
Abstract
The main histopathology of Alzheimer’s disease (AD) is featured by the extracellular accumulation of amyloid-β (Aβ) plaques and intracellular tau neurofibrillary tangles (NFT) in the brain, which is likely to result from co-pathogenic interactions among multiple factors, e.g., aging or genes. The link between defective autophagy/mitophagy and AD pathologies is still under investigation and not fully established. In this review, we consider how AD is associated with impaired autophagy and mitophagy, and how these impact pathological hallmarks as well as the potential mechanisms. This complicated interplay between autophagy or mitophagy and histopathology in AD suggests that targeting autophagy or mitophagy probably is a promising anti-AD drug candidate. Finally, we review the implications of some new insights for induction of autophagy or mitophagy as the new therapeutic way that targets processes upstream of both NFT and Aβ plaques, and hence stops the neurodegenerative course in AD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
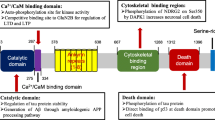
Alzheimer disease (AD) is a slow progressive and irreversible neurodegenerative disease, affecting about 45 million people worldwide [1]. Clinically, AD is characterized by an insidious onset and a gradual deterioration of cognitive functions involving memory loss to impairment of higher executive functions [2,3,4]. The subjects of AD have a profound influence on the quality of life, of both the affected patients and their careers or relatives [5]. This represents a major global public health issue and socioeconomic burden with cost of dementia care exceeding $800 billion [1, 6]. The main histopathological hallmarks of AD are extracellular amyloid-β (Aβ) plaques and intracellular tau neurofibrillary tangles (NFT) (Fig. 1). These hallmarks are accompanied by neuroinflammation, synaptic dysfunction, and mitochondrial dysfunction leading to neuronal loss, thereby resulting in brain atrophy [7,8,9]. Despite decades of extensive research, the precise cause of AD is still not fully understood, and many attempts in treating AD by drugs targeting the proteins Aβ plaque or p-tau have been unsuccessful [10,11,12,13,14]. Exploring new targets and mechanism is urgent in front of these conditions.

Summaries of the etiologies of AD: The histopathological hallmarks of AD are extracellular senile plaques, composed of accumulation of amyloid-β (Aβ), and intracellular NFT, containing aberrantly hyperphosphorylated microtubule associated protein tau (MAPT/p-tau). These hallmarks are accompanied by neuroinflammation, synaptic dysfunction, and mitochondrial dysfunction leading to neuronal loss, thereby resulting in brain atrophy. Mutations in APP, PSEN1, or PSEN2, which respectively encode APP and the APP-processing enzymes presenilin 1 and 2, alter amyloidogenic processing of APP and cause familial AD. For sporadic AD, polymorphisms in genes that regulate microglial immune activation (APOE, TREM2, CR1, MS4A6A, MS4A6E, CD33), lipid metabolism (APOE, TREM2, CLU, ABCA7, WWOX), and endocytosis (APOE, PICALM, BIN1, SORL1, CD2AP) are risk factors for sporadic AD. These factors contribute to Aβ and tau pathology-mediated neurodegeneration. AD, Alzheimer’s disease; Aβ, amyloid-β; NFT, neurofibrillary tangle; APP, Aβ precursor protein; PSEN, presenilin; APOE, apolipoprotein E; TREM2, Triggering Receptor Expressed On Myeloid Cells 2; CR1, the complement component receptor 1 gene; MS4A6A, Membrane Spanning 4-Domains A6A; CLU, the clusterin; ABCA7, ATP Binding Cassette Subfamily A Member 7; WWOX, WW Domain Containing Oxidoreductase; PICALM, Phosphatidylinositol Binding Clathrin Assembly Protein; BIN1, Bridging integrator 1; SORL1, the sortilin-related receptor 1; CD2AP, CD2-associated protein
Mitochondria are double-membrane bound organelles that perform essential roles in several cellular processes, ranging from energy production through oxidative phosphorylation (OXPHOS) to initiation of cell death [15, 16]. Despite the brain accounting for about 2% of body weight, it utilizes up to 20% of oxygen at rest [17]. The energy requirement is largely dependent on the cellular demand to perform and/or maintain function. Thus, mitochondria are considered as the “powerhouses” of cells that play a key role in developmental and neuronal survival [16, 18]. Mitochondrial maintenance and the timely clearance of damaged mitochondria are a necessity to maintain neuronal function, synaptic plasticity, and neuronal survival. Thus, there are a number of quality control systems in place for the sake of maintaining mitochondrial health and efficient functioning, under physiological conditions. In particular mitochondrial autophagy, termed mitophagy, predominantly regulates mitochondrial maintenance. Autophagy is the process by which intracellular redundant components are degraded and recycled. Age-related diseases, including neurodegeneration, have been associated with reduced autophagy [19]. Mitophagy, the mitochondria-specific autophagy, can either specifically clear impaired mitochondria or eliminate all mitochondria during specialized developmental stages or starvation [19]. Accumulating evidences presented have indicated that compromised mitophagy contributes to aging and neurodegeneration observed in models of premature aging disease and AD [18, 20,21,22,23]. This review summarizes the recent developments on the role of autophagy, which also includes mitophagy, in the etiology of AD and the key mechanisms underlying its cross-talk interplay with the AD-associated neuropathological hallmarks. We review the reasons for the failures of current therapeutic approaches in AD and suggest possible new approaches.
Autophagy: General
Autophagy is an elaborately controlled stepwise quality control mechanism, which is active at basal level under physiological conditions; however, it can be activated in response to cellular stresses, such as DNA damage, nutrient deprivation, toxic stimulation, oxidative stress, and protein aggregation [24]. Autophagy degrades cellular products that can result in cytotoxicity upon accumulation, such as oxidative stress protein aggregates or impaired mitochondria, which is necessary for cellular homeostasis, organelle quality control, and organismal accommodation to environmental changes [25]. The resulting degradation products of autophagy can be used for various purposes including, but not limited to, protein synthesis and energy production [26]. In addition, autophagy is tightly fit with cell environments to sustain healthy aging [27].
