
Abstract
Ischemic tolerance is the establishment of brain resistance to severe ischemic damage by a mild preconditioning stimulus, insufficient to irreversible tissue damage, but capable of initiating a defense response. We developed the model of focal-focal ischemic tolerance, in which the first local photothrombotic infarct (PTI) in the rat brain cortex reduced the infarct caused by second PTI applied to the contralateral cortex of the same rat 7 days later. Using antibody microarrays, we compared protein profiles in the penumbra surrounding the PTI core after single and double PTI. We observed up- or downregulation of several dozens of proteins that are aimed at neurodegeneration or neuroprotection. Both single and double PTI induced damaging processes in the rat cerebral cortex that included over-expression of various pro-apoptotic and signaling proteins and downregulation of other signaling proteins and regulators of proliferation, some components of actin, intermediate fiber and microtubular cytoskeletons, and proteins involved in vesicle transport and synaptic transmission. The simultaneous protective processes included the upregulation of different signaling and anti-apoptotic proteins, stimulators of proliferation, and proteins involved in remodeling of actin cytoskeleton. The elevated expression of some signaling proteins, such as calcium-dependent PLCγ1, PKVα1, CaMKIIα, calnexin, and calreticulin was preserved after double PTI. Less pro-survival proteins were downregulated in the penumbra after double than single impact.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucose oxidation is the major energy source in the brain, so disruption of the brain supply with oxygen leads to infarct of the nervous tissue. Ischemic brain damage due to vascular occlusion and reduced blood flow is a leading cause of human disability and mortality worldwide. However, despite an intensive search in numerous laboratories, effective neuroprotective drugs are still missing [1, 2]. Therefore, deeper and more comprehensive studies of the mechanisms of neurodegeneration and neuroprotection are needed to develop new approaches to stroke treatment.
There is a high probability of ischemic brain damage exists during the human life. One can, therefore, assume the existence of some endogenous regenerative mechanism for ensuring brain survival after quite mild ischemic attack or trauma. Ischemic preconditioning (IP) is such adaptation mechanism. The essence of IP is that quite soft preconditioning stimulus, insufficient to irreversible tissue damage, but capable of initiating defense response, increases tolerance of cells to the next more powerful or prolonged ischemic effect. Ischemic tolerance (IT) is an ancient form of evolutionary plasticity of the nervous system of invertebrates and vertebrates [3,4,5]. Transient ischemic attack promotes IT occurrence in humans [6, 7].
There are two IT phases found in the brain. The early IT phase that takes several hours does not require additional protein synthesis. It includes phosphorylation and other post-translational modifications of proteins, changes in the permeability of ion channels, etc. It disappears rapidly [8]. The late IT phase requires de novo synthesis of proteins involved in regulation of metabolism, ion homeostasis, proliferation, excitotoxicity, and cell death. It develops for 12–72 h and is maintained up to 1–2 weeks. Namely this late phase may be used for stroke prophylaxis and treatment [9,10,11,12]. There are three basic intervals in this process:(1) Between the preconditioning stimulus and the damaging effects some enzymes, kinases, transcription factors, stress proteins, structural and transport proteins, regulators of the cell cycle, and apoptosis may be over-expressed to protect the cells from further damage.(2) Ischemia, which brings weaker consequences than without a preconditioning stimulus. (3) During post-ischemic reperfusion, the effector mechanisms are designed to stabilize the energy and protein metabolism, reduce the damaging effect of glutamate and reactive oxygen and nitrogen species, and reduce inflammation [8]. The gene expression profile is known to be different for the early and late IT, and different in neurons, or glial, or endothelial cells [3].
Various cellular components such as redox-sensitive enzymes, cytokines, neurotransmitters, neuromodulators, membrane receptors, and ion channels may serve as sensors of preconditioning stimuli [3, 12]. They initiate the adaptive response. Different protein kinase cascades such as Ras/Raf/MEK/ERK [13], phosphatidylinositol 3-kinase/Akt [14], nitric oxide-related signaling [15, 16], and transcription factors HIF-1a (hypoxia inducible factor 1a) [16], STAT (signal transducer and activator of transcription) [17], CREB (cAMP-dependent transcription factor) [18], AP-1 (activator protein-1) [19], and nuclear factor NF-κB [20] serve as transducers. Activation of transducers changes the expression of many effector proteins.
Initial IT stages may vary depending on the inductor nature, but the final steps associated with activation of effectors, are universal for all stimuli. IT may be induced not only by ischemia, but a variety of physical and chemical factors such as hypoxia, hyperoxia, heating, inflammatory mediators, metabolic inhibitors, trauma, physical activity, and others [21]. Preconditioning protects nerve and glial cells from oxidative stress, maintains membrane potential, and activates hypoxia-sensitive anti-apoptotic genes [2, 21]. It improves blood flow to the penumbra [22] and inhibits aggregation of platelets and white blood cells, which causes post-ischemic microvascular occlusion [23].
Several experimental models of ischemic stroke are used in preclinical studies: transient mechanical occlusion of the middle cerebral artery (MCAO) by ligation or by the introduction of the silicon-coated nylon thread; short-term bilateral occlusion of the carotid arteries. Thrombotic occlusion is created by injection or thrombin clots [2, 4, 21]. However, it is not easy to obtain a controlled and reproducible local thrombosis in these cases.
The focal photothrombotic infarct (PTI) based on photodynamic effect and local laser irradiation better satisfies these requirements [24,25,26,27]. In photodynamic effect the energy of photoexcited dye molecules that stain cells is transferred to oxygen and converts it into the highly toxic singlet form. Singlet oxygen and other reactive oxygen species cause oxidative stress and cell death. Based on this effect photodynamic therapy is used in oncology for destruction of malignant tissues including brain tumors [28]. Local photothrombosis of brain vessels is a non-traditional use of photodynamic effect. In this method, the water-soluble photosensitizer Rose Bengal (RB), which does not penetrate cells and remains in the bloodstream, is used. Following local laser irradiation results in oxidative damage to the endothelium and basal membrane, platelet aggregation and occlusion of small cerebral vessels. This causes focal photothrombotic infarction of the cerebral tissue. The advantages of PTI as a stroke model include non-invasiveness, possibility to control location, size and extent of the damage, good reproducibility, possibility of repeated manipulations, and good animal survival [25, 26]. Therefore, PTI may be considered as a relevant inducer of ischemic tolerance. However, its drawback is a small penumbra width due to rapid infarct of the cerebral tissue under intense laser irradiation. Using less intense but longer irradiation, we induced PTI in the rat cerebral cortex with rather broad penumbra (about 1.5 mm), sufficient to obtain enough tissue for the proteomic study [27].
In the present work, we carried out proteomic study of molecular mechanisms of the late (steady) ischemic tolerance in the penumbra surrounding the ischemic core in the rat cerebral cortex. In this study, the primary PTI have served as a preconditioning stimulus, and the secondary, testing PTI was applied to the contralateral cerebral cortex 7 days later. The results of this experiment were compared with the results of a single (primary) PTI treatment.
