
Abstract
We previously demonstrated that arsenic, cadmium, and lead mixture at environmentally relevant doses induces astrocyte apoptosis in the developing brain. Here, we investigated the mechanism and contribution of each metal in inducing the apoptosis. We hypothesized participation of transcription factor, peroxisome proliferator-activated receptor gamma (PPARγ), reported to affect astrocyte survival. We treated cultured rat astrocytes with single metals and their combinations and performed apoptosis assay and measured PPARγ expression levels. We found that cadmium demonstrated maximum increase in PPARγ as well as apoptosis, followed by arsenic and then lead. Interestingly, we observed that the metals mimicked PPARγ agonist, troglitazone, and enhanced PPARγ-transcriptional activity. Co-treatment with PPARγ-siRNA or PPARγ-antagonist, GW9662, suppressed the astrocyte apoptosis, suggesting a prominent participation of PPARγ in metal(s)-induced astrocyte loss. We explored PPARγ-transcriptional activity and identify its target gene in apoptosis, performed in silico screening. We spotted PPARγ-response elements (PPREs) within poly(ADP-ribose) polymerase (PARP) gene, and through gel-shift assay verified metal(s)-mediated increased PPARγ binding to PARP-PPREs. Chromatin-immunoprecipitation and luciferase-reporter assays followed by real-time PCR and Western blotting proved PPRE-mediated PARP expression, where cadmium contributed most and lead least, and the effects of metal mixture were comparable with troglitazone. Eventually, dose-dependent increased cleaved-PARP/PARP ratio confirmed astrocyte apoptosis. Additionally, we found that PPARγ and PARP expressions were c-Jun N-terminal kinases and cyclin-dependent kinase5-dependent. In vivo treatment of developing rats with the metals corroborated enhanced PPARγ-dependent PARP and astrocyte apoptosis, where yet again cadmium contributed most. Overall, our study enlightens a novel PPARγ-dependent mechanism of As-, Cd-, and Pb-induced astrocyte apoptosis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Astrocytes regulate several key functions within the brain, particularly, blood-brain barrier (BBB) maintenance, immune responsiveness, neurotransmission, etc. [1, 2]. Increased astrocyte expression stimulates astrogliosis and inflammation [3]. Alternatively, the loss of astrocytes impedes the normal central nervous system functioning [4, 5]. Therefore, a balanced astrocyte count is essential for sustaining the healthy brain.
Arsenic (As), cadmium (Cd), and lead (Pb) are major environmental contaminants of several developing countries [6–9]. We previously proved that exposure to a mixture of As, Cd, and Pb at ground water concentrations of our country, i.e., India, caused white matter and myelin damage and prompted premature Alzheimer’s disease (AD)-like pathology, where Pb and its combinations contributed the highest [7, 10]. We also found that the metal mixture enhanced astrocyte apoptosis that triggered a decrease in astrocyte marker, glial fibrillary acidic protein (GFAP), and attenuated glial-neuronal interactions in the developing rat brain [11, 12]. However, the mechanism stimulating astrocyte apoptosis, for the individual metals and their combinations, remained less investigated.
The nuclear receptor, peroxisome proliferator-activated receptor-gamma (PPARγ), is a major regulator of cellular lipid homeostasis and participates in vital metabolic functions within the brain [13, 14]. PPARγ protein is expressed in the astrocytes [15, 16], and our earlier study revealed that As + Cd + Pb-mediated modulation in the rat brain GFAP levels involved the participation of PPARγ [12]. We further reported that PPARγ agonist, troglitazone, was capable of attenuating the GFAP levels [12]. In the primary human astrocytes, modulation of the apoptotic process has been described as an important function of PPARγ [15]. For primary murine astrocytes, contradictory observations have been presented. While two studies found the primary rat astrocytes to be relatively resistant to the cytotoxic effects of PPARγ agonists [17, 18], findings of Perez-Ortiz et al. revealed an increased PPARγ agonist, particularly glitazone-mediated apoptosis in the rat astrocytes [19]. In the current study, we checked whether As-, Cd-, and Pb-induced apoptosis in rat brain astrocytes involved the PPARγ.
PPARγ is a transcription factor, and its ligands and agonists promote PPARγ binding to specific PPAR-responsive elements (PPRE) of the target genes [20]. Astrocyte apoptosis involves modulations of key apoptotic mediators, viz., caspases, poly(ADP-ribose) polymerase (PARP), B cell lymphoma 2 (Bcl2), bcl-2-associated x protein (Bax), B cell lymphoma-extra large (Bcl-xl), bcl-2-associated death promoter (BAD), etc. [21, 22]. However, transactivating role of PPARγ in astrocyte apoptosis remains unreported. In addition, the expression and activity of PPARγ are strongly controlled by the mitogen-activated protein kinases (MAPK), particularly c-Jun N-terminal kinases (JNK) and P38 MAPK, regulating astrocyte functions [23]. We also reported that As, Cd, and Pb stimulated JNK and cyclin-dependent kinase-5 (CDK5) pathways, which resulted in an altered GFAP expression within the astrocytes [12].
