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381(6584):706–709. https://doi.org/10.1038/381706a0
Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 92(19):8856–8860. https://doi.org/10.1073/pnas.92.19.8856
Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16(6):1137–1145. https://doi.org/10.1016/s0896-6273(00)80140-3
Baj G, Pinhero V, Vaghi V, Tongiorgi E (2016) Signaling pathways controlling activity-dependent local translation of BDNF and their localization in dendritic arbors. J Cell Sci 129(14):2852–2864. https://doi.org/10.1242/jcs.177626
Barbacid M (1993) Nerve growth factor: a tale of two receptors. Oncogene 8(8):2033–2042
Dinsmore CJ, Soriano P (2018) MAPK and PI3K signaling: at the crossroads of neural crest development. Dev Biol 444(Suppl 1):S79–S97. https://doi.org/10.1016/j.ydbio.2018.02.003
Chapleau CA, Pozzo-Miller L (2012) Divergent roles of p75NTR and Trk receptors in BDNF’s effects on dendritic spine density and morphology. Neural Plast 2012:578057. https://doi.org/10.1155/2012/578057
Einat H, Yuan P, Gould TD, Li J, Du J, Zhang L, Manji HK, Chen G (2003) The role of the extracellular signal-regulated kinase signaling pathway in mood modulation. J Neurosci 23(19):7311–7316
Peng CH, Chiou SH, Chen SJ, Chou YC, Ku HH, Cheng CK, Yen CJ, Tsai TH et al (2008) Neuroprotection by Imipramine against lipopolysaccharide-induced apoptosis in hippocampus-derived neural stem cells mediated by activation of BDNF and the MAPK pathway. Eur Neuropsychopharmacol 18(2):128–140. https://doi.org/10.1016/j.euroneuro.2007.05.002
Kuzniewska B, Rejmak E, Malik AR, Jaworski J, Kaczmarek L, Kalita K (2013) Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol Cell Biol 33(11):2149–2162. https://doi.org/10.1128/MCB.00008-13
Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8(10):776–789. https://doi.org/10.1038/nrn2150
Van Horck FP, Holt CE (2008) A cytoskeletal platform for local translation in axons. Sci Signal 1(8):pe11. https://doi.org/10.1126/stke.18pe11
Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME (2006) A brain-specific microRNA regulates dendritic spine development. Nature 439(7074):283–289. https://doi.org/10.1038/nature04367
Troca-Marin JA, Alves-Sampaio A, Tejedor FJ, Montesinos ML (2010) Local translation of dendritic RhoA revealed by an improved synaptoneurosome preparation. Mol Cell Neurosci 43(3):308–314. https://doi.org/10.1016/j.mcn.2009.12.004
Messaoudi E, Kanhema T, Soule J, Tiron A, Dagyte G, da Silva B, Bramham CR (2007) Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J Neurosci 27(39):10445–10455. https://doi.org/10.1523/JNEUROSCI.2883-07.2007
Gooney M, Messaoudi E, Maher FO, Bramham CR, Lynch MA (2004) BDNF-induced LTP in dentate gyrus is impaired with age: analysis of changes in cell signaling events. Neurobiol Aging 25(10):1323–1331. https://doi.org/10.1016/j.neurobiolaging.2004.01.003
Panja D, Bramham CR (2014) BDNF mechanisms in late LTP formation: a synthesis and breakdown. Neuropharmacology 76 Pt C:664–676. doi:https://doi.org/10.1016/j.neuropharm.2013.06.024
Lessmann V, Heumann R (1998) Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience 86(2):399–413. https://doi.org/10.1016/s0306-4522(98)00035-9
Stoop R, Poo MM (1996) Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci 16(10):3256–3264
Vicario-Abejon C, Collin C, McKay RD, Segal M (1998) Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. J Neurosci 18(18):7256–7271
Takei N, Numakawa T, Kozaki S, Sakai N, Endo Y, Takahashi M, Hatanaka H (1998) Brain-derived neurotrophic factor induces rapid and transient release of glutamate through the non-exocytotic pathway from cortical neurons. J Biol Chem 273(42):27620–27624. https://doi.org/10.1074/jbc.273.42.27620
Levine ES, Crozier RA, Black IB, Plummer MR (1998) Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci U S A 95(17):10235–10239. https://doi.org/10.1073/pnas.95.17.10235
Lau CG, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Murphy J, Bennett MV, Zukin RS (2009) Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem Soc Trans 37(Pt 6):1369–1374. https://doi.org/10.1042/BST0371369