Mechanisms of Autophagy
During the initiation of autophagy, the initial vesicles are derived from a variety of membrane sources, including the endoplasmic reticulum, Golgi apparatus, mitochondria, and certain endosomal intermediates et al. [28]. Nutrient deprivation, growth factor depletion, or low cellular energy levels are documented inducers of primordial autophagy [24], which mediates mechanistic target of rapamycin complex 1 (mTORC1) inhibition and AMP-activated protein kinase (AMPK) activation, and then upregulates the Unc-51 like kinase-1 (ULK1) serine threonine kinase complex, including autophagy-related 101 (ATG101), ATG13, and focal adhesion kinase family-interacting protein of 200 kDa (FIP200) through a range of phosphorylation events [29]. Phagophore nucleation is a complicated process, and its underlying mechanisms are still not fully characterized. According to contemporary knowledge, induction of the ULK1 complex following activates the class III PI3K VPS34 at the phagophore initiation sites, where VPS34 generates PI3P, whereas in a complex with VPS15, ATG14L, and Beclin1 to act in phagophore nucleation, which leads to the development of cup-shaped pre-autophagosome structures following the activation of ATG9 [30]. PI3P also recruits the WIPI and DFCP1 by binding domains and subsequently defines the microtubule-associated protein LC3 lipidation sites for autophagosome precursors by facilitating the supplementation of the ATG5-ATG12-ATG16L1 complex and enabling the membrane prolongation [31]. The ATG5-ATG12-ATG16L1 complex is recruited to the phagophore assembly site via the interaction of ATG16L1 with WIPI2. This complex is necessary for the linkage of LC3-I to phosphatidylethanolamine (PE) in membranes by a mechanism that is dependent on ATG3 and ATG7 for the formation of LC3-II, which sustains autophagosome membrane expansion and completion [32]. The isolation membrane gradually elongates and finally seals into a double-membraned vesicle structure, called the autophagosome. During the entire autophagy process, ATG proteins can interact with other ATG proteins that reside outside of their “classic” complex. For example, FIP200 combines with ATG16L1 to correctly guide it to the isolation phagophore of the nascent autophagosome [33]. The ATG13 HORMA domain interacts with ATG9, a multi-spanning membrane protein, to hierarchically recruit ATG9 vesicles to the pre-autophagosomal structure (PAS) [33]. Finally, following elongation and closing of the phagophore, the autophagosome achieves maturation, which recruits the STX17 and SNAP29 on the autophagosome membrane and VAMP8 on the lysosome membrane together with the HOPS complex to complete fusion [34]. Moreover, adequate lysosome quantity is also needed for fusion with autophagosomes due to this reaction expends the lysosomes. TFEB has been established as a pivotal regulator of lysosomal biogenesis and plays a role in the control of the autolysosome degradation stage [35]. In brief, the autophagosome outer membrane must fuse with the lysosome to form an autolysosome and engulf a variety of substrates that are ultimately degraded; this leads to nutrient recycling via the release of amino acids, glucose, and lipids [36]. An overview of the autophagy molecular pathway and some targeting involved in the autophagy machinery is shown in Fig. 2.

Overview of the autophagy molecular pathway and the genes implicated in AD pathogenesis: Nutrient deprivation, oxidative stress, and/or low cellular energy levels are well-established primordial autophagy inducers, which is mediated mTORC1 inhibition and AMPK activation, then positively adjust the ULK1 complex bearing with ATG13, ATG101, ULK1, and FIP200 through a ranges of phosphorylation events. Induction of the ULK1 complex following activates the class III PI3K VPS34 to the phagophore initiation sites where VPS34 generates PI3P while in a complex with p115, VPS15, AMBRA1, ATG14L, and BECN1 to act in phagophore nucleation, which leads to developing a cup-shaped pre-autophagosome structures with the activation of ATG9. PI3P also recruits the WIPI and DFCP1 via bind domains and subsequently defines the LC3 lipidation sites for autophagosome precursors by helping in the supplement of the ATG5-ATG12-ATG16L1 complex, and enabling the membrane prolongation. This complex is essential for the conjugation of LC3-I to PE in membranes by a mechanism dependent on ATG3, ATG7 to the formation of LC3-II, which sustains autophagosome membrane expansion and completion. The isolation membrane gradually elongates and finally seals into a double-membraned vesicle, termed the autophagosome. Finally, following elongation and closing of the phagophore, the autophagosome experiences maturation, which implicates the STX17 and SNAP29 on the autophagosome membrane and VAMP8 on the lysosome membrane, with the help of HOPS complex to finish fusion together. TFEB has been established as a pivotal regulator for lysosomal biogenesis which plays an important role in the control of autolysosome degradation stage. Mutations in core autophagy genes that are implicated in AD occur. PICALM plays a role in autophagosome formation and maturation, disruption cargo recognition, and inhibition of autolysosome formation. In addition, the mutation of PS-1, which is involved in lysosomal v-ATPase, alters the way the APP protein is processed and contributes to AD. Moreover, mutations of the BECN1, Dynein, ATG7, TMED10, and TFEB all have been suggested to be involved in AD pathogenesis. mTORC1, mechanistic target of rapamycin complex 1; AMPK, AMP-activated protein kinase; ULK1, Unc-51 like kinase-1; ATG, autophagy-related; FIP200, family-interacting protein of 200 kDa; VPS34, vacuolar protein sorting 34; PI3P, phosphatidylinositol 3-phosphate; AMBRA1, autophagy and beclin 1 regulator 1; BECN1, Beclin1; WIPI, WD repeat domain phosphoinositide-interacting proteins; DFCP1, zinc-finger FYVE domain-containing protein 1; LC3, microtubule-associated protein light-chain 3; PE, phosphatidylethanolamine; STX17, syntaxin 17; SNAP29, synaptosomal-associated protein 29; VAMP8, vesicle-associated membrane protein 8; HOPS, homotypic fusion and protein sorting; TFEB, transcription factor EB
Autophagy: a Key Contributor to AD Pathogenesis
Autophagy is a conserved valid neuroprotective process that actively results in the elimination of pathogenic misfolded aggregates. Various lines of evidence suggest that aging regulates the autophagy process [37]. Indeed, autophagy plays a key role during development and disease, and evidences assembled over the past decades indicate that autophagy has a direct role in modulating aging, especially in AD [38]. In cross-species AD animal models, damaged autophagy is closely connected with the collection of Aβ plaques and NFT. Meanwhile, AD main pathologies can also serve as upstream events to impaired autophagy. Actually, there is a little complicated relationship between Aβ and autophagy. On one hand, Aβ can be degraded by autophagy in multiple systems and opposite also be generated in autophagosomes, which appear to contain both APP and PS-1, an enzyme involved in the cleavage of APP to Aβ [39, 40]. Hyperphosphorylated tau co-localizes with LC3-positive vesicles and the autophagy cargo-receptor p62 to disrupt the autophagy flux [41]. Moreover, tau has also been shown to bind lysosomal membranes and perturb lysosomal permeability in AD models to impair autophagy process [42]. Decline in autophagic functions in AD models can be primary or secondary to other potential causes. For instance, PS mutation proteins in AD are primary defects, which can cause autophagy impairment that due to their physiological competence is lost [43]. In addition, autophagosomes produced in the distal end of the axon utilize the retrograde transport way for the soma to arrive, where they fuse with lysosomes [44]. Several studies have shown that abnormal binding of Aβ oligomers to the cytoskeletal motor protein dynein would interfere with autophagosome trafficking and fusion, which decreases autophagosome motility in neurons and contributes to their accumulation in the distal part of the axons in patients with AD [45]. In terms of autophagy induction, rapamycin, the best known mTOR inhibitor, has been displayed to avert protein accumulation and neurodegeneration in experimental models of AD [46]. Moreover, temsirolimus could enhance Aβ clearance by an autophagy-dependent manner and improved spatial learning and memory abilities in APP/PS1 transgenic mice [47].