Materials and Methods
Chemicals
The Panorama Ab Microarray–Cell Signaling Kits (CSAA1, Sigma-Aldrich Co) and other chemicals were obtained from Sigma-Aldrich-Rus (Moscow, Russia). Cy3™ or Cy5™ monofunctional reactive dyes were supplied by GE Healthcare.
Animals
The experiments were performed on adult male Wistar rats (200–250 g). The animal holding room was maintained at a temperature of 22–25 °C, 12-h light/dark schedule, and an air exchange rate of 18 changes per hour. All experimental procedures were carried out in accordance with the European Union guidelines 86/609/ЕЕС for the use of experimental animals and local legislation for ethics of experiments on animals. The animal protocols were evaluated and approved by the Animal Care and Use Committee of Southern Federal University (Approval No 02/2014).
Focal Photothrombotic Infarct in the Rat Cerebral Cortex
The unilateral focal photothrombotic infarction in the rat somatosensory cortex was induced as previously described [27]. The rats were anesthetized with chloral hydrate (300 mg/kg, i.p.). After the longitudinal incision of the skull skin, the periosteum was gently removed. Bengal Rose (20 mg/kg) was injected in the v. subclavia. The unilateral irradiation of the somatosensory cortex was performed through the cranial bone using a 532-nm diode laser (64 mW/cm2, Ø 3 mm, 30 min). Irradiation zone was located according to stereotactic coordinates into representative forelimbs in the sensorimotor cortex: 0.5 mm anterior to bregma, 0.5 mm lateral to the midline (Fig. 1). The animals were euthanized with the chloral hydrate overdose (600 mg/kg, i.p.) and decapitated 4 or 24 h after single or double PTI. The sham-operated animals, which were underwent to the same procedures but not irradiated, were used as a control. A single PTI was considered as a preconditioning impact. In this case, the wound was sutured after PTI. Animals well survived this operation without functional disorders. In the case of double PTI, the second testing PTI with the same parameters was applied in 7 days after the first preconditioning PTI to the contralateral cortex of the same rats. These animals were similarly euthanized and decapitated 4 or 24 h after the second PTI. The data of the experiments with single unilateral and double bilateral PTI were compared with control results obtained on sham-operated rats, which were similarly treated, but not irradiated.

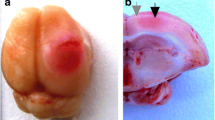
The location of the photothrombotic infarct (PTI) in the sensomotory cortex of a rat. I. The scheme. II. The rat brain in 1 day after PTI (A), 7 days (B), 14 days (C), and 21 days (D). Scale bar: 10 mm
Infarct Volume Determination
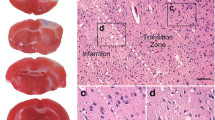
At various intervals after PTI mice were deeply anesthetized with chloral hydrate and decapitated. The brains were rapidly extracted and placed into a freezer (−80 °C) for 5 min and sliced into 1-mm-thick coronal sections. The sections were stained 30 min with 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich Co) at 37 °C in dark. The hemisphere and infarction areas were traced and measured on each section using the Image J software (NIH, USA).
For immunohistochemical study, the rats were perfused transcardially with 10% buffered formalin (pH 7.2) under chloral hydrate anesthesia. The extracted brains were post-fixed with formalin and embedded into paraffin using the standard technique. Five-micrometer sections of the experimental and control cortex tissues were made. The sections were demasked in Tris-EDTA buffer with 0.05% Tween-20 (pH 9.0) in the programmed barocamera Pascal (Dako). Reactions were visualized using the REVEAL Polyvalent HRP-DAB Detection System (Spring Bioscience, SPD-060, USA) according to the manufacturer’s instructions. The primary antibodies against PAR4 (SAB4502078, Sigma-Aldrich, 1:3000) or cofilin, (SAB4500148, Sigma-Aldrich, 1:3000) were used. The sections were counterstained with Mayer’s hematoxylin and imaged on the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, 40×/0.60). Omission of the primary antibody was routinely used to certify the absence of nonspecific labeling. All observations were performed in lamina granularis externa, lamina pyramidalis and lamina granularis interna. Twelve images were acquired per section, and studied under the Eclipse FN1 microscope (Nikon, Japan; objective lens Plan Fluor ELWD, 40×/0.60). The image analysis was performed using the ImageJ software (NIH, USA). For quantification, the immunoreactivity coefficients were determined in each section in control (n = 3) and experimental (n = 3) samples according the formula: I = N ip/N t * 100%, where N ip and N t are the numbers of immunopositive pixels and total number of pixels, respectively. Statistical analysis was performed with Student’s t test for independent experiments. Quantitative data were presented as mean ± SEM.
Western Blot
The tissue extracts containing 10 μg of protein in 15 μl of the buffered saline were subjected to electrophoresis in 7.5% polyacrylamide gel in Mini-PROTEAN Tetra cell (Bio-Rad). Then proteins were electro-transferred on the polyvinyl-difluoride membrane Immun-Blot PVDF Membrane (162-0177, Bio-Rad), using mini Trans-Blot transfer cell (Bio-Rad). After washing the membrane was incubated 1 h in a blocking solution (PBS, 5% dry skimmed milk). Then the membranes were consecutively incubated overnight with the primary rabbit anti-Bcl-xL antibody and with the secondary antibody anti-rabbit IgG-peroxidase (both from Sigma-Aldrich, USA). Then the membranes were washed in TTBS and incubated with Сlarity Western ECL Substrate (Bio-Rad). The chemiluminescence was registered using the gel-documentation system Fusion SL (Vilber Lourmat, France) and the software Vision Capt. Quantitative data are presented as mean ± SEM.
Proteomic Study
The Panorama Antibody Array–Cell Signaling kit (CSAA1, Sigma-Aldrich Co) contains two identical microarrays, in which the nitrocellulose-coated glass slides containing 448 microdroplets with immobilized antibodies against 224 signaling proteins in duplicate. Non-labeled bovine serum albumin was used as a negative control, and single spots with Cy3 and Cy5-conjugated BSA were used as a positive control. After photothrombotic treatment, the rat cortex was extracted, and the PTI core region was excised using the Ø3 mm circular knife. Then the surrounding 2 mm width ring-shape cortex area around the PTI core (penumbra) was cut out by another Ø7 mm circular knife (the experimental sample). The similar piece from the somatosensory cortex of sham-operated rats was used as control. Each sample contained tissues from three rats. The pieces were weighed, homogenized, and lysed in the extraction/labeling buffer supplemented with protease and phosphatase inhibitor cocktails and nuclease bensonase (components of CSAA1). Then the control and experimental lysates were centrifuged in the cooled centrifuge (10,000 rpm, 4 min, 4 °C). The supernatants were frozen in liquid nitrogen and stored at − 80 °C for further analysis. After thawing, the protein contents in both experimental and control samples were determined using Bradford reagent. Then the experimental and control samples were diluted to 1 mg/ml protein content and incubated 30 min in darkness at room temperature with Cy3 or Cy5, respectively. The unbound dye was removed by centrifugation (4000 rpm; 4 min) of the SigmaSpin Columns (CSAA1 components) filled with 200 μl of the labeled protein samples. The eluates were collected, and protein concentration was detected again. In another set, these samples were stained oppositely, by Cy5 and Cy3, respectively.