Yamada MK, Nakanishi K, Ohba S, Nakamura T, Ikegaya Y, Nishiyama N, Matsuki N (2002) Brain-derived neurotrophic factor promotes the maturation of GABAergic mechanisms in cultured hippocampal neurons. J Neurosci 22(17):7580–7585
Rutherford LC, DeWan A, Lauer HM, Turrigiano GG (1997) Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci 17(12):4527–4535
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E (2005) Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci 25(5):1089–1094. https://doi.org/10.1523/JNEUROSCI.3741-04.2005
Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S (2005) Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 192(2):348–356. https://doi.org/10.1016/j.expneurol.2004.11.016
Yang T, Nie Z, Shu H, Kuang Y, Chen X, Cheng J, Yu S, Liu H (2020) The role of BDNF on neural plasticity in depression. Front Cell Neurosci 14:82. https://doi.org/10.3389/fncel.2020.00082
Kuipers SD, Trentani A, Tiron A, Mao X, Kuhl D, Bramham CR (2016) BDNF-induced LTP is associated with rapid Arc/Arg3.1-dependent enhancement in adult hippocampal neurogenesis. Sci Rep 6:21222. doi:https://doi.org/10.1038/srep21222
Leal G, Bramham CR, Duarte CB (2017) BDNF and hippocampal synaptic plasticity. Vitam Horm 104:153–195. https://doi.org/10.1016/bs.vh.2016.10.004
Bamji SX, Rico B, Kimes N, Reichardt LF (2006) BDNF mobilizes synaptic vesicles and enhances synapse formation by disrupting cadherin-beta-catenin interactions. J Cell Biol 174(2):289–299. https://doi.org/10.1083/jcb.200601087
Valenta T, Hausmann G, Basler K (2012) The many faces and functions of beta-catenin. EMBO J 31(12):2714–2736. https://doi.org/10.1038/emboj.2012.150
Seong E, Yuan L, Arikkath J (2015) Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adh Migr 9(3):202–213. https://doi.org/10.4161/19336918.2014.994919
Arikkath J, Reichardt LF (2008) Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci 31(9):487–494. https://doi.org/10.1016/j.tins.2008.07.001
Zhang L, Yang X, Yang S, Zhang J (2011) The Wnt /beta-catenin signaling pathway in the adult neurogenesis. Eur J Neurosci 33(1):1–8. https://doi.org/10.1111/j.1460-9568.2010.7483.x
Wuhanqimuge AM (2013) Lysophosphatidylcholine potentiates BDNF-induced TrkB phosphorylation and downstream signals in cerebellar granule neurons. Biosci Biotechnol Biochem 77(12):2510–2513. https://doi.org/10.1271/bbb.130622
Shimizu T, Kagawa T, Inoue T, Nonaka A, Takada S, Aburatani H, Taga T (2008) Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol 28(24):7427–7441. https://doi.org/10.1128/MCB.01962-07
Maguschak KA, Ressler KJ (2008) Beta-catenin is required for memory consolidation. Nat Neurosci 11(11):1319–1326. https://doi.org/10.1038/nn.2198
Umschweif G, Alexandrovich AG, Trembovler V, Horowitz M, Shohami E (2013) The role and dynamics of beta-catenin in precondition induced neuroprotection after traumatic brain injury. PLoS ONE 8(10):e76129. https://doi.org/10.1371/journal.pone.0076129
Chen BY, Wang X, Wang ZY, Wang YZ, Chen LW, Luo ZJ (2013) Brain-derived neurotrophic factor stimulates proliferation and differentiation of neural stem cells, possibly by triggering the Wnt/beta-catenin signaling pathway. J Neurosci Res 91(1):30–41. https://doi.org/10.1002/jnr.23138
Liyanli WM, Yang J, Jin R, Ren Z, Gao W, Li X, Wang T, Luo T et al (2016) The relationship between BDNF and the Wnt signaling pathway in the growth of human neural stem. Neurology 86(16 Supplement):5148
Yang JW, Ru J, Ma W, Gao Y, Liang Z, Liu J, Guo JH, Li LY (2015) BDNF promotes the growth of human neurons through crosstalk with the Wnt/beta-catenin signaling pathway via GSK-3beta. Neuropeptides 54:35–46. https://doi.org/10.1016/j.npep.2015.08.005
Zhang F, Liu CL, Tong MM, Zhao Z, Chen SQ (2019) Both Wnt/beta-catenin and ERK5 signaling pathways are involved in BDNF-induced differentiation of pluripotent stem cells into neural stem cells. Neurosci Lett 708:134345. https://doi.org/10.1016/j.neulet.2019.134345
Zhang W, Shi Y, Peng Y, Zhong L, Zhu S, Zhang W, Tang SJ (2018) Neuron activity-induced Wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J Biol Chem 293(40):15641–15651. https://doi.org/10.1074/jbc.RA118.002840