Defective Autophagy in AD
The accumulation of lysosomes is a well-established neuropathologic feature of AD. In the human AD cortex, striking accumulations of immature autophagic vacuole (AV) forms in dystrophic neurites when using immuno-electron microscopy suggest that the transport of AVs and their maturation to lysosomes may be impaired, thereby impeding the function of autophagy [48]. Of note, based on genetic studies over recent decades, several gene mutations in AD have been reported that are also involved in different steps of autophagy and impair autophagy efficiency. One study has demonstrated that phosphatidylinositol-binding clathrin assembly protein (PICALM) plays a role in AD etiology, which is a clathrin adaptor protein that plays a role in AD etiology, and ablation of this protein has been shown to prohibit autophagy at various steps, including autophagosome formation and maturation, disruption of cargo recognition and inhibition of autolysosome formation [49]. Similarly, PICALM is aberrantly cleaved in the brain samples of AD patients, and the quantity of the un-split 65–75-kDa full-length PICALM species is clearly decreased, leading to tau pathology and endocytic dysfunction in AD [50]. PICALM is also indispensable for the endocytosis of VAMP2, VAMP3, and VAMP8, which are important for autophagosome–lysosome fusion. Autophagosome precursor vesicles incapable of fusing are observed without PICALM, contributing to damaged autophagosome biogenesis and a decrease in the formation of autolysosome and autophagic flux, and this may influence AD in a variety of ways comprising regulation of tau turnover [51]. In addition, there is a mass of evidence showing that the mutation of PS-1, as the catalytic subunit of the γ-secretase enzyme complex, alters the way the APP protein is processed and contributes to AD. PS-1 has been exhibited to function as an endoplasmic reticulum (ER) chaperone for the V0a1 subunit of the lysosomal v-ATPase and is essential for lysosomal turnover of autophagic and endocytic protein substrates [52]. Mutations in PS1 cause early-onset AD due to decreased lysosome acidification and maturation of the lysosomal v-ATPase, thus increasing lysosomal pH, which would be forecasted to decrease autophagosome clearance [53]. Although the established mechanism that generates this reduction in turnover is dimness, there seems to be a consensus concerning the idea that PS1 has a role in AD due to the defective autophagy [54]. Meanwhile, in AD models, the increase in the level of acid sphingomyelinase (ASM) in fibroblasts and the brain can cause a decrease in TFEB levels, leading to defective autophagic degradation due to lysosomal depletion. In contrast, genetic inhibition of ASM in heterozygous knockout mice is able to ameliorate AD clinical performance and pathological findings (e.g., Aβ deposition) because it reduces the accumulation of LC3-II and p62 by restoring lysosomal biogenesis [55]. The level of BECN1 mRNA is reduced in AD brain tissue following caspase-cleavage, resulting in impaired autophagosome formation. Inhibition of autophagy through conditional knockout of the ATG7 in the forebrains of mice results in phospho-tau accumulation and aberrant intraneuronal Aβ accumulation [56]. In parallel, meta-analyses of existing genome-wide association studies have shown that mutations in autophagic receptor p62 were associated with AD [57]. In summary, the function of autophagy in AD is complicated and controversial due to the different stages of autophagy implicated in the disease.
Defective Mitophagy in AD
It is now explicit that mitochondria perform various important roles in the cellular homeostasis and are implicated in nearly all fields of cell function by forming a highly interconnected network throughout the cells, including oxidative phosphorylation, calcium buffering in synapses, metabolism, and the biosynthesis of intermediates for cell growth or death et al. [16]. Consequently, mitochondrial dysfunction in bioenergetic and metabolism can contribute to the etiology of neurodegenerative diseases by affecting several pathways, suggesting that strategies aimed at restoring mitochondrial function are a significant drug target for these highly widespread diseases [58]. Mitochondria have long been studied as a key organelle in the pathogenesis of AD, as the subjects show early mitochondrial dysfunction prior to the appearance of any histopathological or clinical symptoms [59]. Recently, there have been hundreds of studies recording mitochondrial abnormalities and the accumulation of damaged mitochondria in AD models in vitro and in vivo and illuminating the potential molecular circadian mechanisms and cellular consequences of mitochondrial insufficiency [22]. Cytochrome c, cytochrome oxidase 1, and Bax, as markers of mitochondrial dysfunction, were observed in dystrophic neurites in transgenic AD mice and progressively increased with age [60]. To uncover the condition of mitophagy impairment and the accumulation of damaged mitochondria in AD brain, Fang found excessive mitochondrial damage and small-sized mitochondrial morphology by electron microscopy in postmortem hippocampal tissues from AD patients compared to healthy controls, indicating the mitophagy dysfunction in AD subjects [23]. A pathway that links nuclear DNA damage to mitochondrial homeostasis is the PAPR1-NAD+-SIRT1-PGC1α axis, in which PAPR1 is activated in AD and subsequently inhibits various DNA repair pathways and NAD+ depletion, leading to impaired nuclear to mitochondrial signaling [61, 62]. As a result, dysfunctional mitochondria gather in neurons, leading to reduced cellular ATP levels and superfluous production of reactive oxygen species (ROS) production, which can create a “vicious circle” that worsens mitochondrial damage, resulting in the formation Aβ deposition and tau phosphorylation and finally contributing to AD.