In the present study, we investigated the effects of environmentally relevant doses of As, Cd, and Pb on rat astrocyte apoptosis and examined the mechanism involved. We examined the contribution of individual metals and their binary and tertiary combinations on astrocyte apoptosis. We assessed involvement of PPARγ in the apoptosis and probed its transactivating function by comparing with PPARγ agonist, troglitazone. We examined whether MAPK and CDK5 activities affected PPARγ and an ultimate apoptosis in As-, Cd-, and Pb-treated astrocytes. Overall, the present study aims at identifying the mechanism involved and relative contribution of As, Cd, and Pb in astrocyte apoptosis, specifically focusing upon the participation of PPARγ.
Materials and Methods
Reagents and Kits
Poly-L-lysine, sodium-arsenite, lead-acetate, cadmium-chloride, sodium-orthovanadate, sodium chloride, ethanol, methanol, acetone, glycerol, tris-HCl, TWEEN 20, phenylmethylsulfonyl fluoride (PMSF), bromophenol blue, isopropanol, chloroform, dithiothreitol (DTT), protease inhibitor cocktail, p-formaldehyde (PFA), bovine serum albumin (BSA), acrylamide, bis-acrylamide, agarose, nonylphenoxypolyethoxylethanol (NP-40), sodium dodecyl sulfate (SDS), MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide], 3-aminobenzamide (3-ABA), ampicillin, cell lytic reagent, and all other fine chemicals were procured from Sigma Chemical Co. (St. Louis, MO). Ponceau S stain, citric acid, sodium fluoride, and sodium citrate were purchased from Texas Biogene Inc. (Woodland, TX). Sodium bicarbonate was procured from Sisco research laboratories Pvt. Ltd. (Mumbai, India). Trypsin-EDTA, G-5 supplement, Dulbecco’s Modified Eagle medium: F12 (DMEM/F12), fetal bovine serum (FBS), phosphate-buffered saline (PBS), antibiotic-antimycotic, MAX Efficiency® DH5α™ competent cells, JNK inhibitor (SP600125), P38 MAPK inhibitor (SB203580), and protein markers were purchased from Invitrogen (Carlsbad, CA). Electrophoretic mobility shift assay (EMSA) kit, Biotin 3′ End Labeling kit, nuclear and cytoplasmic extraction kit (NE-PER), Biodyne B Nylon membrane, and SuperSignal West Femto Maximum Sensitivity Substrate were purchased from Thermo Scientific (Rockford, IL). PPARγ agonist, troglitazone (Trog), PPARγ inhibitor (GW9662), CDK5 inhibitor (R-CR8), and caspase inhibitor (Z-VAD-FMK) were purchased from Tocris biosciences (Bristol, United Kingdom). In Situ Cell Death Detection Kit, Fluorescein for terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was purchased from Roche (Mannheim, Gemany). Chromatin Immunoprecipitation (ChIP) kit, ApopNexin™FITC Apoptosis detection kit, Bicinchoninic acid (BCA) protein assay kit, propidium iodide (PI), and polyvinylidenefluoride (PVDF) membrane were procured from Millipore (Temecula, CA). PPARγ-siRNA and non-targeting (NT)-siRNA were purchased from Dharmacon (Dharmacon Inc., Lafayette, CO). Plasmid midi kit, mini elute gel extraction kit, DNA ladder, and gel loading dye were from Qiagen (Valencia, CA). The pM vector, pM-PPARγ, Gal4-tk-Luc, and pCMX-PPARγ plasmids were kindly gifted by Dr. S. Sanyal (Drug Target Discovery and Development Division, CSIR-Central Drug Research Institute, Lucknow, INDIA). Amaxa™ Basic Glial Cells Nucleofector™kit and pmaxGFP™ were procured from Lonza (Allendale, NJ). T4 DNA ligase, KpnI, and SacI restriction enzymes were procured from New England Biolabs (Ipswich, MA). Steady-Glo Luciferase Assay System was procured from Promega Corporation (Madison, WI).
Antibodies
Rabbit polyclonal PPARγ (cat no. ab19481) and TATA-binding protein and TBP (cat no. ab63766) antibodies were purchased from Abcam (Cambridge, MA). Rabbit monoclonal PARP (cat no. 9532), rabbit polyclonal cleaved PARP (cat no. 9545), rabbit polyclonal cleaved caspase-3 (cat no. 9661), rabbit monoclonal cleaved caspase-7 (cat no. 8438), and normal rabbit IgG (cat no. 2729) antibodies were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal β-actin, peroxidise-conjugated secondary IgG rabbit (cat no. A0545), and peroxidise-conjugated secondary IgG mouse (cat no. A9044) antibodies were procured from Sigma Chemical Co. (St. Louis, MO). Mouse monoclonal GFAP (cat no. MAB360), rabbit polyclonal Histone H3 (cat no. 06-755), and anti-acetyl Histone H3 (cat no. 06-599) antibodies were purchased from Millipore (Temecula, CA).