Gomes C, Smith SC, Youssef MN, Zheng JJ, Hagg T, Hetman M (2011) RNA polymerase 1-driven transcription as a mediator of BDNF-induced neurite outgrowth. J Biol Chem 286(6):4357–4363. https://doi.org/10.1074/jbc.M110.170134
Li XT, Liang Z, Wang TT, Yang JW, Ma W, Deng SK, Wang XB, Dai YF et al (2017) Brain-derived neurotrophic factor promotes growth of neurons and neural stem cells possibly by triggering the phosphoinositide 3-kinase/ AKT/glycogen synthase kinase-3beta/beta-catenin pathway. CNS Neurol Disord Drug Targets 16(7):828–836. https://doi.org/10.2174/1871527316666170518170422
Cohen-Cory S, Kidane AH, Shirkey NJ, Marshak S (2010) Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev Neurobiol 70(5):271–288. https://doi.org/10.1002/dneu.20774
Lin G, Bella AJ, Lue TF, Lin CS (2006) Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: part 2. J Sex Med 3(5):821–829. https://doi.org/10.1111/j.1743-6109.2006.00292.x
Lin G, Zhang H, Sun F, Lu Z, Reed-Maldonado A, Lee YC, Wang G, Banie L et al (2016) Brain-derived neurotrophic factor promotes nerve regeneration by activating the JAK/STAT pathway in Schwann cells. Transl Androl Urol 5(2):167–175. https://doi.org/10.21037/tau.2016.02.03
Hixson KM, Cogswell M, Brooks-Kayal AR, Russek SJ (2019) Evidence for a non-canonical JAK/STAT signaling pathway in the synthesis of the brain’s major ion channels and neurotransmitter receptors. BMC Genomics 20(1):677. https://doi.org/10.1186/s12864-019-6033-2
Yoshimatsu T, Kawaguchi D, Oishi K, Takeda K, Akira S, Masuyama N, Gotoh Y (2006) Non-cell-autonomous action of STAT3 in maintenance of neural precursor cells in the mouse neocortex. Development 133(13):2553–2563. https://doi.org/10.1242/dev.02419
Oatley JM, Kaucher AV, Avarbock MR, Brinster RL (2010) Regulation of mouse spermatogonial stem cell differentiation by STAT3 signaling. Biol Reprod 83(3):427–433. https://doi.org/10.1095/biolreprod.109.083352
Yadav A, Kalita A, Dhillon S, Banerjee K (2005) JAK/STAT3 pathway is involved in survival of neurons in response to insulin-like growth factor and negatively regulated by suppressor of cytokine signaling-3. J Biol Chem 280(36):31830–31840. https://doi.org/10.1074/jbc.M501316200
Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M (2012) Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199(3):437–451. https://doi.org/10.1083/jcb.201203109
Quarta S, Baeumer BE, Scherbakov N, Andratsch M, Rose-John S, Dechant G, Bandtlow CE, Kress M (2014) Peripheral nerve regeneration and NGF-dependent neurite outgrowth of adult sensory neurons converge on STAT3 phosphorylation downstream of neuropoietic cytokine receptor gp130. J Neurosci 34(39):13222–13233. https://doi.org/10.1523/JNEUROSCI.1209-13.2014
Bouret SG, Bates SH, Chen S, Myers MG Jr, Simerly RB (2012) Distinct roles for specific leptin receptor signals in the development of hypothalamic feeding circuits. J Neurosci 32(4):1244–1252. https://doi.org/10.1523/JNEUROSCI.2277-11.2012
Tang QP, Shen Q, Wu LX, Feng XL, Liu H, Wu B, Huang XS, Wang GQ et al (2016) STAT3 signal that mediates the neural plasticity is involved in willed-movement training in focal ischemic rats. J Zhejiang Univ Sci B 17(7):493–502. https://doi.org/10.1631/jzus.B1500297
Radin DP, Patel P (2017) BDNF: an oncogene or tumor suppressor? Anticancer Res 37(8):3983–3990. https://doi.org/10.21873/anticanres.11783
Batista CM, Mariano ED, Barbosa BJ, Morgalla M, Marie SK, Teixeira MJ, Lepski G (2014) Adult neurogenesis and glial oncogenesis: when the process fails. Biomed Res Int 2014:438639. https://doi.org/10.1155/2014/438639
Li Z, Zhang Y, Tong Y, Tong J, Thiele CJ (2015) Trk inhibitor attenuates the BDNF/TrkB-induced protection of neuroblastoma cells from etoposide in vitro and in vivo. Cancer Biol Ther 16(3):477–483. https://doi.org/10.1080/15384047.2015.1016659
Lv X, Gu C, Guo S (2020) Activation of BDNF-AS/ADAR/p53 positive feedback loop inhibits glioblastoma cell proliferation. Neurochem Res 45(2):508–518. https://doi.org/10.1007/s11064-019-02943-w
Lai PC, Chiu TH, Huang YT (2010) Overexpression of BDNF and TrkB in human bladder cancer specimens. Oncol Rep 24(5):1265–1270. https://doi.org/10.3892/or_00000981