One microarray was incubated 40 min at room temperature on a rocking shaker in 5 ml of the mixture of the control and experimental samples (10 μg/ml each) labeled with Cy3 and Cy5, respectively. Another microarray was incubated with the oppositely labeled samples: Cy5 and Cy3, respectively. Such swapped staining provided verification of results and compensation of a potential bias in binding of Cy3 or Cy5 dyes to protein samples. This provides the double test and full control of the experiment. After following triple washing in the washing buffer (CSAA1 component) and triple washing in the pure water, the microarray slides were air dried overnight in darkness.
The microarrays were scanned using the GenePix 4100A Microarray Scanner (Molecular Devices, USA) at 532 and 635 nm (fluorescence maximums of Cy3 and Cy5, respectively). The integrated fluorescence intensity in each antibody spot was proportional to the quantity of the bound protein. The fluorescence images of the antibody microarrays were analyzed and normalized (ratio-based normalization) using the software GenePix Pro 6.0. Local fluorescence values in the rings around each spot were used as a background. The median fluorescence value determined over all spot pixels was used for estimation of the protein content in each spot, and the ratios of the experimental to control values characterized the difference in the protein level between photothrombotic and normal cortical tissue in the contralateral cortex. Two samples labeled independently and reversely in duplicate provided 4 Exp/Ctr (or Ctr/Exp) ratio values for relative expression of each protein (two microarrays with two microdroplets of each antibody) in each experiment. The experiments, in which penumbra tissues from three rats were united, were repeated two or three times as indicated in the headings of Tables 2 and 3. The obtained 8 or 12 Exp/Ctr (or Ctr/Exp) ratio values were averaged. The standard statistical treatment based on Student’s t test and 95% significance level was used. Quantitative data in Tables 2 and 3 were presented as mean ± SD. Only > 30% differences (the cut-off level) in the protein levels between irradiated and control animals (i.e. experimental/control ratios > 1.3) are displayed in Table 1 and discussed. The reliability of the proteomic microarray data was provided by the self-control experiment using the swapped staining in the second microarray [29, 30].
Results
Infarct Volume Changes in the Rat Cerebral Cortex After Single and Double PTI
The infarct volume of animals subjected to single PTI was maximal at 24 h after photothrombotic treatment: 27.8 ± 1.3 mm3. It decreased to 21.5 ± 2.1 mm3 during next 13 days (Table 1). In the rat groups subjected to double PTI, the mean volume of primary infarct was the same as in the groups subjected to single PTI. The volumes of secondary infarct did not differ from that after a single infarct at 1 or 7 days after the treatment. However, the difference became significant at 14 days after the second PTI: 15.9 ± 1.5 versus 21.5 ± 2.1 (p < 0.05) (Table 1). Thus, primary PTI as a preconditioning factor provided ischemic tolerance in the rat cerebral cortex that was observed 14 days after the secondary damage.
The Changes in the Expression of Signaling Proteins in the Penumbra After Single PTI
The present data on changes of the protein expression profile in the penumbra that surrounds the infarction core in the cerebral cortex of rats subjected to single unilateral PTI were compared with the protein profile in the cortex of sham-operated rats (Table 2). These data, as a whole, were similar to the results of the previous experiments, in which the protein profile in the penumbra was compared with that in the untreated contralateral cortex of the same animals 1, 4, and 24 h after PTI [27]. That experiments showed up- or downregulation of proteins involved in various cellular subsystems in the penumbra tissue such as intracellular signaling, regulation of apoptosis, cell cycle, cytoskeleton, vesicular transport and synaptic processes. The changes in the protein profile were maximal 4 h after PTI and decreased later, at 24 h. In the present experiments, 48 and 27 proteins were upregulated, whereas 48 and 17 proteins were downregulated at 4 and 24 h after single PTI, respectively (Table 2). Double PTI (first preconditioning PTI + second testing PTI applied in the contralateral cortex in 7 days after the first impact) induced upregulation of 52 and 40 proteins, and downregulation of 31 and 23 proteins in 4 and 24 h, respectively (Table 3).
4 h After Single PTI
At 4 h after single PTI, diverse pro-apoptotic proteins were upregulated by 37–94% in the penumbra (Table 2). These included various apoptosis execution proteins: caspases 3, 6, 7, and 11, activated caspase 3, SMAC/DIABLO, and PSR (phosphatidylserine receptor), which recognizes apoptotic cells for further elimination. It was of interest that many proteins that can initiate diverse apoptosis pathways in specific situations were simultaneously over-expressed in the penumbra. These are AIF that induces caspase-independent apoptosis, Bcl-10 that activates expression of p38, JNK and NF-κB; p38 that stimulates apoptosis under stressful impacts and activates expression of p53 and E2F1;c-myc, which regulates many cellular processes including transcription, proliferation and apoptosis [31]; transcription factors p53 and E2F1 that regulate expression of various apoptosis-associated genes [32, 33]; Par4 (prostate apoptosis response 4) that initiates p53-independent apoptosis of ischemic neurons [34]; stress-activated transcription factor GADD153(CHOP10) that inhibits proliferation and stimulates apoptosis [35]; pro-apoptotic neurotrophin receptor p75 [36]; glutamate receptor NMDA2a that mediates excitotoxicity and apoptosis [37]. Glutamate decarboxylase (GAD65/67), which converts l-glutamate into GABA [38], and CUG-binding protein 1 (CUGBP1), which controls mRNA stability and alternative splicing of some genes, can stimulate apoptosis of neurons [39]. Amyloid precursor protein APP and nicastrin were over-expressed after PTI. Nicastrin is known to split APP and to release the peptide AICD, which enhances p53-mediated apoptosis [40].
At the same time, a number of anti-apoptotic proteins such as Bcl-2 family proteins Bcl-x, Bcl-xL, and Mcl-1; p53 antagonist p63; p53-dependent inhibitor of proliferation and apoptosis p21Waf-1 [41]; anti-apoptotic neuroprotectors ERK5 that mediates regeneration of injured axons [42]; protein phosphatase MKP-1, which stops the pro-apoptotic signaling of p38 and JNK [43]; and estrogen receptor, the anti-inflammatory, antioxidant and anti-apoptotic factor [44], were upregulated by 1.3–2.2 times in the penumbra at 4 h after single PTI (Table 2).
The 1.5- to 2-fold downregulation of diverse pro-apoptotic proteins such as caspase 9; extracellular pro-apoptotic ligand Fas (CD95/Apo-1); death-associated domain FADD; DAP kinase (death-associated protein kinase) that controls cytoskeleton remodeling during apoptosis [45], transcription factor PML that is involved in apoptosis of ischemic neurons [46]; mitochondrial protein ARTS (apoptosis-related protein in the TGF-β signaling pathway) that induces apoptosis after translocation into the nucleus [47], and p14arf which inhibits MDM2 and thereby prevents proteolysis of p53 [48], also contributed into neuroprotection in the penumbra.