Mechanisms of the Mitophagy Pathway
Insufficient degradation of damaged or superfluous mitochondria is likely to be responsible for the aggregation of defective mitochondria in AD [22]. Recently, mitophagy has been found to be one of the most common forms of selective autophagy in scientific fields that degrades mitochondria within lysosomes, and which is often mediated by autophagy receptors such as PTEN-induced putative kinase 1 (PINK1), Parkin and BNIP3L, and mitophagy impairment resulting in the disability of mitochondrial cargo recognition machinery, thus inhibiting the clearance of damaged mitochondria and leading to cell and tissue injuries [59, 63]. In addition, compromised mitophagy in AD may result from dysfunctional fusion between autophagosomes and lysosomes, which is the final step of mitophagy. Molecular pathways of mitophagy are intricate, including PINK1–Parkin-dependent or independent pathways [64]. Following oxidative stress and membrane potential dissipation, leading to stabilization of the PINK1 at the outer mitochondrial membrane (OMM). Afterwards, PINK1 is activated first by auto-phosphorylation and then phosphorylates mitofusin 2 (MFN2) and ubiquitin, leading to the recruitment of Parkin to the OMM surface. Phosphorylated poly-ubiquitin chains serve as a “swallow me” signal for the autophagic machinery. Meanwhile, parkin ubiquitylates several outer membrane components that are following recognized by the adaptor proteins ubiquitin-binding proteins optineurin (OPTN), p62, nuclear dot protein 52 (NDP52), and neighbor of BRCA1 gene 1 (NBR1) et al., which recruit the impaired mitochondria to the autophagy pathway and initiate autophagosome formation through binding with LC3. In addition to Parkin, BNIP3, NIX/BNIP3L, and FUNDC1 are referred to as mitophagy receptors, localize to the OMM, and connect directly with LC3 to induce mitochondrial elimination [65] (Fig. 3).

Overview of the mitophagy molecular pathway and implicated in AD pathological hallmarks. A damaged mitochondrion is marked and recognized by the autophagic machinery, forming a mature mitophagosome, and then fuses to a lysosome to be degraded. Following oxidative stress and membrane potential dissipation, leading to stabilization of the PINK1 at the OMM. PINK1 is activated by auto-phosphorylation and then phosphorylates MFN2 and ubiquitin, leading to recruiting Parkin to the OMM surface. Phosphorylated poly-ubiquitin chains serving as a “swallow me” signal for the autophagic machinery. Meanwhile, parkin ubiquitylates several outer membrane components, such as FUNDC1, AMBRA1, TAX1, NIX/BNIP3L, BNIP3, and FKBP8, that are following recognized by the adaptor proteins ubiquitin-binding proteins OPTN, p62, NDP52, and NBR1 et al., which recruit the impaired mitochondria to the autophagy pathway and initiate autophagosome formation through binding with LC3. Neurons affected in AD undergo mitochondrial abnormalities and a bioenergetic deficit that may generate the disease-defining Aβ pathologies and NFT, leading to brain atrophy and AD. PINK1, PTEN-induced putative kinase 1; OMM, outer mitochondrial membrane; MFN2, mitofusin 2; FUNDC1, FUN14 Domain Containing 1, AMBRA1, autophagy and beclin 1 regulator 1; NDP52, Nuclear Dot Protein 52 kDa; NIX, BCL2 Interacting Protein 3 Like; FKBP8, FKBP Prolyl Isomerase 8; OPTN, optineurin; NBR1, neighbor of BRCA1 gene 1; LC3, microtubule-associated protein light-chain 3
Pathogenic Aβ Interfering with Defective Mitophagy
Neurons affected by AD undergo mitochondrial abnormalities and a bioenergetic deficit that may generate the disease-defining Aβ pathologies. Oppositely, Aβ deposition can also deteriorate mitochondrial homeostasis [66]. Studies of cultured neurons and those in brain specimens from AD patients have demonstrated that mitochondrial dysfunction accelerates Aβ production through oxidative stress production [67], and restoring mitochondrial function by administration of superoxide dismutase-2 (SOD2) might protect the aging brain against hAPP/Aβ-induced impairments [68]. Accumulation of unrepaired damaged nuclear due to dysfunction of base excision repair and DNA double-strand break repair and mitochondrial DNA may occur early in the AD, leading to Aβ increase by inhibition of the nicotinamide adenine dinucleotide (NAD+)/sirtuin–PGC-1a pathway [61]. Simultaneously, a growing number of evidence report that Aβ peptides exert a critical role in mediating mitochondrial toxicity. The results of one cross-species study offer robust evidence that mitophagy defects play a robust role in AD development and progression, which can be ascribed to both Aβ1–42 and p-tau pathologies, which is a major factor of AD progression and memory disability range from Caenorhabditis elegans and mice to humans [23]. Accumulation of Aβ in neurons expressing mutant APP diminished mitochondrial ATP production and mitochondrial membrane potential decreased and enzyme competence, as well as increasing levels of mitochondrial ROS [69]. Inhibition of mitochondrial permeability transition pores and reduction in mitochondrial injury would be expected to protect neurons against Aβ neurotoxicity [70]. Moreover, mitophagy failure was observed because of diminished autophagy degradation function in fibroblasts and iPSC-derived neurons harboring the PS1 mutation A246E, thus leading to the accumulation of damaged mitochondria [71]. In terms of synaptic mitochondria, especially Aβ-rich region, may be especially apt to damage by Aβ result from their high ATP demand and constant Ca2+ influx during synaptic activation, emphasizing the critical role of synaptic mitochondrial dysfunction ascribe to the development of synaptic degeneration in AD [72].
Pathogenic Tau Interfering with Defective Mitophagy
Tau belongs to a family of microtubule-associated proteins (MAPs) that is essential for maintenance of cell structure and integrity. It is bound to microtubules, performing an important role in regulating the assembly and stabilization of microtubules, which is necessary in order to facilitate diverse process, including axonal transport, synaptogenesis, and neurite outgrowth [73,74,75,76]. Phosphorylation of tau protein is reported to decrease its capacity to bind and stabilize microtubule; though at the same time, it is also critical for the dynamic regulation of microtubules in order to perform their function efficiently. In a group of neurodegenerative disease, referred to as “tauopathies,” tau is aberrantly hyperphosphorylated and aggregated, which results in changes to its structure and function. The most common of which is AD, where p-tau constitutes to paired helical filaments (PHFs), which facilitate formation of one of the pathological hallmarks, namely NFT [73]. Accumulating evidence stemming from AD human post-mortem tissue as well as models of AD, namely drosophila, murine, and human cell lines overexpressing wild-type (WT) and/or mutant tau, implicate impairments in mitochondrial morphology, function, dynamics, and transport [73, 77,78,79,80,81]. In particular, localization of p-tau on the outer mitochondrial membrane, within the inner mitochondrial membrane, as well as in the mito-autophagosomes has become evident [82, 83]. Mitochondrial localization of p-tau has been associated with impairments in mitochondrial Ca2+ homeostasis, mitochondrial-ER communication, and mitophagy, in particular [82, 83]. Further evidence shows overexpressing mutant tau implicates impaired mitophagy which leads to accumulation of damaged mitochondria and functionally to cognitive deficits [21, 23, 84]. Overexpression of tau in human cell lines revealed reduced levels of Parkin in mitochondrial fraction accompanied by localization of tau in the outer membrane of the mitochondria [84]. Moreover, upon upregulation of Parkin, the mitophagy deficits were rescued. Götz et al. reported that disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria in cell and also in C. elegans models. Thus, implicating the insertion of tau in the outer membrane may reduce the interaction between Parkin and the mitochondria [73, 84]. Collectively, these studies suggest that an overexpression of tau impairs the recognition of damaged mitochondria owed to impaired Parkin-mitochondria interaction, resulting in accumulation of dysfunctional mitochondria.