Vanhecke E, Adriaenssens E, Verbeke S, Meignan S, Germain E, Berteaux N, Nurcombe V, Le Bourhis X et al (2011) Brain-derived neurotrophic factor and neurotrophin-4/5 are expressed in breast cancer and can be targeted to inhibit tumor cell survival. Clin Cancer Res 17(7):1741–1752. https://doi.org/10.1158/1078-0432.CCR-10-1890
Yin B, Ma ZY, Zhou ZW, Gao WC, Du ZG, Zhao ZH, Li QQ (2015) The TrkB+ cancer stem cells contribute to post-chemotherapy recurrence of triple-negative breast cancers in an orthotopic mouse model. Oncogene 34(6):761–770. https://doi.org/10.1038/onc.2014.8
Au CW, Siu MK, Liao X, Wong ES, Ngan HY, Tam KF, Chan DC, Chan QK et al (2009) Tyrosine kinase B receptor and BDNF expression in ovarian cancers - effect on cell migration, angiogenesis and clinical outcome. Cancer Lett 281(2):151–161. https://doi.org/10.1016/j.canlet.2009.02.025
Lam CT, Yang ZF, Lau CK, Tam KH, Fan ST, Poon RT (2011) Brain-derived neurotrophic factor promotes tumorigenesis via induction of neovascularization: implication in hepatocellular carcinoma. Clin Cancer Res 17(10):3123–3133. https://doi.org/10.1158/1078-0432.CCR-10-2802
Guo D, Hou X, Zhang H, Sun W, Zhu L, Liang J, Jiang X (2011) More expressions of BDNF and TrkB in multiple hepatocellular carcinoma and anti-BDNF or K252a induced apoptosis, supressed invasion of HepG2 and HCCLM3 cells. J Exp Clin Cancer Res 30:97. https://doi.org/10.1186/1756-9966-30-97
Okugawa Y, Tanaka K, Inoue Y, Kawamura M, Kawamoto A, Hiro J, Saigusa S, Toiyama Y et al (2013) Brain-derived neurotrophic factor/tropomyosin-related kinase B pathway in gastric cancer. Br J Cancer 108(1):121–130. https://doi.org/10.1038/bjc.2012.499
Fujikawa H, Tanaka K, Toiyama Y, Saigusa S, Inoue Y, Uchida K, Kusunoki M (2012) High TrkB expression levels are associated with poor prognosis and EMT induction in colorectal cancer cells. J Gastroenterol 47(7):775–784. https://doi.org/10.1007/s00535-012-0532-0
Tanaka K, Okugawa Y, Toiyama Y, Inoue Y, Saigusa S, Kawamura M, Araki T, Uchida K et al (2014) Brain-derived neurotrophic factor (BDNF)-induced tropomyosin-related kinase B (Trk B) signaling is a potential therapeutic target for peritoneal carcinomatosis arising from colorectal cancer. PLoS ONE 9(5):e96410. https://doi.org/10.1371/journal.pone.0096410
Li Z, Oh DY, Nakamura K, Thiele CJ (2011) Perifosine-induced inhibition of Akt attenuates brain-derived neurotrophic factor/TrkB-induced chemoresistance in neuroblastoma in vivo. Cancer 117(23):5412–5422. https://doi.org/10.1002/cncr.26133
Lee J, Jiffar T, Kupferman ME (2012) A novel role for BDNF-TrkB in the regulation of chemotherapy resistance in head and neck squamous cell carcinoma. PLoS ONE 7(1):e30246. https://doi.org/10.1371/journal.pone.0030246
Zhang S, Guo D, Luo W, Zhang Q, Zhang Y, Li C, Lu Y, Cui Z et al (2010) TrkB is highly expressed in NSCLC and mediates BDNF-induced the activation of Pyk2 signaling and the invasion of A549 cells. BMC Cancer 10:43. https://doi.org/10.1186/1471-2407-10-43
Yuan Y, Ye HQ, Ren QC (2018) Upregulation of the BDNF/TrKB pathway promotes epithelial-mesenchymal transition, as well as the migration and invasion of cervical cancer. Int J Oncol 52(2):461–472. https://doi.org/10.3892/ijo.2017.4230
Jia S, Wang W, Hu Z, Shan C, Wang L, Wu B, Yang Z, Yang X et al (2015) BDNF mediated TrkB activation contributes to the EMT progression and the poor prognosis in human salivary adenoid cystic carcinoma. Oral Oncol 51(1):64–70. https://doi.org/10.1016/j.oraloncology.2014.10.008
Brunelli A, Dimauro I, Sgro P, Emerenziani GP, Magi F, Baldari C, Guidetti L, Di Luigi L et al (2012) Acute exercise modulates BDNF and pro-BDNF protein content in immune cells. Med Sci Sports Exerc 44(10):1871–1880. https://doi.org/10.1249/MSS.0b013e31825ab69b
Moalem G, Gdalyahu A, Shani Y, Otten U, Lazarovici P, Cohen IR, Schwartz M (2000) Production of neurotrophins by activated T cells: implications for neuroprotective autoimmunity. J Autoimmun 15(3):331–345. https://doi.org/10.1006/jaut.2000.0441
Camara ML, Corrigan F, Jaehne EJ, Jawahar MC, Anscomb H, Baune BT (2015) Tumor necrosis factor alpha and its receptors in behaviour and neurobiology of adult mice, in the absence of an immune challenge. Behav Brain Res 290:51–60. https://doi.org/10.1016/j.bbr.2015.04.040