Serine/threonine protein kinases activate proteins by phosphorylation of these residues. In normal tissue the basic levels of phosphoserines and phosphothreonines are low but increase under injury. The observed 64–71% increase in the levels phosphoserines and phosphothreonines (Table 2) indicates the activation of different proteins in the penumbra. Serine/threonine protein kinases ERK1/2, SGK, RAF1, PKCα that regulate diverse cellular processes such as metabolism, proliferation, neurotransmission survival and apoptosis were upregulated in the penumbra by 36–78%. The level of transcription activator SMAD4 increased by 36%. However, the NF-κB-mediated signaling pathway was not involved in the penumbra response to PTI. In fact, both NF-κB and NF-κB-activating kinase NAK were downregulated (Table 2). Activator proteins AP-2α, AP-2β, and AP-2γ, transcription factors that regulate proliferation, differentiation, and activity of the monoaminergic systems [49] were downregulated in the penumbra by 2.3–2.7 times (Table 2).
Phospholipases PLA2 group V and PLCγ1 were upregulated in the penumbra by 56–58%. Upon binding of growth factors, PLCγ1 produces inositol triphosphate, which stimulates Ca2 +release from endoplasmic reticulum (ER). Cytosolic Ca2+activates various signaling proteins. The Ca2+-dependent proteins calmodulin, CaMKIIα, and CaMKIV, which regulate ionotropic receptors, gene expression, synthesis and release of neurotransmitters, axonal transport, memory formation, and other neuronal functions, were upregulated in the penumbra by 36–47% (Table 2). These processes are pro-survival. The level of another Ca2+-binding protein S-100, which regulates Ca2+ homeostasis, activity of various enzymes and transcription factors, decreased 3.3 times (Table 2). The upregulation of Ca2+-dependent ER chaperones calnexin and calreticulin and protein kinase MAKAPK2, which activates small ER chaperones hsp25/hsp27, was also pro-survival. However, downregulation of mitochondrial antioxidant protein AOP-1, chaperone HSP90 and cystatin A, which inhibits cathepsins, lysosomal proteases, reduced the protective potential in the penumbra tissue.
Various regulators of the cell cycle were differently expressed in the penumbra 4 h after single PTI. The downregulation of Cdc7 kinase that is involved in the initiation of DNA replication, cyclin D1 and Cdk6 that regulate G1/S transition in the cell cycle, Cdk-7/CAK, which activates cyclin-dependent kinases, topoisomerase-1 that is involved in DNA replication, and Trf-1 that controls the telomere length (Table 2), indicated the suppression of diverse proliferation processes. However, the expression of CDC6, which organizes the prereplicative complex and controls G1/S transition, increased. Possibly, various components of the cell cycle were differently regulated in different penumbra regions.
Diverse proteins associated with actin cytoskeleton were also up- or downregulated in the penumbra after single PTI (Table 2). The over-expression of catenin α and catenin p120CTN, which bind fibrillar actin to E-and N-cadherins that link cells to each other, and p35, which together with Cdk5 binds to β-catenin/N-cadherin adhesion complex and regulates the growth of injured axons (+ 83,+ 55, and + 66%, respectively; Table 2) could stabilize the neurovascular units in the penumbra and participate in neuroprotection [50]. The level of actopaxin, a component of the protein scaffold, which links the intracellular actin cytoskeleton with integrins that bind cells to the extracellular matrix [51] increased by 48%. Cofilin depolymerizes fibrillar actin. Its two-fold over-expression could be associated with remodeling of the actin cytoskeleton [52]. However, spectrin (α + β), which maintains the plasma membrane integrity and cytoskeleton structure, ezrin that links actin filaments to the plasma membrane, and tropomyosin that regulates binding of various proteins to the actin cytoskeleton, were downregulated 5, 1.6, and 3-fold, respectively. The levels of focal adhesion kinase FAK, associated with the actin-binding platform, and its phosphorylated form pFAK also decreased 1.5–2.5 times (Table 2). Such bidirectional changes in the expression of cytoskeleton proteins could be associated either with cell death and tissue destruction, or with physiological remodeling that changes the cell shape during intracellular movements and cell motility. Simultaneously, the downregulation of the microtubule components βI-tubulin, βIV tubulin, and polyglutamylated β-tubulin, microtubule-associated protein MAP-1, cytokeratin 7, the component of vascular intermediate fibers, and CNPase that mediates myelin formation [53] (Table 2) indicated tissue destruction in the penumbra.
The single PTI violated transport processes in the penumbra. The levels of adaptin (β1 + β2) that mediates formation of clatrin vesicles and GRP1 (or ARNO3, or cytohesin-3) that controls Golgi structure and regulates protein sorting and membrane trafficking [54] decreased by 2.4 and 1.6 times, respectively (Table 2). Expression of myosins IIA and Va that mediate transport of vesicles, mRNA and organelles along actin filaments in neurons and glial cells decreased 1.5 times. The proteins Ran and NTF2 involved in the nuclear import of cytoplasmic proteins [55] were also downregulated by 1.5 and 2.3 times, respectively (Table 2).
The levels of proteins involved in the synthesis of neuromediators, dopamine (tyrosine hydroxylase and DOPA decarboxylase), and serotonin (tryptophan decarboxylase) decreased by 1.7–2.8 times. The most significant was 14-fold decrease in the level of syntaxin that mediates docking of synaptic vesicles and neurotransmitter release (Table 2).
24 h After Single PTI
The changes in the protein profile in the penumbra at 24 h after single PTI resembled that at 4 h but were weaker and included fewer proteins: 27 proteins were upregulated and 17 downregulated. The values of changes in the protein levels were also lower (Table 2).
Among pro-apoptotic proteins, we observed only over-expression of caspase 3, SMAC/DIABLO, Par4, p53, GAD65/67, and CUGBP1. However, the expression of pro-apoptotic proteins, which were downregulated in the penumbra at 4 h after single PTI (except FADD), did not change relatively to control at 24 h. On the other hand, almost all anti-apoptotic proteins except p21Waf that were upregulated at 4 h, were also upregulated at 24 h. The levels of protective proteins AOP-1, cystatin A, and chaperone Hsp 90 that were downregulated at 4 h, did not change relatively to control at 24 h; only chaperon Hsp 70 was downregulated (Table 2).
Fewer changes were also observed in the expression of signaling proteins. The levels of phosphoserines and phosphothreonines as well as ERK1, GRB2, RAF1, and MAKAPK2 remained elevated at 24 h and that of synthrophin α1 and S-100 reduced. The expression of other signaling proteins did not differ significantly from control. Different regulators of the cell cycle that were downregulated at 4 h also did not change at 24 h. However, cyclin B1 was upregulated by 35% at 24 h after single PTI (Table 2).
The levels of various components of the actin cytoskeleton, which were upregulated at 4 h, remained elevated at 24 h. The levels of α-catenin, spectrin (α + β), ezrin and tropomyosin did not change at 24 h. β-Tubulins remained downregulated. Additionally, the levels of γ-tubulin and cytokeratins 13 and 8.12 decreased (Table 2). The levels of adaptin (β1 + β2) and NTF2 involved in the intracellular transport processes remained decreased, but the expression of other transport proteins did not differ from control. The levels of proteins that mediate synthesis of neuromediators did not differ anymore from that in the control tissue (Table 2).