Interventional Strategies of Targeting Autophagy/Mitophagy to AD Treatment
Autophagy Inducers: Preclinical Studies
Among the different compounds to induce autophagy, rapamycin is the best-known mTOR inhibitor, and has been shown to avoid protein aggregation and ameliorates memory features in experimental models of AD [85]. Besides, rapamycin administration also plays beneficial effects confronting mitochondrial dysfunction by maintaining energy metabolism homeostasis and stress resistance possibly through mitophagy in murine cells [86]. Temsirolimus can enhance Aβ and p-tau clearance in an autophagy-dependent manner in various AD models, as well as improve spatial learning and memory abilities [47]. Metformin, as a biguanides antidiabetic, administration either acts on tau dephosphorylation via mTOR/protein PP2A signaling in AD [87] or induces mitophagy by facilitating Parkin activity through p53 downregulation [88]. Moreover, many mTOR-independent autophagy inducers signal via AMPK. For example, trehalose, a natural disaccharide, has shown neuroprotective properties and exhibits therapeutic effects in the fields of cognitive and learning ability and concomitant with autophagy induction in AD mouse [89]. Carbamazepine, an inhibitor of inositol synthesis that activates autophagy in an mTOR-independent manner, can significantly improve spatial learning and memory deficits as well as reduce the cerebral amyloid plaque burden in the APP/PS1 mice [90]. Strategies that target the lysosomal compartment are also gaining attention. For instance, regulation of TFEB, known to control lysosome biogenesis and autophagy, has produced momentous interest as an elementary for against NDDs. Overexpression of TFEB based on adeno-associated virus vector-mediated can effectively reduce NFT and rescue behavioral deficit in experimental mouse models of AD by targeting toxic tau [91]. A synthesized curcumin derivative termed compound C1 specifically binds to TFEB and promotes TFEB nuclear translocation without influencing mTOR activity, as well as enhances autophagy and lysosome biogenesis, and can be a candidate agent for the treatment of AD [92]. The E3-ubiquitin ligase Parkin expression ubiquitinates intracellular proteins and motivates BECN-dependent molecular cascade of autophagy/mitophagy in triple transgenic AD mouse [93].
Autophagy Inducers: Clinical Studies
Latrepirdine (Dimebon), which is a mTOR-dependent autophagy targeted drug, showed divergent results in several clinical trials in AD [94]. A large controlled phase 2 trial in 183 mild-to-moderate AD patients shows apparently therapeutic benefit over placebo after 24 weeks in terms of cognitive symptoms, daily living, and also for neuropsychiatric symptoms [95]. This positive effect was not reproduced in several subsequent larger trials where both groups improved to about the same extent with no significant difference (NCT00675623; NCT00838110). Furthermore, nilotinib is currently being tested in a phase 2 trial in mild to moderate AD (NCT02947893) because of the strong preclinical evidence about the effects on AD models, including inhibiting brain Aβ deposition, decreasing p-tau, modulating brain and peripheral immune profiles, and alleviating cognitive decline [96]. Nilotinib strengthens the autophagic machinery by increasing endogenous parkin levels and ubiquitination, which may enhance parkin recycling and interaction with BECN-1 that plays a critical role in clearance of misfolded and damaged protein [97]. A pilot study of metformin was carried out among persons with amnestic mild cognitive impairment (MCI) with the goal of collecting preliminary data on feasibility, safety, and efficacy (NCT00620191). There were no differences between metformin and placebo in terms of efficacy, as well as no serious adverse event discrepancy [98]. In addition, a phase 2 trial to study the effect of metformin on AD Biomarkers (NCT01965756) is finished but not published yet. Participants received metformin for 8 weeks as follows: 500 mg daily for 1 week, then daily dose increased by 500 mg per week until a maximum of 2000 mg/day was reached. The final analysis data concerning on the outcome measures are still not public right now. The most well-known compound that can enhance autophagy through inositol signaling pathway is lithium. A couple of ongoing trials are recruiting to assess the effect of lithium on AD (NCT02129348; NCT03185208). Collectively, significant progress has been made in ascertaining the roles of autophagy in the AD etiology, while interventions that stimulate autophagy can be a potential therapeutic strategy for AD patients.
Mitophagy Inducers
The accurate mechanisms of protein misfolding in AD are poorly understood; and hence, there are currently no effective treatments that block the progression of AD. Recently, one study reported that boosting neuronal mitophagy by augmentation with nicotinamide mononucleotide (NMN), a NAD+ precursor, could reduce insoluble Aβ1–42 levels and against cognitive performances in both C. elegans and APP/PS1 AD mouse models, suggesting impaired removal of damaged mitochondria is a critical etiology in AD, and induction mitophagy via NAD+-precursor can reverse the AD pathological hallmarks and clinical phenotypes [23]. Analogically, treatment of 3xTgAD mice with nicotinamide (NAM) for 8 months improved cognitive performance, as well as reduced Aβ and p-tau pathologies in cerebral by a mechanism involving NAD+ biosynthesis, PI3K-AKT signaling, MAPK/ERK1/2, SIRT1, and autophagy [99]. NAM could hold mitochondrial integrity and dynamics, and improve lysosome procession by promoting autophagy or mitophagy and lysosomal acidification to reduce autophagosome accumulation [99, 100]. In addition, several energy modulators, including metformin, pifithrin-a, resveratrol, spermidine, p62-mediated mitophagy inducer (PMI), urolithin A (UA), and actinonin (AC), to some degree, have been shown to maintain mitochondrial integrity and boost mitochondrial biogenesis through mitophagy induction [22, 64]. These small compound treatments induce mitophagy-related cytoprotective and anti-aging effects through recovering energy metabolism in several model organisms, considered to be promising anti-AD drug candidates in theory [101]. However, the underlying molecular mechanisms of their effects and detailed functions in AD require further exploration. Among them, some modulators have been tested in AD cross-species models. Induction of neuronal mitophagy with UA (200 mg/kg/day) and AC (30 mg/kg/day) relieved cognitive decline, p-tau, and Aβ plaque by promoting mitophagy machinery in the APP/PS1 mouse model. The effects of UA and AC are in part dependent on key mitophagy genes, such as DCT-1, PDR-1, and PINK1 [23]. There are some additional pharmacological compounds that show protection against mitochondrial injury in animals, including mitochondria-targeted antioxidants (e.g., MitoQ) [102], mitochondrial uncoupling agents (e.g., DNP) [103], and mitochondria-targeted peptides (e.g., Bendavia) [104]. Moreover, therapies that strengthen mitochondrial biogenesis by increasing the AMPK-PGC1α-BDNF-NGF activities with 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) [105] or 2-deoxyglucose [106] are also effective in limited AD models. Collectively, all aforementioned results provide a rationale for future thorough investigation of compounds that induce mitophagy as potential candidates in preclinical AD models. Recently, a 24-week, randomized, double-blind, placebo-controlled phase 2 trial was launched, recruiting in 2018 to evaluate the safety, tolerability, and effects of treatment with AMX0035 (a combination of sodium phenylbutyrate and tauroursodeoxycholic acid (TUDCA) and phenylbutyrate) in subjects with MCI or early-stage AD (NCT03533257). TUDCA is a bile acid that protects mitochondrial dynamics by reducing mitochondrial permeability and increasing mitophagy by Parkin signal [107]. The summary of the aforementioned discussed autophagy/mitophagy inducers is in Table 1.