Xu D, Lian D, Wu J, Liu Y, Zhu M, Sun J, He D, Li L (2017) Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J Neuroinflammation 14(1):156. https://doi.org/10.1186/s12974-017-0930-6
Han R, Liu Z, Sun N, Liu S, Li L, Shen Y, Xiu J, Xu Q (2019) BDNF alleviates neuroinflammation in the hippocampus of type 1 diabetic mice via blocking the aberrant HMGB1/RAGE/NF-kappaB pathway. Aging Dis 10(3):611–625. https://doi.org/10.14336/AD.2018.0707
Stillman J, Martin A, Miguez MJ, McDaniel HR, Konefal J, Woolger JM, Lewis JE (2020) Relationship between brain-derived neurotrophic factor and immune function during dietary supplement treatment of elderly with Alzheimer’s dementia. J Clin Transl Res 5(2):68–75
Weinstock-Guttman B, Zivadinov R, Tamano-Blanco M, Abdelrahman N, Badgett D, Durfee J, Hussein S, Feichter J et al (2007) Immune cell BDNF secretion is associated with white matter volume in multiple sclerosis. J Neuroimmunol 188(1–2):167–174. https://doi.org/10.1016/j.jneuroim.2007.06.003
Linker RA, Lee DH, Demir S, Wiese S, Kruse N, Siglienti I, Gerhardt E, Neumann H et al (2010) Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: therapeutic implications in a model of multiple sclerosis. Brain 133(Pt 8):2248–2263. https://doi.org/10.1093/brain/awq179
Islas-Hernandez A, Aguilar-Talamantes HS, Bertado-Cortes B, Mejia-delCastillo GJ, Carrera-Pineda R, Cuevas-Garcia CF, Garcia-delaTorre P (2018) BDNF and Tau as biomarkers of severity in multiple sclerosis. Biomark Med 12(7):717–726. https://doi.org/10.2217/bmm-2017-0374
Hu ZL, Luo C, Hurtado PR, Li H, Wang S, Hu B, Xu JM, Liu Y et al (2021) Brain-derived neurotrophic factor precursor in the immune system is a novel target for treating multiple sclerosis. Theranostics 11(2):715–730. https://doi.org/10.7150/thno.51390
Yoshimura S, Ochi H, Isobe N, Matsushita T, Motomura K, Matsuoka T, Minohara M, Kira J (2010) Altered production of brain-derived neurotrophic factor by peripheral blood immune cells in multiple sclerosis. Mult Scler 16(10):1178–1188. https://doi.org/10.1177/1352458510375706
Li L, Shui QX, Zhao ZY (2003) Regulation of brain-derived neurotrophic factor (BDNF) expression following antibiotic treatment of experimental bacterial meningitis. J Child Neurol 18(12):828–834. https://doi.org/10.1177/088307380301801202
Alvin Z, Laurence GG, Coleman BR, Zhao A, Hajj-Moussa M, Haddad GE (2011) Regulation of L-type inward calcium channel activity by captopril and angiotensin II via the phosphatidyl inositol 3-kinase pathway in cardiomyocytes from volume-overload hypertrophied rat hearts. Can J Physiol Pharmacol 89(3):206–215. https://doi.org/10.1139/Y11-011
Xu D, Lian D, Zhang Z, Liu Y, Sun J, Li L (2017) Brain-derived neurotrophic factor is regulated via MyD88/NF-kappaB signaling in experimental Streptococcus pneumoniae meningitis. Sci Rep 7(1):3545. https://doi.org/10.1038/s41598-017-03861-z
Mitchell JA, Paul-Clark MJ, Clarke GW, McMaster SK, Cartwright N (2007) Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J Endocrinol 193(3):323–330. https://doi.org/10.1677/JOE-07-0067
Lian D, He D, Wu J, Liu Y, Zhu M, Sun J, Chen F, Li L (2016) Exogenous BDNF increases neurogenesis in the hippocampus in experimental Streptococcus pneumoniae meningitis. J Neuroimmunol 294:46–55. https://doi.org/10.1016/j.jneuroim.2016.03.014
Bifrare YD, Kummer J, Joss P, Tauber MG, Leib SL (2005) Brain-derived neurotrophic factor protects against multiple forms of brain injury in bacterial meningitis. J Infect Dis 191(1):40–45. https://doi.org/10.1086/426399
Han BH, D’Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A et al (2000) BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis 7(1):38–53. https://doi.org/10.1006/nbdi.1999.0275
Han BH, Holtzman DM (2000) BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 20(15):5775–5781
Hisatomi T, Sakamoto T, Murata T, Yamanaka I, Oshima Y, Hata Y, Ishibashi T, Inomata H et al (2001) Relocalization of apoptosis-inducing factor in photoreceptor apoptosis induced by retinal detachment in vivo. Am J Pathol 158(4):1271–1278. https://doi.org/10.1016/S0002-9440(10)64078-3