Primary Astrocyte Culture
Astrocytes from the rat brain were isolated and cultured as described previously [12]. Briefly, cerebral cortices of 1-day-old neonatal rat pups were dissected and gently transferred to a Petri dish containing PBS and rinsed in serum-free modified DMEM/F12 culture medium. After removal of meninges, the cortices were dissociated into a cell suspension by using 10- and 2-ml syringe needles and digested with 0.05% trypsin/EDTA at 37 °C for 15 min. Cells were re-suspended in DMEM/F12 with 10% heat-inactivated FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin and passed through 80 μm nylon mesh followed by centrifugation at 1000 rpm for 10 min. The pellet obtained was suspended in complete (DMEM)/F12 media, and cells were seeded on poly-L-lysine-coated culture flasks and grown until confluent in humidified CO2 incubator at 37 °C with 5% CO2–95% air. At the third day, cells were supplemented with G5 growth supplement. The culture flasks were shaken on orbital shaker at 200 rpm and 37 °C for 18 h to detach loosely attached microglia and oligodendrocytes. Immediately after shaking, the cells were washed with PBS and fresh culture medium added. The confluent cells were rinsed with PBS and then detached with 0.05% trypsin/EDTA and sub-cultured. Purity of astrocyte culture was confirmed through GFAP immunostaining (Suppl. 1A).
MTT Assay
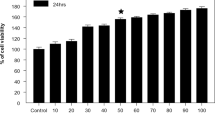
Astrocyte viability following As, Cd, and Pb treatments was assessed through MTT assay. The 70–80% confluent astrocytes were pre-incubated (2 h) in reduced serum (0.5% FBS) medium and then incubated with As, Cd, or Pb at a concentration range of 0.01 to 200 μM for 18 h in humidified CO2 incubator at 37 °C with 5% CO2–95% air. The astrocytes were incubated with MTT (10.4 mg/ml) and cell viability determined following a standardized protocol [11]. Absorbance was determined at 595 nm, and the background at 655 nm was subtracted. The lethal concentration-50 (LC-50) of As, Cd, and Pb on astrocytes was calculated using GraphPad Prism 3.0 software (Table 1).
Astrocyte Treatments
Astrocyte treatments, in vitro, have been provided in Table 2.
The 70–80% confluent rat primary astrocytes were pre-incubated in reduced serum medium for 2 h. The astrocytes were then treated for 18 h with As, Cd, and Pb at LC-5, LC-10, and LC-15, individually and in mixtures. The LC-5 values closely matched the metal concentrations reaching the brain (reported earlier) [11] upon treating developing rats with environmentally relevant doses of the metals, i.e., the in vivo treatment doses (Table 3). The astrocytes were treated with troglitazone (10 μM) for 18 h [19]. To assess the partici-pation of PPARγ in astrocyte apoptosis, the cells were treated with PPARγ antagonist, GW9662 (10 μM) [24, 25] or silenced with PPARγ-siRNA. To examine the participation of PARP and caspases in astrocyte apoptosis, the astrocytes were treated with respective inhibitors, 3-aminobenzamide (1 mM) [26, 27] and Z-VAD-FMK (50 μM) [28]. The involvement of kinase molecules like P38 MAPK, JNK, and CDK5 in PPARγ and PARP expressions was determined by pre-incubating the astrocytes with SB203580 (10 μM), SP600125 (10 μM), and (R)-CR8 (0.25 μM) that are inhibitors to P38 MAPK, JNK, and CDK5, respectively.
Annexin V/PI Assay
Analysis of apoptosis was performed using the ApopNexin™FITC Apoptosis Detection kit and according to the manufacturer’s instructions, following an earlier published protocol [29]. Briefly, cells were trypsinized and centrifuged at 1000 rpm for 5 min. The cell pellets were washed twice with ice-cold PBS and re-suspended in ice-cold 1× binding buffer. Annexin conjugate ApopNexin™ FITC reagent was added to the cell suspension, followed by the addition of PI reagent. The mixture was incubated on ice for 15 min in dark. FACS analysis was performed on a BD FACScanto II Flow cytometer (BD Biosciences, San Jose, CA). For each experiment, 10,000 cells per sample were analyzed.
Protein Extraction and Western Blotting
Protein extraction and Western blotting of astrocyte cells were performed as described before [11]. Briefly, after specific treatments, cells were washed in cold PBS and lysed with CellLytic MT mammalian cell lysis buffer containing protease inhibitor cocktail and 1 mM DTT and kept on ice for 10 min. Cells were scraped using cell scraper, centrifuged at 15,000 rpm for 15 min, and clear supernatant was collected. Protein concentration was determined through BCA protein assay, as per manufacturer’s recommended protocol. SDS-PAGE and Western blotting of samples were then performed as described earlier [12]. Briefly, protein samples were prepared at 5-μg/μl concentration with sample loading dye and denatured at 85 °C for 15 min. Equal amounts of protein (50–80 μg) were loaded on 10–12% tris-glycine polyacrylamide gel and run in running buffer at 100 V until bands were properly separated. After electrophoresis, gels were transferred onto activated PVDF membrane (methanol activated) in semi-dry transfer assembly at 16 V for 90 min. Membranes were then blocked in 5% non-fat milk or 5% BSA in TBST (Tris buffer [10 mM Tris, pH 8.0, 150 mM NaCl, 0.01% Tween 20] for 1 h and washed with TBST. Membranes were incubated overnight at 4 °C with PPARγ (1:1000), PARP (1:2000), cleaved PARP (1:2000), cleaved caspase-3 (1:3000), cleaved caspase-7 (1:3000), TBP (1:5000), and β-actin (1:10,000) antibodies. Membranes were then washed three times with TBST and incubated for 2 h with horseradish peroxidise-conjugated secondary antibody. Proteins were visualized through Amersham Imager 600 (GE Healthcare Life Sciences, Pittsburgh, PA) using Supersignal West Femto Maximum Sensitivity Substrate. Protein expression relative to β-actin was quantified using Bio-Rad Quantity One image analysis software (Bio-Rad, Hercules, CA).