The Changes in the Expression of Signaling Proteins in the Penumbra After Double PTI
As shown above, the second, testing PTI applied 7 days after the first, preconditioning impact resulted in a smaller infarction volume than after only one PTI. That was the manifestation of the preconditioning effect. What is the biochemical mechanism of this effect? We have studied the changes in the protein profile in the penumbra around the core of the second photothrombotic infarct at 4 and 24 h after the second PTI.
4 h After Double PTI
At 4 h after second PTI, we observed the over-expression of 52 proteins in the penumbra, whereas 31 proteins were downregulated (Table 3), fewer than after single PTI: 48 (Table 2). The majority of the upregulated proteins in the penumbra were the same as after the single impact (Tables 2 and 3). Nevertheless, some difference in the protein profile should be noted. Unlike single PTI, at 4 h after second PTI pro-apoptotic MAP kinase JNK and phosphatidylserine receptor PSR were over-expressed by 37 and 62%, respectively (Table 3). At the same time, we did not observe the downregulation of pro-apoptotic proteins Fas, FADD, ARTS, caspases 4, 5 and 9.These data indicated the shift of the life/death balance to damage of the penumbra tissue. On the other hand, the upregulation of anti-apoptotic protein MDM2, and the absence of expression of glutamate decarboxylase GAD65/67 and CUGBP1, which promote apoptosis in some situations, indicated the simultaneous pro-survival tendency.
The changes in the expression of intracellular signaling in the penumbra at 4 h after double PTI (Table 3) were, in some respect, lower than after a single impact (Table 2). In fact, overall phosphorylation of serines and threonines in proteins did not increase, but decreased as compared to the control levels. This indicated less activation of cellular proteins. Phospholipases PLA2 (group V) and PLCγ1 were not upregulated, but 3.3- to 3.4-fold downregulated in the penumbra after double PTI (Table 3). The level of SMAD4 did not increase. On the other hand, unlike single PTI, double PTI caused upregulation of transcription factor ATF2 and protein kinase Cβ (+ 44 and + 52%, respectively; Table 3). The expression of PML, NF-κB, NAK, DAP kinase, synthrophin α1, focal adhesion kinase FAK and its phosphorylated form did not decrease as after a single PTI (compare Tables 2 and 3).
The over-expression of cyclin B1 and cyclin-dependent kinase Cdk4, the downregulation of cell cycle inhibitor p19INK4D, along with the absence of downregulation of Cdk6, cyclin D1, and Cdc-7/CAK (Table 3) after double but not single PTI evidences the higher proliferation activity that was probably directed to recovery of the penumbra tissue.
The PTI-induced changes in the actin skeleton were almost the same after double and single PTI (Table 3). DAP kinase that regulates cytoskeleton remodeling was not downregulated as after single PTI. The levels of other cytoskeleton components, tubulins and intermediate fibers, such as β-tubulins I and IV, microtubule-associated protein MAP1, pan cytokeratin, and cytokeratin 13 did not decrease in the penumbra after double PTI that indicates better safety of the cytoskeleton. Moreover, the levels of cytokeratin 8.13, cytokeratin 19, and α-internexin increased after second but not first PTI. Therefore, double PTI damaged microtubules and intermediate filaments weaker than the single impact.
The increased levels of the vesicular protein bCOP and clathrin light chain protein (+ 30%, Table 3) indicated activation of the vesicular transport. The levels of Ran and NTF2 that import proteins into the cell nucleus were not reduced, and this transport was not so impaired as after single PTI (Table 2). CNPase involved in the myelin formation was not downregulated. These data indicate weaker damage of the intracellular transport processes and probably higher protective potential in the penumbra tissue.
As a whole, at 4 h after the preconditioning impact, second PTI weaker damaged the penumbra tissue around the infarction core in the rat cerebral cortex.
24 h After Double PTI
The characteristic time for development of the late ischemic tolerance is 24 h. At 24 h after single PTI, 27 proteins were upregulated and 17 proteins downregulated (Table 2), whereas at the same interval after double PTI, 40 proteins were upregulated and 23 down regulated (Table 3). Unlike the rats subjected to single PTI, in the preconditioned animals the apoptotic processes in the penumbra were more pronounced. Pro-apoptotic proteins caspase 3, caspase 6, SMAC/DIABLO, Bcl-10, AIF, Par4, E2F1, p38, p53, p75, c-myc, GADD153, NMDAR2, NGFR p75were upregulated at 24 h after double PTI (Table 3) like at 4 h after either single or double PTI (Tables 2 and 3). Unlike, only Par4, p53, GAD 65/67, CUGBP1, caspase 3, and SMAC/DIABLO were over-expressed after single PTI (Table 2). Almost the whole sets of the anti-apoptotic proteins over-expressed at 4 h after PTI were upregulated in both cases (Tables 2 and 3).
Unlike single PTI, nicastrin and Ca2+-dependent signaling proteins PLCγ1, PKCα, and CaMKIIα were upregulated in the penumbra by 30–38% at 24 h after double PTI (Table 3). However, the levels of activator proteins AP2α, β, and γ, and synthrophin α1decreased by 30–35% at 24 h after second PTI. The changes in the expression of other signaling proteins and regulators of the cell cycle were the same or absent after single and double PTI: increase in the overall threonine and serine phosphorylation and upregulation of ERK1/2, RAF1, GRB-2, and MAKAPK2 (Tables 2 and 3).
The only difference between the expression of proteins associated with actin cytoskeleton at 24 h after single and double PTI was 38% upregulation of α-catenin (Table 3) instead of 33% increase in the level of β-catenin (Table 2).
We also observed 48% upregulation of glutamate decarboxylase (GAD65/67) that converts l-glutamate into GABA after double but not single PTI (Table 3). On the other hand, double but not single PTI decreased the expression of DOPA decarboxylase and tyrosine hydroxylase, which mediate dopamine synthesis, in the penumbra by 31 and 47%, respectively (Table 3). This shows increase in the role of GABAergic processes, and decrease of the role of dopaminergic processes in the post-ischemic penumbra at 24 h after double PTI.
Immunohistochemical and Western Blot Studies of Expression of Par4, Cofilin, and Bcl-xL in Penumbra
The immunohistochemical analysis showed the significant increase in the expression of Par4 in the penumbra at 4 and 24 h after single PTI as compared with the cerebral cortex of the sham-operated animals (+ 90 and + 77%, p < 0.05, respectively) and at 4 h after double PTI (+ 46%). Its expression at 24 h after double PTI did not differ significantly from control, but was 36% lesser than after single PTI (p < 0.05) (Fig. 2).