Galvin KA, Oorschot DE (2003) Continuous low-dose treatment with brain-derived neurotrophic factor or neurotrophin-3 protects striatal medium spiny neurons from mild neonatal hypoxia/ischemia: a stereological study. Neuroscience 118(4):1023–1032. https://doi.org/10.1016/s0306-4522(03)00066-6
Ventimiglia R, Mather PE, Jones BE, Lindsay RM (1995) The neurotrophins BDNF, NT-3 and NT-4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci 7(2):213–222. https://doi.org/10.1111/j.1460-9568.1995.tb01057.x
Qiao LY, Shen S, Liu M, Xia C, Kay JC, Zhang QL (2016) Inflammation and activity augment brain-derived neurotrophic factor peripheral release. Neuroscience 318:114–121. https://doi.org/10.1016/j.neuroscience.2016.01.018
Lai NS, Yu HC, Huang Tseng HY, Hsu CW, Huang HB, Lu MC (2021) Increased serum levels of brain-derived neurotrophic factor contribute to inflammatory responses in patients with rheumatoid arthritis. Int J Mol Sci 22 (4). doi:https://doi.org/10.3390/ijms22041841
Reddy MS (2010) Depression: the disorder and the burden. Indian J Psychol Med 32(1):1–2. https://doi.org/10.4103/0253-7176.70510
Ahmed S, Malemud CJ, Koch AE, Athar M, Taub DD (2014) Cytokines and chemokines: disease models, mechanisms, and therapies. Mediators Inflamm 2014:296356. https://doi.org/10.1155/2014/296356
Galic MA, Riazi K, Pittman QJ (2012) Cytokines and brain excitability. Front Neuroendocrinol 33(1):116–125. https://doi.org/10.1016/j.yfrne.2011.12.002
Riazi K, Galic MA, Pittman QJ (2010) Contributions of peripheral inflammation to seizure susceptibility: cytokines and brain excitability. Epilepsy Res 89(1):34–42. https://doi.org/10.1016/j.eplepsyres.2009.09.004
Duman RS, Malberg J, Nakagawa S, D’Sa C (2000) Neuronal plasticity and survival in mood disorders. Biol Psychiatry 48(8):732–739. https://doi.org/10.1016/s0006-3223(00)00935-5
D’Sa C, Duman RS (2002) Antidepressants and neuroplasticity. Bipolar Disord 4(3):183–194. https://doi.org/10.1034/j.1399-5618.2002.01203.x
Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R (2014) Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci 8:430. https://doi.org/10.3389/fncel.2014.00430
Molendijk ML, Bus BA, Spinhoven P, Penninx BW, Kenis G, Prickaerts J, Voshaar RC, Elzinga BM (2011) Serum levels of brain-derived neurotrophic factor in major depressive disorder: state-trait issues, clinical features and pharmacological treatment. Mol Psychiatry 16(11):1088–1095. https://doi.org/10.1038/mp.2010.98
Hsieh MT, Lin CC, Lee CT, Huang TL (2019) Abnormal brain-derived neurotrophic factor exon IX promoter methylation, protein, and mRNA levels in patients with major depressive disorder. J Clin Med 8 (5). doi:https://doi.org/10.3390/jcm8050568
Schroter K, Brum M, Brunkhorst-Kanaan N, Tole F, Ziegler C, Domschke K, Reif A, Kittel-Schneider S (2020) Longitudinal multi-level biomarker analysis of BDNF in major depression and bipolar disorder. Eur Arch Psychiatry Clin Neurosci 270(2):169–181. https://doi.org/10.1007/s00406-019-01007-y
Chiou YJ, Huang TL (2017) Serum brain-derived neurotrophic factors in Taiwanese patients with drug-naive first-episode major depressive disorder: effects of antidepressants. Int J Neuropsychopharmacol 20(3):213–218. https://doi.org/10.1093/ijnp/pyw096
Chiou YJ, Huang TL (2019) Accuracy of brain-derived neurotrophic factor levels for differentiating between Taiwanese patients with major depressive disorder or schizophrenia and healthy controls. PLoS ONE 14(2):e0212373. https://doi.org/10.1371/journal.pone.0212373
Hsieh M, Lin CC, Huang TL (2019) Increased brain-derived neurotrophic factor exon IV histone 3 lysine 9 dimethylation in patients with schizophrenia. Taiwan Journal of Psychiatry 33(2):99–104. https://doi.org/10.4103/TPSY.TPSY_18_19
Caldieraro MA, McKee M, Leistner-Segal S, Vares EA, Kubaski F, Spanemberg L, Brusius-Facchin AC, Fleck MP et al (2018) Val66Met polymorphism association with serum BDNF and inflammatory biomarkers in major depression. World J Biol Psychiatry 19(5):402–409. https://doi.org/10.1080/15622975.2017.1347713