PPARγ-siRNA and pCMX-PPARγ Plasmid Transfection
To examine the participation of PPARγ in astrocyte apoptosis and to verify the specificity of PPARγ antibody, we transfected the astrocytes with PPARγ-siRNA or NT-siRNA and examined PPARγ levels through Western blotting. To re-validate the specificity of PPARγ antibody, we transfected the astrocytes with pCMX-PPARγ plasmid, which served as a positive control as described before [30]. The rat primary astrocytes (70–80% confluent) were transfected with NT-siRNA (25 nM), PPARγ-siRNA (25 nM), or pCMX-PPARγ plasmid (2 μg) using TurboFect reagent as per manufacturer’s instructions and as the previously described protocol for transfection [7]. Briefly, 70–80% confluent astrocytes were incubated in serum-free medium for 2 h. The siRNA, pCMX-PPARγ plasmid, and TurboFect reagents along with pmax-GFP plasmid were suspended in serum-free media for 30 min at 37 °C and allowed to form a complex. The complex was then incubated with astrocytes in a serum-free medium overnight, and medium was replaced with complete media for another 24 h. We detected attenuated PPARγ (Suppl. 1B) and increased PPARγ expressions (Suppl. 1C) using siRNA and pCMX-PPARγ plasmid, respectively.
Isolation of Nuclear and Cytoplasmic Proteins
Nuclear and cytosolic fractions from astrocytes cells were isolated using NE-PER reagents (Pierce-ThermoScientific), as per manufacturer’s protocol. Briefly, 70–80% confluent astrocyte cells were scraped and washed in cold PBS, centrifuged at 3000 rpm and 4 °C for 5 min. The supernatant was discarded, and pellets were re-suspended in cytoplasmic extraction reagen-1, vortexed five times for 15 s at 10-min intervals, and incubated on ice. Cytoplasmic extraction reagent 2 was then added, vortexed for 15 s, and incubated on ice, followed by centrifugation for 10 min at 14,000 rpm at 4 °C. The centrifuged pellet was suspended in ice-cold nuclear extraction reagent, vortexed for 15 s, incubated on ice, and re-vortexed four times at 10-min intervals for 40 min, followed by centrifugation at 14,000 rpm for 10 min. Supernatant containing the nuclear extract was transferred to a clean pre-chilled micro-centrifuge for further use. The nuclear extract was then used for Western blotting and EMSA.
Immunocytochemistry
Cultured astrocytes were treated with metals or troglitazone, and immunocytochemistry was performed as described earlier [31]. Treated primary astrocytes were washed with PBS and fixed with 4% PFA. Cells were then permeabilized with methanol. After blocking with 5% serum, astrocytes were incubated overnight with PPARγ and GFAP (1:200) primary antibodies. Following three washings, cells were incubated with Alexa Fluor goat anti-rabbit IgG and anti-mouse IgG-conjugated secondary antibodies for 2 h followed by counterstaining with Hoechst 33258 (0.2 mM). Astrocytes were then mounted in VECTASHIELD medium (Vector Laboratories, Burlingame, CA), and photomicrography performed under a fluorescence microscope (Nikon Instech Co. Ltd., Kawasaki, Kanagawa, Japan). Images were imported into Image-J 1.42q (http://rsb.info.nih.gov/ij/., WayneRasband, National Institutes of Health, Bethesda, MD) for image analysis.
PPARγ Trans-activation Function Through Luciferase Reporter Assay
To assess the PPARγ trans-activation function, astrocytes were transfected with pM-PPARγ (0.1 μg) or pM vector (0.1 μg) along with Gal4-responsive tk-Luc reporter (Gal4-tk-Luc) (0.1 μg) as previously described [32, 33]. The pmax-GFP plasmid was co-transfected in the astrocytes for normalizing the transfection efficiency [34]. Luciferase assay was then performed with the transfected cells using Steady-Glo-Luciferase assay Kit as per manufacturer’s instructions. Briefly, the cells were equilibrated to room temperature, media aspirated from the cells, and the cells were then washed with PBS. Luciferase substrate (100 μl) was added to each well and incubated for 5–10 min for sufficient cell lysis. The luciferase activity was determined by measuring luminescence in a Micro-plate reader (Tecan, infinite 200 PRO, Switzerland).
Identification of PPRE Sequences
The PPRE sequences on target genes were identified using NUBIScan, an in silico approach for predicting nuclear receptor response elements as described before [12, 35]. Briefly, major apoptotic genes, such as PARP (Gene ID-25591, NC_005112.4), BAX (Gene ID: 24887, NC_005100.4), BAD (Gene ID: 64639, NC_005100.4), BCl-2 (Gene ID: 24224, NC_005112.4), BCL-XL (Gene ID: 24888, NC_005102.4), caspase-3 (Gene ID: 25402, NC_005115.4), and caspase-7 (Gene ID: 64026, NC_005100.4) were scanned. The PPRE sequences on PARP gene were selected based on P values (Table 4).