The immunoreactivity of Par4 in the sensomotory cerebral cortex of rats after single and double photothrombotic infarct (PTI). a–f: Immunohistochemistry of Par4. a The control cortex region of the sham-operated rat 4 h after the operation. b The penumbra region 4 h after the single PTI. c The penumbra region 4 h after the double PTI, when the second (testing) PTI was applied in 7 days after the first (preconditioning) PTI. d The control cortex region of the sham-operated rat 24 h after the operation. e The penumbra region 24 h after the single PTI. f The penumbra region 24 h after the double PTI, when the second (testing) PTI was applied in 7 days after the first (preconditioning) PTI. The scale bar: 100 μm. g Immunoreactivity coefficient of Par4 (M ± SEM; n = 3) in the penumbra and the cortex of sham-operated control rats 4 and 24 h after single (ischemia) and double (preconditioning + ischemia) PTI. *p < 0.05 relatively to sham-operated animals. # p < 0.05 relatively to single PTI and the same time
The expression of cofilin in the penumbra increased at 4 and 24 h after single (+ 130 and + 73%, p < 0.05, respectively) and double PTI (+ 48 and +39 %, p < 0.05, respectively) as compared with the cerebral cortex of the sham-operated animals. Its expression at 4 h after double PTI was 37% lesser than after single PTI (p < 0.05) (Fig. 3).

The immunoreactivity of cofilin in the sensomotory cerebral cortex of rats after single and double photothrombotic infarct (PTI). a–f: Immunohistochemistry of cofilin. a The control cortex region of the sham-operated rat 4 h after the operation. b The penumbra region 4 h after the single PTI. c The penumbra region 4 h after the double PTI, when the second (testing) PTI was applied in 7 days after the first (preconditioning) PTI. d The control cortex region of the sham-operated rat 24 h after the operation. e The penumbra region 24 h after the single PTI. f The penumbra region 24 h after the double PTI, when the second (testing) PTI was applied in 7 days after the first (preconditioning) PTI. The scale bar: 100 μm. g Immunoreactivity coefficient of cofilin (M ± SEM; n = 3) in the penumbra and the cortex of sham-operated control rats 4 and 24 h after single (ischemia) and double (preconditioning + ischemia) PTI. *p < 0.05 relatively to sham-operated animals. # p < 0.05 relatively to single PTI and the same time
The western blot analysis showed the 2.3-fold decrease in the level of the anti-apoptotic protein Bcl-xL in the penumbra at 4 h after single (p < 0.05), but not double PTI (Fig. 4). At 24 h after single or double PTI, its expression showed the tendency of decrease, but it was not significant (Fig. 4).

Western blot analysis of the expression of Bcl-xL in the penumbra at 4 and 24 h after single (1PTI) and double (2PTI) photothrombotic infarct in the rat cerebral cortex. In the case of double PTI, the second (testing) PTI was applied in 7 days after the first (preconditioning) PTI. M ± SEM. (n = 6) *p < 0.05 relatively to sham-operated animals. # p < 0.05 relatively to a single PTI
Discussion
In order to reveal the biochemical basis of the preconditioning effect, we compared the protein profiles in the penumbra around the infarct core in the rat brain cortex at 4 or 24 h after the single photothrombotic impact with the protein profile in the penumbra surrounding the second infarct zone in the cerebral cortex of rats, which suffered from the same preconditioning photothrombotic infarct in the contralateral cortex that happened 7 days earlier (Tables 2 and 3, respectively).
The present data showed that double PTI induced lesser infarct volume than single PTI. This effect became visible at 14 days after the second impact (Table 1). This was possibly the result of remote ischemic tolerance induced in the rat brain by first non-lethal PTI. Thus, we developed a new model of focal-focal ischemic tolerance, in which the photothrombotic impact to the cerebral cortex can serve as a remote preconditioning stimulus, which increases a tolerance to the subsequent photothrombotic injury in another brain region.
The responses of the cerebral tissue to photothrombotic impact were complex and involved reactions of various cellular subsystems. Several dozens of signaling proteins, whose levels increased or decreased 4 or 24 h after single or double PTI (the early or late reperfusion periods), participated in pro- and anti-apoptotic processes; different intracellular signaling pathways; remodeling and destruction of various cytoskeleton elements, such as microtubules, actin filaments and intermediate filaments; suppression of vesicular transport and synaptic processes; impairment of the cell cycle, etc. The role of some proteins in the response of the cerebral cortex to ischemic stroke and development of ischemic tolerance has been discussed in the literature [30] while the roles of others are not well known.
The revealed changes in the protein profile were bidirectional, aimed at both neurodegeneration and neuroprotection. After the first, preconditioning PTI, the cerebral cortex in the contralateral hemisphere became both more vulnerable and more resistant to the forthcoming lesion. Indeed, both pro- and anti-apoptotic proteins were expressed simultaneously in the penumbra. The second PTI induced the greater apoptotic processes in the rat cortical tissue than the single impact. The number of over-expressed pro-apoptotic proteins was almost the same at 4 h after single and double PTI. However, at 24 h after double PTI, the majority of pro-apoptotic proteins, 13, remained over-expressed (Table 3), whereas only 6 of them were upregulated after single PTI (Table 2). Both, single and double PTI destructed the brain tissue, damaged vesicle transport and synaptic processes, impaired cell adhesion, cytoskeleton and cell proliferation.
At the same time, different protective processes developed in the cerebral cortex after PTI. These included the upregulation of various anti-apoptotic proteins. They were almost the same at 4 and 24 h after single or double PTI, and included Bcl-2 family proteins (Bcl-x, Bcl-xL, Mcl-1), p53 antagonists (p63, p21Waf, MDM2), and some signaling proteins (ERK5, MKP-1, estrogen receptor). Some other signaling proteins could be either pro- or anti-apoptotic depending on the situation. The upregulation of the cell cycle regulators such as cyclin B1 and Cdk4, and downregulation of the proliferation inhibitor p19INK4D could stimulate proliferation in the penumbra at 24 h after double but not single PTI. The proliferation processes could be associated with angiogenesis, gliosis, and scar formation. The upregulation of actin-binding proteins could display the remodeling of the actin cytoskeleton associated with changes in the cell shape and mobility. Lesser changes in the levels of different cytokeratins and tubulin isoforms indicate the weaker damage of microtubules and intermediate fibers after double than after single PTI. The upregulation of bCOP and clathrin light chain protein and the absence of downregulation of Ran and NTF2 showed stimulation of the vesicular transport and weaker damage to nuclear protein import after double PTI. Ca2+ ions are involved in both cell degeneration and cell protection. The increased expression of calmodulin and calmodulin-dependent kinases II and IV, calcium-dependent chaperones calnexin and calreticulin, and MARKARK2 could be associated with cell protection and increase of the resistance of cortical tissue to ischemic damage.
Possibly, the opposite, damaging and survival processes dominated in different parts of the penumbra that are located either close to the infarct core or at the penumbra periphery, respectively. They could be also differently developed in different cell types: neurons, astro-, micro- and oligodendroglia, vascular endothelium, smooth vascular muscles, different blood cell, etc. [27, 30]. So, more detailed analysis is needed for investigation of the IT mechanism. It should be noted that the over-expression of some proteins is not always required for cell responses to external impacts. Only if the present proteins are not sufficient for the cell response, the signaling pathways and transcription factors trigger the synthesis of additional molecules. Simultaneously, some proteins are subjected to proteolysis and degrade. The proteomic technique evaluates the changes in the protein levels, but does not reveal their causes, namely, the signaling cascades or transcription factors, which regulate the expression of various proteins. The mechanisms of these changes should be examined at the next stage of the study.