Hosang GM, Shiles C, Tansey KE, McGuffin P, Uher R (2014) Interaction between stress and the BDNF Val66Met polymorphism in depression: a systematic review and meta-analysis. BMC Med 12:7. https://doi.org/10.1186/1741-7015-12-7
Chen ZY, Bath K, McEwen B, Hempstead B, Lee F (2008) Impact of genetic variant BDNF (Val66Met) on brain structure and function. Novartis Found Symp 289:180–188. https://doi.org/10.1002/9780470751251.ch14 (discussion 188-195)
Groves JO (2007) Is it time to reassess the BDNF hypothesis of depression? Mol Psychiatry 12(12):1079–1088. https://doi.org/10.1038/sj.mp.4002075
Chai HH, Fu XC, Ma L, Sun HT, Chen GZ, Song MY, Chen WX, Chen YS et al (2019) The chemokine CXCL1 and its receptor CXCR2 contribute to chronic stress-induced depression in mice. FASEB J 33(8):8853–8864. https://doi.org/10.1096/fj.201802359RR
Walker AK, Budac DP, Bisulco S, Lee AW, Smith RA, Beenders B, Kelley KW, Dantzer R (2013) NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38(9):1609–1616. https://doi.org/10.1038/npp.2013.71
Francija E, Petrovic Z, Brkic Z, Mitic M, Radulovic J, Adzic M (2019) Disruption of the NMDA receptor GluN2A subunit abolishes inflammation-induced depression. Behav Brain Res 359:550–559. https://doi.org/10.1016/j.bbr.2018.10.011
Crupi R, Cuzzocrea S (2016) Neuroinflammation and immunity: a new pharmacological target in depression. CNS Neurol Disord Drug Targets 15(4):464–476. https://doi.org/10.2174/1871527315666160321105339
Jeon SW, Kim YK (2016) Neuroinflammation and cytokine abnormality in major depression: cause or consequence in that illness? World J Psychiatry 6(3):283–293. https://doi.org/10.5498/wjp.v6.i3.283
Sen S, Duman R, Sanacora G (2008) Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry 64(6):527–532. https://doi.org/10.1016/j.biopsych.2008.05.005
Autry AE, Monteggia LM (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64(2):238–258. https://doi.org/10.1124/pr.111.005108
Duman RS (2014) Pathophysiology of depression and innovative treatments: remodeling glutamatergic synaptic connections. Dialogues Clin Neurosci 16(1):11–27
Gold PW (2021) The PPARg system in major depression: pathophysiologic and therapeutic implications. Int J Mol Sci 22 (17). doi:https://doi.org/10.3390/ijms22179248
Lee BH, Kim YK (2010) The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment. Psychiatry Investig 7(4):231–235. https://doi.org/10.4306/pi.2010.7.4.231
Kariharan T, Nanayakkara G, Parameshwaran K, Bagasrawala I, Ahuja M, Abdel-Rahman E, Amin AT, Dhanasekaran M et al (2015) Central activation of PPAR-gamma ameliorates diabetes induced cognitive dysfunction and improves BDNF expression. Neurobiol Aging 36(3):1451–1461. https://doi.org/10.1016/j.neurobiolaging.2014.09.028
Zhou C, Zhang H, Qin Y, Tian T, Xu B, Chen J, Zhou X, Zeng L et al (2018) A systematic review and meta-analysis of deep brain stimulation in treatment-resistant depression. Prog Neuropsychopharmacol Biol Psychiatry 82:224–232. https://doi.org/10.1016/j.pnpbp.2017.11.012
Dandekar MP, Fenoy AJ, Carvalho AF, Soares JC, Quevedo J (2018) Deep brain stimulation for treatment-resistant depression: an integrative review of preclinical and clinical findings and translational implications. Mol Psychiatry 23(5):1094–1112. https://doi.org/10.1038/mp.2018.2
Martis LS, Wiborg O, Holmes MC, Harris AP (2019) BDNF(+/-) rats exhibit depressive phenotype and altered expression of genes relevant in mood disorders. Genes Brain Behav 18(2):e12546. https://doi.org/10.1111/gbb.12546
Tadic A, Muller-Engling L, Schlicht KF, Kotsiari A, Dreimuller N, Kleimann A, Bleich S, Lieb K et al (2014) Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol Psychiatry 19(3):281–283. https://doi.org/10.1038/mp.2013.58
Lieb K, Dreimuller N, Wagner S, Schlicht K, Falter T, Neyazi A, Muller-Engling L, Bleich S et al (2018) BDNF plasma levels and BDNF Exon IV promoter methylation as predictors for antidepressant treatment response. Front Psychiatry 9:511. https://doi.org/10.3389/fpsyt.2018.00511
Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, Sacchetti S, Lembo F et al (2010) Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry 67(3):258–267. https://doi.org/10.1001/archgenpsychiatry.2010.9