Outstanding Questions and Conclusions
Notably, Aβ plaque and p-tau were virtually decreased by anti-Aβ/tau therapies, but did not show any cognitive benefit in clinical trials over the past decade [2]. Hence, an alternative promising option for AD therapeutics is to address the dysfunction of autophagy and mitophagy for developing AD. However, the roles of autophagy in AD may be more complex and are not simply cell-autonomous, but interacting with other molecular processes. For instance, different theories such as “amyloid plaques, NFT, neuroinflammation, mitochondrial dysfunction, and comprised autophagy” result in AD etiologies that fuse with each other [111]. The “chicken and egg” relationships between different hallmarks of AD needs to be established, and specific therapy should be directed to targeting the reason of the neuronal insult and not the host response. Meanwhile, we can pick up some clues from other diseases of aging, including heart disease, cancers, and hypertension et al. that combinations of administrations based on different theories are essential and reasonable for AD patients.
This complex interplay between autophagy or mitophagy and histopathology in AD suggests that targeting autophagy or mitophagy is a promising anti-AD drug candidate. We discuss the implications of these new insights for induction of autophagy or mitophagy as the new therapeutic strategies that target processes upstream of both Aβ and tau, and therefore forestall the neurodegenerative process in AD.
Data Availability
Not applicable.
References
Prince M, Comas-Herrera A, Knapp M, Guerchet M, Karagiannidou M (2016) World Alzheimer Report 2016. In: Improving healthcare for people living with dementia. London, pp 140
Panza F, Lozupone M, Logroscino G, Imbimbo BP (2019) A critical appraisal of amyloid-beta-targeting therapies for Alzheimer disease. Nat Rev Neurol 15(2):73–88
Nelson PT, Braak H, Markesbery WR (2009) Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68(1):1–14
Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20(3):148–160
Sinha RN (2011) Make dementia a public health priority in India. Indian J Public Health 55(2):67–69
As A (2018) 2018 Alzheimer's disease facts and figures. Alzheimers Dement 14(3):367–425
Jagust W (2018) Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci 19(11):687–700
Kivipelto M, Mangialasche F, Ngandu T (2018) Lifestyle interventions to prevent cognitive impairment, dementia and Alzheimer disease. Nat Rev Neurol 14(11):653–666
Perl DP (2010) Neuropathology of Alzheimer's disease. Mt Sinai J Med 77(1):32–42
Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H et al (2018) Trial of solanezumab for mild dementia due to Alzheimer's disease. New Engl J Med 378(4):321–330
Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, Sur C, Mukai Y, Voss T, Furtek C et al (2018) Randomized Trial of verubecestat for mild-to-moderate Alzheimer's disease. New Engl J Med 378(18):1691–1703
Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Rabe C, Brunstein F et al (2018) ABBY A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90(21):E1889–E1897
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun XY, Aisen PS et al (2014) Phase 3 Trials of solanezumab for mild-to-moderate Alzheimer's disease. New Engl J Med 370(4):311–321
Cummings JL, Morstorf T, Zhong K (2014) Alzheimer's disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6(4):37
Mattson MP, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60(5):748–766
Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G (2010) Mitochondrial dysfunction is a trigger of Alzheimer's disease pathophysiology. Biochim Biophys Acta 1802(1):2–10
Camandola S, Mattson MP (2017) Brain metabolism in health, aging, and neurodegeneration. EMBO J 36(11):1474–1492
Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA (2014) Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 157(4):882–896
Fivenson EM, Lautrup S, Sun N, Scheibye-Knudsen M, Stevnsner T, Nilsen H, Bohr VA, Fang EF (2017) Mitophagy in neurodegeneration and aging. Neurochem Int 109:202–209
Fang EF, Kassahun H, Croteau DL, Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S, Bollineni RC, Wilson MA et al (2016) NAD+ replenishment improves lifespan and healthspan in ataxia telangiectasia models via mitophagy and DNA repair. Cell Metab 24(4):566–581
Cummins N, Tweedie A, Zuryn S, Bertran-Gonzalez J, Gotz J (2018) Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J 38(3):e99360
Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF (2017) Mitophagy and Alzheimer's disease: cellular and molecular mechanisms. Trends Neurosci 40(3):151–166
Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X et al (2019) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci 22(3):401–412
Hurley JH, Young LN (2017) Mechanisms of autophagy initiation. Annu Rev Biochem 86:225–244
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333(6046):1109–1112
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147(4):728–741
Sica V, Galluzzi L, Bravo-San Pedro JM, Izzo V, Maiuri MC, Kroemer G (2015) Organelle-specific initiation of autophagy. Mol Cell 59(4):522–539
Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141(4):656–667
Kim J, Kundu M, Viollet B, Guan KL (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13(2):132-U171
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C et al (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93(5):1015–1034
Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 55(2):238–252
Tsuboyama K, Koyama-Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354(6315):1036–1041
Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N (2013) FIP200 regulates targeting of Atg16L1 to the isolation membrane. Embo Rep 14(3):284–291
Itakura E, Kishi-Itakura C, Mizushima N (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151(6):1256–1269
Frake RA, Ricketts T, Menzies FM, Rubinsztein DC (2015) Autophagy and neurodegeneration. J Clin Invest 125(1):65–74
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI et al (2017) Molecular definitions of autophagy and related processes. EMBO J 36(13):1811–1836
Hansen M, Rubinsztein DC, Walker DW (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19(9):579–593
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997
Vingtdeux V, Chandakkar P, Zhao HT, d’Abramo C, Davies P, Marambaud P (2011) Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. Faseb J 25(1):219–231