Gel-Shift Assay
EMSA was performed using a Light Shift Chemiluminescent EMSA Kit as per manufacturer’s instructions and following a previously published protocol [12]. Repeat sequences of direct repeat-5 (DR5) and inverted repeat-3 (IR3) oligonucleotides (Table 5) were synthesized by Eurofins MWG Operon (Huntsville, AL). Biotin labeling of oligonucleotides was performed using Biotin 3′ End Labeling kit. Briefly, 5 μg of nuclear protein was incubated with 50-fmole biotin labeled oligonucleotide probes in a 20-μl reaction buffer, containing 50 ng/μl poly dI.dC, 5 mM MgCl2, 0.05% NP40, and 1× binding buffer, for 30 min at room temperature. For competition experiments, unlabeled oligonucleotides were added at 100-fold molar excess. For super-shift assays, nuclear extracts were pre-incubated with anti-PPARγ for 1 h at 4 °C, followed by addition of biotin-labeled oligonucleotide probes. After incubation, the sample loading dye was added to the reaction mixture and subjected to native PAGE on 6% TBE gel at 90 V, and then, the protein-DNA complex was transferred onto Biodyne B nylon membrane. The transferred labeled DNA was then cross-linked to the membrane by exposure to 254 nm UV radiation on a UV transilluminator for 1 min. The cross-linked membrane was blocked in blocking solution for 15 min with gentle shaking, incubated in conjugated/blocking solution, and supplemented with stabilized streptavidin-HRP conjugate for 15 min. The membrane was then washed in 1× washing buffer four times for 5 min, incubated in substrate equilibration buffer and then in substrate working solution (equal amount of luminal/enhancer solution and stable peroxide solution) for 5 min, and visualized using chemiluminescence on Amersham Imager 600 (GE Healthcare Life Sciences).
ChIP Assay
ChIP assay was carried out using its specific kit according to the manufacturer’s instructions, and following a previously published protocol [12]. Briefly, 1–2 × 106 astrocyte cells were cross-linked with 1% formaldehyde at 37 °C for 10 min. Cross-linking reactions were then attenuated by the addition of 12 mM glycine. Cells were then washed twice with ice-cold PBS, scraped, and centrifuged at 2000 rpm for 4 min. The cells were then re-suspended in cell lysis buffer (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris (pH 8.0), 5 mM EDTA) with protease inhibitors (1 μg/ml aprotinin, 1 μg/ml pepstatin A, and 1 mM PMSF) and kept on ice for 15 min. Cell lysates were sonicated to yield chromatin fragments of approximately 200–1000 base pairs and centrifuged at 13,000 rpm for 10 min at 4 °C. The sonicated supernatant was diluted by 10-fold in the ChIP dilution buffer. One percent of the diluted supernatant was kept as input/starting material (input represents PCR amplification of the total sample). The supernatant was pre-cleared with protein A agarose/salmon sperm DNA (50% slurry) for 30 min at 4 °C, centrifuged, and supernatant fraction was collected. Further, the supernatant fractions were immunoprecipitated with anti-PPARγ antibody or anti-acetyl histone H3 antibody (positive control) or normal rabbit IgG1 (negative control) overnight at 4 °C. Protein A agarose/salmon sperm DNA (50% slurry) was added to the precipitate and incubated for 1 h at 4 °C with rotations to collect the antibody/histone complex. The complex was then centrifuged at 1000 rpm for 1 min at 4 °C and washed with wash buffer for 5 min. DNA fraction was eluted by adding freshly prepared elution buffer (1% SDS and 0.1 M NaHCO3). To reverse the formaldehyde cross-linking, 5 M NaCl was added in eluted fraction and incubated at 65 °C for 4 h. Eluted fraction containing DNA fragments was purified by phenol-chloroform extraction, followed by ethanol precipitation, and then dissolved in DNAse and RNAse free water. Purified DNA was amplified using specific primers (Table 6) and quantified by qPCR using SYBR Green PCR master mix in a PRISM 7900 HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA).
Plasmid Construct Generation, Transient Transfection, and Luciferase Reporter Assay
The designed three repeat sequences of DR5 and IR3 (Table 7) were cloned into pGL3-basic promoter plasmid vectors (Promega Corporation, Madison, WI) at KpnI-SacI restriction site, following the protocol described before [12, 33]. Briefly, pGL3-basic promoter plasmid vector was double digested with KpnI and SacI restriction enzymes at 37 °C for 2 h. DR5 and IR3-PPRE sequences were cloned into digested pGL3-basic promoter plasmid vector in a ratio of (3:1) by T4 DNA ligase and incubated at 16 °C overnight. The pGL3-DR5 and pGL3-IR3 plasmid constructs were amplified using competent DH5α bacterial cells (Invitrogen, Carlsbad CA) and transformed in ampicillin-containing LB media. Finally, the DNA construct was isolated using Qiagen Plasmid Midi kit, according to the manufacturer’s protocol. The integrity of plasmid DNA was validated by running on 1% agarose gel, and band size was confirmed using KpnI and SacI restriction enzymes.