Sakata K, Woo NH, Martinowich K, Greene JS, Schloesser RJ, Shen L, Lu B (2009) Critical role of promoter IV-driven BDNF transcription in GABAergic transmission and synaptic plasticity in the prefrontal cortex. Proc Natl Acad Sci U S A 106(14):5942–5947. https://doi.org/10.1073/pnas.0811431106
Castren E (2014) Neurotrophins and psychiatric disorders. Handb Exp Pharmacol 220:461–479. https://doi.org/10.1007/978-3-642-45106-5_17
Castren E, Kojima M (2017) Brain-derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol Dis 97(Pt B):119–126. https://doi.org/10.1016/j.nbd.2016.07.010
Green MJ, Matheson SL, Shepherd A, Weickert CS, Carr VJ (2011) Brain-derived neurotrophic factor levels in schizophrenia: a systematic review with meta-analysis. Mol Psychiatry 16(9):960–972. https://doi.org/10.1038/mp.2010.88
Noto MN, Maes M, Vargas Nunes SO, Ota VK, Cavalcante D, Oliveira G, Rossaneis AC, Verri WA Jr et al (2021) BDNF in antipsychotic naive first episode psychosis: effects of risperidone and the immune-inflammatory response system. J Psychiatr Res 141:206–213. https://doi.org/10.1016/j.jpsychires.2021.07.011
Lin Z, Su Y, Zhang C, Xing M, Ding W, Liao L, Guan Y, Li Z et al (2013) The interaction of BDNF and NTRK2 gene increases the susceptibility of paranoid schizophrenia. PLoS ONE 8(9):e74264. https://doi.org/10.1371/journal.pone.0074264
Dong Z, Sun X, Pan C, Lu T, Han Y, Wang L, Yan H, Dong L et al (2016) Association of DISC1, BDNF, and COMT polymorphisms with exploratory eye movement of schizophrenia in a Chinese Han population. Psychiatr Genet 26(6):258–265. https://doi.org/10.1097/YPG.0000000000000138
Farkas K, Réthelyi J, Polgár P, Benkovits J, Fábián Á, Czobor P, Bitter I (2011) BDNF and DISC1 are associated with cognitive dysfunction but not with schizophrenia in a hungarian sample. Eur Psychiatry 26(S2):805–805. https://doi.org/10.1016/S0924-9338(11)72510-X
Yi H, Hu J, Qian J, Hackam AS (2012) Expression of brain-derived neurotrophic factor is regulated by the Wnt signaling pathway. NeuroReport 23(3):189–194. https://doi.org/10.1097/WNR.0b013e32834fab06
Mallei A, Ieraci A, Corna S, Tardito D, Lee FS, Popoli M (2018) Global epigenetic analysis of BDNF Val66Met mice hippocampus reveals changes in dendrite and spine remodeling genes. Hippocampus 28(11):783–795. https://doi.org/10.1002/hipo.22991
Stoyanov D, Maes MH (2021) How to construct neuroscience-informed psychiatric classification? Towards nomothetic networks psychiatry. World J Psychiatry 11(1):1–12. https://doi.org/10.5498/wjp.v11.i1.1
Maes M, Vojdani A, Galecki P, Kanchanatawan B (2020) How to construct a bottom-up nomothetic network model and disclose novel nosological classes by integrating risk resilience and adverse outcome pathways with the phenome of schizophrenia. Brain Sci 10(9):645. https://doi.org/10.3390/brainsci10090645
Maes M, Rachayon M, Jirakran K, Sodsai P, Klinchanhom S, Gałecki P, Sughondhabirom A, Basta-Kaim A (2022) The immune profile of major dysmood disorder: proof of concept and mechanism using the precision nomothetic psychiatry approach. Cells 11(7):1183. https://doi.org/10.3390/cells11071183
Funding
The work was supported by the National University Complex for Biomedical and Applied Research, with participation in BBMRI-ERIC (NUCBPI-BBMRI.BG), within the national road map for research infrastructure (Contract No. DO1-395/18.12.2020).
Author information
Authors and Affiliations
Contributions
All contributing authors have participated in the preparation of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Research Involving Human Participants and/or Animals
Not applicable.
Informed Consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nikolay Mehterov and Danail Minchev shared first authorship.
Rights and permissions
About this article

Cite this article
Mehterov, N., Minchev, D., Gevezova, M. et al. Interactions Among Brain-Derived Neurotrophic Factor and Neuroimmune Pathways Are Key Components of the Major Psychiatric Disorders. Mol Neurobiol 59, 4926–4952 (2022). https://doi.org/10.1007/s12035-022-02889-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-02889-1