The list of proteins involved in the penumbra response to PTI and IT development is not short. It includes several dozens of enzymes. Many important proteins remained beyond the present study. The brain reaction to injury is very complex, and it is impossible to find a single critical pathway that being modulated can protect the nervous tissue. A complex of protecting medications should be possibly used for protection of the penumbra tissue. The phenomenon of ischemic tolerance is of importance in this aspect because it is based on the intrinsic mobilization of such complex response. On the base of the obtained data, one can select several signaling pathways, which seem to be important for responses of the penumbra tissue to focal thrombosis and development of ischemic tolerance. These pathways include: (a) proteins that initiate and regulate the concerted pro-apoptotic response such asBcl-10, p38, E2F1, and p53; (b) anti-apoptotic proteins Bcl-x, p63, MDM2, p21Waf, and ERK5; (c) signaling regulators ERK1/2, RAF1, MAPKAP2, calmodulin, and calmodulin-dependent kinases; (d) amyloid-related proteins APP, nicastrin, and others. This list is of course incomplete. It includes both proteins involved in tissue injury, which activities have to be inhibited, and those involved in tissue protection, which should be activated. We believe that such study will help us to find medications that reduce the injurious effects and assist the development of ischemic tolerance.
References
Bramlett HM, Dietrich WD (2004) Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 24:133–150
Iadecola C, Anrather J (2011) Stroke research at a crossroad: asking the brain for directions. Nat Neurosci 14:1363–1368
Gidday JM (2006) Cerebral preconditioning and ischaemic tolerance. Nat Rev Neurosci 7:437–448
Obrenovitch TP (2008) Molecular physiology of preconditioning-induced brain tolerance to ischemia Physiol. Rev 88:211–247
Thushara Vijayakumar N, Sangwan A, Sharma B, Majid A, Rajanikant GK (2016) Cerebral ischemic preconditioning: the road so far. Mol Neurobiol 53:2579–2593. https://doi.org/10.1007/s12035-015-9278-z
Weih M, Kallenberg K, Bergk A, Dirnagl U, Harms L, Wernecke KD, Einhaupl KM (1999) Attenuated stroke severity after prodromal TIA: a role for ischemic tolerance in the brain? Stroke 30:1851–1854
Moncayo J, de Freitas GR, Bogousslavsky J, Altieri M, van Melle G (2000) Do transient ischemic attacks have a neuroprotective effect? Neurology 54:2089–2094
Wang Y, Reis C, Applegate R 2nd, Stier G, Martin R, Zhang JH (2015) Ischemic conditioning-induced endogenous brain protection: Applications pre-, per- or post-stroke. Exp Neurol 272:26–40
Dhodda VK, Sailor KA, Bowen KK, Vemuganti R (2004) Putative endogenous mediators of preconditioning-induced ischemic tolerance in rat brain identified by genomic and proteomic analysis. J Neurochem 89:73–89
Stenzel-Poore MP, Stevens SL, King JS, Simon RP (2007) Preconditioning reprograms the response to ischemic injury and primes the emergence of unique endogenous neuroprotective phenotypes: a speculative synthesis. Stroke 38:680–685. https://doi.org/10.1161/01.STR.0000251444.56487.4c
Tang Y, Pacary E, Fréret T, Divoux D, Petit E, Schumann-Bard P, Bernaudin M (2006) Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: identification of potential neuroprotective candidates for stroke. Neurobiol Dis 21:18–28
Karikó K, Weissman D, Welsh FA (2004) Inhibition of toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. J Cereb Blood Flow Metab 24:1288–1304
Miao B, Yin XH, Pei DS, Zhang QG, Zhang GY (2005) Neuroprotective effects of preconditioning ischemia on ischemic brain injury through down-regulating activation of JNK1/2 via N-methyl-D-aspartate receptor-mediated Akt1 activation. J Biol Chem 280:21693–21699
Zhang QG, Han D, Xu J, Lv Q, Wang R, Yin XH, Xu TL, Zhang GY (2006) Ischemic preconditioning negatively regulates plenty of SH3s-mixed lineage kinase 3-Rac1 complex and c-Jun N-terminal kinase 3 signaling via activation of Akt. Neuroscience 143:431–444
Atochin DN, Clark J, Demchenko IT, Moskowitz MA, Huang PL (2003) Rapid cerebral ischemic preconditioning in mice deficient in endothelial and neuronal nitric oxide synthases. Stroke 34:1299–3303
Li QF, Zhu YS, Jiang H (2008) Isoflurane preconditioning activates HIF-1alpha, iNOS and Erk1/2 and protects against oxygen-glucose deprivation neuronal injury. Brain Res 1245:26–35
Thompson JW, Narayanan SV, Koronowski KB, Morris-Blanco K, Dave KR, Perez-Pinzon MA (2015) Signaling pathways leading to ischemic mitochondrial neuroprotection. J Bioenerg Biomembr 47(1–2):101–110. https://doi.org/10.1007/s10863-014-9574-8
Kitagawa K, Sasaki T, Terasaki Y, Yagita Y, Mochizuki H (2012) CREB activation is a key player for ischemic tolerance in the brain. Rinsho Shinkeigaku 52:904–907
Kapinya K, Penzel R, Sommer C, Kiessling M (2000) Temporary changes of the AP-1 transcription factor binding activity in the gerbil hippocampus after transient global ischemia, and ischemic tolerance induction. Brain Res 872:282–293
Watters O, O'Connor JJ (2011) A role for tumor necrosis factor-α in ischemia and ischemic preconditioning. J Neuroinflammation 8:87. https://doi.org/10.1186/1742-2094-8-87
Dirnagl U, Becker K, Meisel A (2009) Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 8:398–412
Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C (2007) Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 27:7083–7093. https://doi.org/10.1523/JNEUROSCI.1645-07.2007
Zhang Y, Park TS, Gidday JM (2007) Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol 292:H2573–H2581
Watson BD, Dietrich WD, Busto R, Wachtel MS, Ginsberg MD (1985) Induction of reproducible brain infarction by photochemically initiated thrombosis. Ann Neurol 17:497–504. https://doi.org/10.1002/ana.410170513
Pevsner PH, Eichenbaum JW, Miller DC, Pivawer G, Eichenbaum KD, Stern A, Zakian KL, Koutcher JA (2001) A photothrombotic model of small early ischemic infarcts in the rat brain with histologic and MRI correlation. J Pharmacol Toxicol Methods 45:227–233
Schmidt A, Hoppen M, Strecker JK, Diederich K, Schäbitz WR, Schilling M, Minnerup J (2012) Photochemically induced ischemic stroke in rats. Exp Transl Stroke Med 4:13. https://doi.org/10.1186/2040-7378-4-13
Uzdensky A, Demyanenko S, Fedorenko G, Lapteva T, Fedorenko A (2016) Protein profile and morphological alterations in penumbra after focal photothrombotic infarction in the rat cerebral cortex. Mol Neurobiol 53:1–17. https://doi.org/10.1007/s12035-016-9964-5