Barbero-Camps E, Roca-Agujetas V, Bartolessis I, de Dios C, Fernandez-Checa JC, Mari M, Morales A, Hartmann T, Colell A (2018) Cholesterol impairs autophagy-mediated clearance of amyloid beta while promoting its secretion. Autophagy 14(7):1129–1154
Piras A, Collin L, Gruninger F, Graff C, Ronnback A (2016) Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun 4:22
Collin L, Bohrmann B, Gopfert U, Oroszlan-Szovik K, Ozmen L, Gruninger F (2014) Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer's disease. Brain 137(Pt 10):2834–2846
Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, Lie PP, Mohan P, Coffey EE, Kompella U et al (2015) Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep 12(9):1430–1444
Maday S, Holzbaur ELF (2014) Autophagosome biogenesis in primary neurons follows an ordered and spatially regulated pathway. Dev Cell 30(1):71–85
Tammineni P, Cai Q (2017) Defective retrograde transport impairs autophagic clearance in Alzheimer disease neurons. Autophagy 13(5):982–984
Ulland TK, Song WM, Huang SCC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou YY, Caims NJ, Kambal A et al (2017) TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell 170(4):649-663.e13
Jiang T, Yu JT, Zhu XC, Zhang QQ, Cao L, Wang HF, Tan MS, Gao Q, Qin H, Zhang YD et al (2014) Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 85:121–130
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64(2):113–122
Moreau K, Fleming A, Imarisio S, Ramirez AL, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F et al (2014) PICALM modulates autophagy activity and tau accumulation. Nat Commun 5:4998
Ando K, Brion JP, Stygelbout V, Suain V, Authelet M, Dedecker R, Chanut A, Lacor P, Lavaur J, Sazdovitch V et al (2013) Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol 125(6):861–878
Moreau K, Fleming A, Imarisio S, Lopez Ramirez A, Mercer JL, Jimenez-Sanchez M, Bento CF, Puri C, Zavodszky E, Siddiqi F et al (2014) PICALM modulates autophagy activity and tau accumulation. Nat Commun 5:4998
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A et al (1997) Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med 3(1):67–72
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7):1146–1158
Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H (2013) Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem 288(2):1295–1306
Lee JK, Jin HK, Park MH, Kim BR, Lee PH, Nakauchi H, Carter JE, He X, Schuchman EH, Bae JS (2014) Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer's disease. J Exp Med 211(8):1551–1570
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC (2013) Abeta secretion and plaque formation depend on autophagy. Cell Rep 5(1):61–69
Cuyvers E, van der Zee J, Bettens K, Engelborghs S, Vandenbulcke M, Robberecht C, Dillen L, Merlin C, Geerts N, Graff C et al (2015) Genetic variability in SQSTM1 and risk of early-onset Alzheimer dementia: a European early-onset dementia consortium study. Neurobiol Aging 36(5):2005.e15–22
Murphy MP, Hartley RC (2018) Mitochondria as a therapeutic target for common pathologies. Nat Rev Drug Discov 17(12):865–886
Martinez-Vicente M (2017) Neuronal mitophagy in neurodegenerative diseases. Front Mol Neurosci 10:64
Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O et al (2003) Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol 184(1):247–263
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA (2016) Nuclear DNA damage signalling to mitochondria in aging. Nat Rev Mol Cell Biol 17(5):308–321
Martire S, Mosca L, d’Erme M (2015) PARP-1 involvement in neurodegeneration: a focus on Alzheimer's and Parkinson's diseases. Mech Aging Dev 146–148:53–64
Cai Q, Tammineni P (2016) Alterations in mitochondrial quality control in Alzheimer's disease. Front Cell Neurosci 10:24
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20(9):1013–1022
Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19(2):93–108
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M et al (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science 304(5669):448–452
Gwon AR, Park JS, Arumugam TV, Kwon YK, Chan SL, Kim SH, Baik SH, Yang S, Yun YK, Choi Y et al (2012) Oxidative lipid modification of nicastrin enhances amyloidogenic gamma-secretase activity in Alzheimer's disease. Aging Cell 11(4):559–568
Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puolivali J, Scearce-Levie K, Masliah E, Mucke L (2006) Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26(19):5167–5179
Leuner K, Schutt T, Kurz C, Eckert SH, Schiller C, Occhipinti A, Mai S, Jendrach M, Eckert GP, Kruse SE et al (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16(12):1421–1433
Hou Y, Ghosh P, Wan R, Ouyang X, Cheng H, Mattson MP, Cheng A (2014) Permeability transition pore-mediated mitochondrial superoxide flashes mediate an early inhibitory effect of amyloid beta1-42 on neural progenitor cell proliferation. Neurobiol Aging 35(5):975–989
Martin-Maestro P, Gargini R (2017) A AS, Garcia E, Anton LC, Noggle S, Arancio O, Avila J, Garcia-Escudero V: Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer's disease-associated presenilin 1 mutation. Front Mol Neurosci 10:291
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci U S A 107(43):18670–18675
Perez MJ, Jara C, Quintanilla RA (2018) Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front Neurosci 12:441
Dixit R, Ross JL, Goldman YE, Holzbaur EL (2008) Differential regulation of dynein and kinesin motor proteins by tau. Science 319(5866):1086–1089
Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu GQ, Mucke L (2015) Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol 209(3):419–433
Yin JA, Gao G, Liu XJ, Hao ZQ, Li K, Kang XL, Li H, Shan YH, Hu WL, Li HP et al (2017) Genetic variation in glia-neuron signalling modulates aging rate. Nature 551(7679):198–203
Schulz KL, Eckert A, Rhein V, Mai S, Haase W, Reichert AS, Jendrach M, Muller WE, Leuner K (2012) A new link to mitochondrial impairment in tauopathies. Mol Neurobiol 46(1):205–216
DuBoff B, Gotz J, Feany MB (2012) Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75(4):618–632
Li XC, Hu Y, Wang ZH, Luo Y, Zhang Y, Liu XP, Feng Q, Wang Q, Ye K, Liu GP et al (2016) Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6:24756
Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol 179(4):2071–2082
Rodriguez-Martin T, Cuchillo-Ibanez I, Noble W, Nyenya F, Anderton BH, Hanger DP (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol Aging 34(9):2146–2157
Cieri D, Vicario M, Vallese F, D’Orsi B, Berto P, Grinzato A, Catoni C, De Stefani D, Rizzuto R, Brini M et al (2018) Tau localises within mitochondrial sub-compartments and its caspase cleavage affects ER-mitochondria interactions and cellular Ca(2+) handling. Biochim Biophys Acta Mol Basis Dis 1864(10):3247–3256