The pGL3-DR5 or pGL3-IR3 plasmid constructs were co-transfected with pmaxGFP™ into rat primary astrocytes using Nucleofector™ Solution in an Amaxa electroporator (AmaxaBiosystems, Gaithersburg, MD) as described before [12]. Briefly, 70–80% confluent primary astrocytes were trypsinized and centrifuged at 1000 rpm for 5 min. The cell pellets were carefully re-suspended in 100-μl room temperature Nucleofector™ Solution. Then, 2 μg of pGL3-DR5, pGL3-IR3, or empty pGL3 vector was electroporated along with pmaxGFP™ using the rat primary astrocyte program specific for the Amaxa Nucleofactor System. After 24 h, the medium was removed and fresh media added. On the second day, 70% transfection efficiency was obtained (data not shown). Luciferase assay was then performed on the transfected cells using Steady-Glo Luciferase Assay System (Promega Corporation, Madison, WI).
qPCR
PARP and GAPDH primer pairs for qPCR were designed using Primer Express version 2.0.0 (Applied Biosystems) (Table 8). Astrocyte RNA was isolated in Trizol reagent (Invitrogen), precipitated by isopropanol, washed with 70% ethanol, and dissolved in nuclease-free water. Then, the quantity and purity of mRNA were determined by UV spectrophotometer and subjected to complementary DNA synthesis using the primer pairs and Omniscript RT Kit (Qiagen, Valencia, CA) as described before [36]. qPCR was carried out using SYBR Green dye over 40 cycles of sequential reactions of 95 °C (10 min), 95 °C (10 s), 60 °C (30 s), and 72 °C (30 s), and mRNA levels quantified in the Real-Time PCR System. Relative expression was calculated using the ΔΔCt method.
Animals and Treatments
Rat treatments, in vivo, have been provided in Table 3.
Guidelines of the Institute (CSIR-IITR) Animal Ethics Committee were followed for animal handling. Timed pregnant Wistar rats were housed in a 12-h day and light cycle environment with the ad libitum availability of diet and water. The dams were treated via oral gavage with As, Cd, Pb, or their tertiary mixture (As + Cd + Pb) at ground water relevant doses of India [6, 7, 11]. The treatment started from gestation-05 (G-05) and continued daily during lactation until postnatal day 16 (PND16) [11]. The rat pups were treated with troglitazone or GW9662 through intraperitoneal injection from PND01 until PND16 [12, 37]. The rat brain cortical tissues were isolated from PND16 rats, as described earlier [12].
Immunohistochemistry
Immunohistochemistry of rat brain cortical tissues was performed following the protocol described before [10]. Briefly, rats were anesthetized and perfused with 4% PFA and 0.2% picric acid in PBS. The whole brain was then isolated and cryoprotected in 25% sucrose. Five-micron sections were made from the cerebral cortex using cryomicrotome (Microm HM 520; Labcon, Munich, Germany). The sections were mounted on 3-aminopropyl-triethoxy-silane-coated slides, antigen-retrieved with citrate buffer, and blocked with 5% BSA. Sections were then probed with PARP or cleaved PARP and GFAP (1:200) antibodies overnight. Sections were then washed thrice in PBS, incubated with Alexa Fluor secondary antibodies, and counterstained with Hoechst 33258 (0.2 mM). Slides were then washed thrice in PBS, dried, mounted in VECTASHIELD mounting medium, and visualized under fluorescence microscope (Nikon Instech Co. Ltd., Kawasaki, Kanagawa, Japan) using 60× objectives. Images were captured using Image-Pro Plus 5.1 software (Media Cybernetics Inc., Silver Spring, MD) and imported into ImageJ 1.42q for image analysis. Fluorescence immunoreactivity quantification was carried out using an RGB plugin of the ImageJ 1.43q software (http://rsb.info.nih.gov/ij/; developed by Wayne Rasband, National Institutes of Health, Bethesda, Maryland) as described earlier [38].
TUNEL Assay
In situ detection of apoptosis was carried out by TUNEL assay as described before [11]. For this, PND16 rats were anesthetized, perfused with 4% PFA, 0.2% picric acid in PBS. The whole brain was then isolated and cryoprotected in 25% sucrose. Five-micron sections were made from the cerebral cortex using cryomicrotome. The sections were mounted on 3-aminopropyl-triethoxy-silane-coated slides and antigen-retrieved with citrate buffer. For TUNEL assay, a labeling reaction was carried out with fluorescein-labeled dUTP in the presence of TdT at 37 °C overnight. Sections were immunostained with GFAP (1:200 dilution in PBS) antibodies and kept overnight. Sections were then washed thrice in PBS and incubated with Alexa Fluor goat anti-rabbit (546) secondary antibody. Slides were then washed in PBS, counterstained with Hoechst 33258 (0.2 mM), dried, mounted in VECTASHIELD mounting medium, and visualized under a fluorescence microscope (Nikon Instech Co. Ltd., Kawasaki, Japan) using 20× objectives. Images were captured using Image-Pro Plus 5.1 software (Media Cybernetics Inc., Silver Spring, MD) and imported into ImageJ 1.42q for image analysis. The TUNEL-positive cells were counted in five randomly selected fields. The apoptotic index was expressed as the number of TUNEL-positive cells per 100 nuclei (Hoechst stained) [39].
Statistics
Statistical analyses were performed using GraphPad PRISM software (GraphPad software, Inc.). Comparisons between two groups of independent samples were carried out using two-tailed and unpaired Studentʼs t tests and for more than two groups by ANOVA using Bonferroni post hoc test.