Uzdensky AB (2010) Cellular and molecular mechanisms of photodynamic therapy. Nauka, Sankt-Petersburg (in Russian)
Demyanenko SV, Panchenko SN, Uzdensky AB (2015) Expression of neuronal and signaling proteins in penumbra around a photothrombotic infarction core in rat cerebral cortex. Biochemistry (Mosc) 80:790–799. https://doi.org/10.1134/S0006297915060152
Demyanenko S, Uzdensky A (2016) Profiling of signaling proteins in penumbra after focal photothrombotic infarct in the rat brain cortex. Mol Neurobiol [Epub ahead of print]. doi:https://doi.org/10.1007/s12035-016-0191-x
Bretones G, Delgado MD, León J (2015) Myc and cell cycle control. Biochim Biophys Acta 1849:506–516. https://doi.org/10.1016/j.bbagrm.2014.03.013
Engelmann D, Pützer BM (2010) Translating DNA damage into cancer cell death—a roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist Updat 13:119–131. https://doi.org/10.1016/j.drup.2010.06.001
Udayakumar T, Shareef MM, Diaz DA, Ahmed MM, Pollack A (2010) The E2F1/Rb and p53/MDM2 pathways in DNA repair and apoptosis: understanding the crosstalk to develop novel strategies for prostate cancer radiotherapy. Semin Radiat Oncol 20:258–266. https://doi.org/10.1016/j.semradonc.2010.05.007
Culmsee C, Zhu Y, Krieglstein J, Mattson MP (2001) Evidence for the involvement of Par-4 in ischemic neuron cell death. J Cereb Blood Flow Metab 21:334–343
Onoue S, Kumon Y, Igase K, Ohnishi T, Sakanaka M (2005) Growth arrest and DNA damage-inducible gene 153 increases transiently in the thalamus following focal cerebral infarction. Brain Res Mol Brain Res 134:189–197
Angelo MF, Aviles-Reyes RX, Villarreal A, Barker P, Reines AG, Ramos AJ (2009) p75 NTR expression is induced in isolated neurons of the penumbra after ischemia by cortical devascularization. J Neurosci Res 87:1892–1903. https://doi.org/10.1002/jnr.21993
Zhou X, Ding Q, Chen Z, Yun H, Wang H (2013) Involvement of the GluN2A and GluN2B subunits in synaptic and extrasynaptic N-methyl-D-aspartate receptor function and neuronal excitotoxicity. J Biol Chem 288:24151–24159. https://doi.org/10.1074/jbc.M113.482000
Jaenisch N, Popp A, Guenther M, Schnabel J, Witte OW, Frahm C (2014) Pro-apoptotic function of GABA-related transcripts following stroke. Neurobiol Dis 70:237–244. https://doi.org/10.1016/j.nbd.2014.06.015
Jin J, Wang GL, Salisbury E, Timchenko L, Timchenko NA (2009) GSK3beta-cyclin D3-CUGBP1-eIF2 pathway in aging and in myotonic dystrophy. Cell Cycle 8:2356–2359
Ozaki T, Li Y, Kikuchi H, Tomita T, Iwatsubo T, Nakagawara A (2006) The intracellular domain of the amyloid precursor protein (AICD) enhances the p53-mediated apoptosis. Biochem Biophys Res Commun 351:57–63
van Lookeren CM, Gill R (1998) Increased expression of cyclin G1 and p21WAF1/CIP1 in neurons following transient forebrain ischemia: comparison with early DNA damage. J Neurosci Res 53:279–296
Su C, Sun F, Cunningham RL, Rybalchenko N, Singh M (2014) ERK5/KLF4 signaling as a common mediator of the neuroprotective effects of both nerve growth factor and hydrogen peroxide preconditioning. Age (Dordr) 36:9685. https://doi.org/10.1007/s11357-014-9685-5
Koga S, Kojima S, Kishimoto T, Kuwabara S, Yamaguchi A (2012) Over-expression of map kinase phosphatase-1 (MKP-1) suppresses neuronal death through regulating JNK signaling in hypoxia/re-oxygenation. Brain Res 1436:137–146. doi:https://doi.org/10.1016/j.brainres.2011.12.004
Hurn PD, Macrae IM (2000) Estrogen as a neuroprotectant in stroke. J Cereb Blood Flow Metab 20:631–652
Wang X, Pei L, Yan H, Wang Z, Wei N, Wang S, Yang X, Tian Q et al (2014) Intervention of death-associated protein kinase 1-p53 interaction exerts the therapeutic effects against stroke. Stroke 45:3089–3091. https://doi.org/10.1161/STROKEAHA.114.006348
Hayashi T, Sasaki C, Iwai M, Sato K, Zhang WR, Warita H, Abe K (2001) Induction of PML immunoreactivity in rat brain neurons after transient middle cerebral artery occlusion. Neurol Res 23:772–776
Larisch S, Yi Y, Lotan R, Kerner H, Eimerl S, Tony Parks W et al (2000) A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nat Cell Biol 2:915–921
Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP (2000) Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res 60:417–424
García MA, Vázquez J, Giménez C, Valdivieso F, Zafra F (1996) Transcription factor AP-2 regulates human apolipoprotein E gene expression in astrocytoma cells. J Neurosci 16:7550–7556
Posada-Duque RA, Barreto GE, Cardona-Gomez GP (2014) Protection after stroke: Cellular effectors of neurovascular unit integrity. Front Cell Neurosci 8:231. https://doi.org/10.3389/fncel.2014.00231
Clarke DM, Brown MC, LaLonde DP, Turner CE (2004) Phosphorylation of actopaxin regulates cell spreading and migration. J Cell Biol 166:901–912
Madineni A, Alhadidi Q, Shah ZA (2016) Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol Neurobiol 53:867–878. https://doi.org/10.1007/s12035-014-9056-3
Myllykoski M, Seidel L, Muruganandam G, Raasakka A, Torda AE, Kursula P (2016) Structural and functional evolution of 2′,3′-cyclic nucleotide 3′-phosphodiesterase. Brain Res 1641:64–78. https://doi.org/10.1016/j.brainres.2015.09.004
Franco M, Boretto J, Robineau S, Monier S, Goud B, Chardin P, Chavrier P (1998) ARNO3, a Sec7-domain guanine nucleotide exchange factor for ADP ribosylation factor 1, is involved in the control of Golgi structure and function. Proc Natl Acad Sci U S A 95:9926–9931
Steggerda SM, Paschal BM (2002) Regulation of nuclear import and export by the GTPase ran. Int Rev Cytol 217:41–91
Acknowledgements
The work was supported by the grant #14-15-00068 of the Russian Science Foundation (the study of single PTI) and the grant #14-04-00741 of the Russian Foundation of Fundamental Research (the study of double PTI). A. Uzdensky was supported by the Ministry of Education and Science of Russian Federation (grant #6.4951.2017/6.7). The authors used the equipment of the Center for Collective Use of Southern Federal University “High Technology” supported by the Ministry of Education and Science of Russian Federation (project RFME_FI59414X0002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Demyanenko, S.V., Uzdensky, A.B. The Focal-Focal Preconditioning Effect of Photothrombotic Impact on the Signaling Protein Profile in the Penumbra Surrounding the Ischemic Core Induced by Another Photothrombotic Impact. Mol Neurobiol 55, 229–248 (2018). https://doi.org/10.1007/s12035-017-0736-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0736-7