Grassi D, Diaz-Perez N, Volpicelli-Daley LA, Lasmezas CI (2019) Palpha-syn* mitotoxicity is linked to MAPK activation and involves tau phosphorylation and aggregation at the mitochondria. Neurobiol Dis 124:248–262
Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, Haynes CM (2016) Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533(7603):416–419
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS ONE 5(4):e9979
Johnson SC, Yanos ME, Kayser EB, Quintana A, Sangesland M, Castanza A, Uhde L, Hui J, Wall VZ, Gagnidze A et al (2013) mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342(6165):1524–1528
Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Kohler A, Glossmann H et al (2010) Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 107(50):21830–21835
Song YM, Lee WK, Lee YH, Kang ES, Cha BS, Lee BW (2016) Metformin restores parkin-mediated mitophagy, suppressed by cytosolic p53. Int J Mol Sci 17(1):122
Du J, Liang Y, Xu F, Sun B, Wang Z (2013) Trehalose rescues Alzheimer's disease phenotypes in APP/PS1 transgenic mice. J Pharm Pharmacol 65(12):1753–1756
Li L, Zhang S, Zhang X, Li T, Tang Y, Liu H, Yang W, Le W (2013) Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer's disease. Curr Alzheimer Res 10(4):433–441
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM et al (2014) Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 6(9):1142–1160
Song JX, Sun YR, Peluso I, Zeng Y, Yu X, Lu JH, Xu Z, Wang MZ, Liu LF, Huang YY et al (2016) A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 12(8):1372–1389
Khandelwal PJ, Herman AM, Hoe HS, Rebeck GW, Moussa CE (2011) Parkin mediates beclin-dependent autophagic clearance of defective mitochondria and ubiquitinated Abeta in AD models. Hum Mol Genet 20(11):2091–2102
Muller WE, Eckert GP, Friedland K, Kolesova N, Gaca J, Eckert SH (2018) Mitochondrial pharmacology of dimebon (Latrepirdine) calls for a new look at its possible therapeutic potential in Alzheimer’s disease. Aging Dis 9(4):729–744
Doody RS, Gavrilova SI, Sano M, Thomas RG, Aisen PS, Bachurin SO, Seely L, Hung D, dimebon i (2008) Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer's disease: a randomised, double-blind, placebo-controlled study. Lancet 372(9634):207–215
Lonskaya I, Hebron ML, Desforges NM, Schachter JB, Moussa CE (2014) Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J Mol Med (Berl) 92(4):373–386
Lonskaya I, Hebron ML, Desforges NM, Franjie A, Moussa CE (2013) Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med 5(8):1247–1262
Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G, Ichise M, Manly J, Devanand DP, Bagiella E (2016) Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis 51(2):501–514
Liu D, Pitta M, Jiang H, Lee JH, Zhang G, Chen X, Kawamoto EM, Mattson MP (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34(6):1564–1580
Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A (2014) Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1–42)-induced rat model of Alzheimer's disease. Free Radic Res 48(2):146–158
Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A et al (2016) Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 22(12):1428–1438
McManus MJ, Murphy MP, Franklin JL (2011) The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci 31(44):15703–15715
Geisler JG, Marosi K, Halpern J, Mattson MP (2017) DNP, mitochondrial uncoupling, and neuroprotection: a little dab'll do ya. Alzheimers Dement 13(5):582–591
Reddy PH, Manczak M, Kandimalla R (2017) Mitochondria-targeted small molecule SS31: a potential candidate for the treatment of Alzheimer's disease. Hum Mol Genet 26(8):1483–1496
Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D’Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S et al (2017) Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552(7684):187–193
Yao J, Chen S, Mao Z, Cadenas E, Brinton RD (2011) 2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease. PLoS ONE 6(7):e21788
Fonseca I, Gordino G, Moreira S, Nunes MJ, Azevedo C, Gama MJ, Rodrigues E, Rodrigues CMP, Castro-Caldas M (2017) Tauroursodeoxycholic acid protects against mitochondrial dysfunction and cell death via mitophagy in human neuroblastoma cells. Mol Neurobiol 54(8):6107–6119
Eckert SH, Gaca J, Kolesova N, Friedland K, Eckert GP, Muller WE (2018) Mitochondrial pharmacology of dimebon (Latrepirdine) calls for a new look at its possible therapeutic potential in Alzheimer's disease. Aging Dis 9(4):729–744
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, Chen SJ, Chen Y, Zhao WL (2012) Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis 3:e275
East DA, Fagiani F, Crosby J, Georgakopoulos ND, Bertrand H, Schaap M, Fowkes A, Wells G, Campanella M (2014) PMI: a DeltaPsim independent pharmacological regulator of mitophagy. Chem Biol 21(11):1585–1596
Mattson MP, Arumugam TV (2018) Hallmarks of brain aging: adaptive and pathological modification by metabolic states. Cell Metab 27(6):1176–1199
Funding
The study was supported by the Projects of National Science Foundation of China (No. 81600977) and the Projects of Wenzhou city Committee of Science and Technology (Y20170067 and Y20180137 and Y2020427) and the Projects of Natural Science Foundation of Zhejiang Province (Y19H090059).
Author information
Authors and Affiliations
Contributions
WWW carried out the idea and searched relevant literatures; HJH and YYG made substantial contributions to conception and design and figures/tables. JC revised the manuscript. RYH and CLX were involved in drafting the manuscript and supervised the process. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
All the authors consent to publication.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key Points
1. Update the interplay between autophagy or mitophagy and histopathology in AD.
2. Interventional strategies of targeting autophagy or mitophagy as promising anti-AD drug candidates.
3. Induction of autophagy or mitophagy as the new therapeutic strategy that targets processes upstream of both Aβ and tau, and therefore forestalls the neurodegenerative process in AD.
Rights and permissions
About this article

Cite this article
Chen, J., He, HJ., Ye, Q. et al. Defective Autophagy and Mitophagy in Alzheimer’s Disease: Mechanisms and Translational Implications. Mol Neurobiol 58, 5289–5302 (2021). https://doi.org/10.1007/s12035-021-02487-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02487-7