Results
Cd and Its Combinations Contribute Most in As + Cd + Pb-Induced Astrocyte Apoptosis
We previously reported that exposure to a mixture of As, Cd, and Pb, at environmentally relevant doses, enhanced apoptosis in astrocytes of the rat brain [11]. Here, we assessed the comparative effects of individual metals (As, Cd, or Pb) and their binary and tertiary combinations on astrocyte apoptosis.
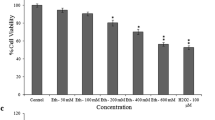
We treated rat primary astrocytes with As, Cd, and Pb at their LC-5, LC-10, and LC-15 doses (Tables 1 and 2) and measured astrocyte apoptosis using Annexin V/PI stains. The LC-5 values closely matched the metal concentrations (as reported earlier [11]) within the As-, Cd-, and Pb-treated developing rat brain, at the environmentally relevant doses. The higher values (LC-10 and LC-15) were selected for studying dose-dependent effects. We detected that the individual metals caused a dose-dependent increase in astrocyte apoptosis, in the order of Cd > As > Pb (Fig. 1a). Accordingly, binary combinations of Cd, such as As + Cd and Cd + Pb, were more apoptotic than As + Pb (Fig. 1b). Moreover, As being more apoptotic than Pb, As + Cd had a higher impact on astrocyte apoptosis compared to Cd + Pb (Fig. 1b). Ultimately, the effect of As + Cd + Pb on astrocyte apoptosis was much greater compared to individual metals and their binary combinations (Fig. 1c).

As, Cd, and Pb induce dose-dependent astrocyte apoptosis individually and in binary and tertiary mixtures, where Cd contributes most. Rat primary astrocytes that were pre-incubated in reduced serum medium and then treated with vehicle (water, V), As, Cd, Pb, or their binary and tertiary mixtures at indicated doses for 18 h. The cells were double stained with annexin V-FITC and PI, and data were assessed by flow cytometry. a Data shown in graph (LHS) and bar diagram (RHS) represent percentage of annexin V-FITC/PI-positive cells for individual As, Cd, and Pb. Graph is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001 and ** P < 0.01 indicate significant difference compared to V. c P < 0.001, b P < 0.01, and a P < 0.05 indicate significant difference compared to its immediate previous dose. $$$ P < 0.001, $$ P < 0.01, and $ P < 0.05 upon Cd indicate significant difference compared to As for the same LC values. $ P < 0.05 upon As indicates significant difference compared to Pb for the same LC values. b Data shown in graph (LHS) and bar diagram (RHS) represent percentage of annexin V-FITC/PI-positive cells for binary As, Cd, and Pb mixtures. Graph is representative of three independent experiments and bar diagram data represent means ± SE of the three independent experiments.*** P < 0.001 and ** P < 0.01 indicate significant difference compared to V. c P < 0.001 and b P < 0.01 indicate significant difference compared to its immediate previous dose. $ P < 0.05 and $$ P < 0.01 upon As + Cd indicate significant difference compared to Cd + Pb at the same LC. $$ P < 0.01 and $$$ P < 0.001 upon Cd + Pb indicate significant difference compared to As + Pb at the same LC. c Data shown in graph (LHS) and bar diagram (RHS) represent percentage of annexin V-FITC/PI-positive cells for tertiary mixtures. Graph is representative of three independent experiments and bar diagram represents means ± SE. *** P < 0.00 indicates significant difference compared to V. c P < 0.001 indicates significant difference compared to its immediate previous dose
As, Cd, and Pb Enhance PPARγ Expression That Induces Astrocyte Apoptosis
We investigated the mechanism of As-, Cd-, and Pb-induced astrocyte apoptosis, particularly focusing upon PPARγ that is reported to participate in the apoptotic process of astrocytes [15, 19]. Examining the effect on PPARγ through Western blotting revealed that individual metals and their mixtures at all three doses, LC-5 (Fig. 2a), LC-10 (Fig. 2b), and LC-15 (Fig. 2c), enhanced PPARγ expression levels. Akin to apoptosis data, the increased PPARγ was in the order of Cd > As > Pb. Combining and re-plotting data from Fig. 2a–c revealed that the metal-induced PPARγ was dose-dependent (Fig. 2d). To understand whether enhanced PPARγ participated in astrocyte apoptosis, we co-treated the As + Cd + Pb with PPARγ-siRNA. Suppressing As+Cd+Pb-induced PPARγ (using the PPARγ-siRNA) to levels close to the vehicle (Suppl. 2) caused a reduction in the annexin V/PI positive cell count (Fig. 2a), indicating increased PPARγ to be participating in astrocyte apoptosis.

As, Cd, and Pb, individually and in binary and tertiary mixtures, induce dose-dependent PPARγ and a PPARγ-dependent apoptosis, where Cd contributes most. Rat primary astrocytes that were pre-incubated in reduced serum medium and then treated with vehicle (water, V), As, Cd, Pb, or their binary and tertiary mixtures at LC-5, LC-10, and LC-15 doses for 18 h. Cell lysates were immunoblotted for PPARγ and β-actin. a, b, c Representative Western blots show PPARγ and β-actin expressions in rat primary astrocytes following individual metal and metal mixture treatments at indicated doses. d Densitometric representation (Western blot data for different doses of a, b, c combined and replotted) reveals dose-dependent increase in PPARγ expressions normalized with β-actin for individual metals, binary and tertiary metal mixture treatments.Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to V. c P < 0.001, b P < 0.01, and a P < 0.05 indicate significant difference compared to immediate previous dose. $$$ P < 0.001, $$ P < 0.01, and $ P < 0.05. # P < 0.001 indicates significant difference compared to all other treatments. e Rat primary astrocytes were pre-incubated in reduced serum medium and then transfected with PPARγ-siRNA or NT-siRNA. The cells were treated with As + Cd + Pb at LC-15 doses for 18 h and double stained with annexin V-FITC and PI. Data shown in graph (LHS) and bar diagram (RHS) demonstrate a reduced percentage of annexin V-FITC/PI staining following PPARγ-siRNA treatment compared to NT-siRNA in As + Cd + Pb-treated astrocytes. NT-siRNA and PPARγ-siRNA transfections in the vehicle-treated cells showed no change. Graph is representative of three independent experiments and bar diagram represent means ± SE. *** P < 0.001
As, Cd, and Pb Like Troglitazone Activate PPARγ
PPARγ is a nuclear receptor and functions as transcription factor [40, 41], and we assessed whether As, Cd, and Pb had any effect on the transactivating function of PPARγ. We compared the effects with PPARγ agonist, troglitazone, that enhances transcriptional activity of PPARγ in astrocytes [12, 42]. We identified increased nuclear levels of PPARγ through Western blotting for As + Cd + Pb as well as troglitazone (Fig. 3a). Immunocytochemistry data corroborated enhanced PPARγ expression in the nucleus (Fig. 3b) (because the vehicles for As + Cd + Pb and troglitazone in Fig. 3a showed no difference in their nuclear levels of PPARγ, a common vehicle has further been used for the two treatments). For proving the transactivating function of PPARγ, we performed a luciferase reporter assay using PPARγ-specific GAL4-luc system, where the wild-type PPARγ-ligand binding domain was fused to the Gal4 DNA-binding domain and the luciferase reporter gene was controlled by GAL4-binding elements, as described before [43]. We transfected astrocytes with the constructs and, upon treatment with As + Cd + Pb or troglitazone, detected increased luciferase activity for both. Thus, our data demonstrate that the metals and troglitzone enhance the transactivation function of PPARγ (Fig. 3c).

As, Cd, and Pb, like troglitazone, upregulate nuclear levels of PPARγ and enhance the PPARγ transactivation function. Rat primary astrocytes that were pre-incubated in reduced serum medium and then co-treated with As + Cd + Pb at LC-15 doses of the metals or troglitazone (Trog, 10 μM) and their vehicles, V As+Cd+Pb and VTrog, respectively, for 18 h. a Nuclear lysates from the astrocytes were immunoblotted for PPARγ and TATA binding protein (TBP). Representative Western blot and densitometric analysis show increased nuclear levels of PPARγ normalized with TBP for As + Cd + Pb and troglitazone treatments compared to respective vehicles. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001 indicates significant difference compared to respective vehicle. b Cultured rat primary astrocytes were co-immunolabeled for PPARγ and GFAP and counter-stained with nuclear Hoechst. Representative fluorescence photomicrograph (40× magnification) show PPARγ (red fluorescence), GFAP (green fluorescence), Hoechst (blue fluorescence), and their merged images in the same field. c Rat primary astrocytes were transfected with pM-Gal4-tk vector (control vector) or pM-PPARγ-Gal4-tk followed by co-treatment with vehicle (water, V), or As + Cd + Pb at LC-15 doses of the metals and troglitazone (Trog, 10 μM) for 18 h. Data indicate that As + Cd + Pb and troglitazone enhanced luciferase activity for pM-PPARγ-Gal4-tk compared to the pM-Gal4-tk vector. Data represent means ± SE of three independent experiments. *** P < 0.001 indicates significant difference compared to V
Activated PPARγ Promotes Astrocyte Apoptosis
To assess participation of the activated PPARγ in astrocyte apoptosis, we co-treated the metal mixture with a PPARγ antagonist, GW9662. We found that GW9662 attenuated As + Cd + Pb-induced astrocyte apoptosis (Fig. 4a). Interestingly, we found that troglitazone also stimulated apoptosis in the astrocytes (Fig. 4a), and co-treatment with GW9662 blocked the increased annexin V/PI-positive staining, proving involvement of activated PPARγ in astrocyte apoptosis (Fig. 4a). We further observed that PPARγ-siRNA decreased the troglitazone-induced percentage of annexin V/PI-positive astrocytes (Fig. 4b), validating participation of increased PPARγ in the troglitazone-mediated astrocyte apoptosis.

As, Cd, and Pb, like troglitazone, induce activated PPARγ-mediated astrocyte apoptosis. a Rat primary astrocytes were pre-incubated in reduced serum medium and PPARγ antagonists, GW9662 (10 μM). The cells were then co-treated with vehicle (water, V), As + Cd + Pb at LC-15 doses of the metals, or troglitazone (Trog, 10 μM) for 18 h. The cells were double stained with annexin V-FITC and PI followed by flow cytometry assay. Data shown in graph (LHS) and bar diagram (RHS) represent GW9662-mediated reduction in the annexin V-FITC/PI staining of As + Cd + Pb and troglitazone. Graph is representative of three independent experiments and bar diagram represent means ± SE. *** P < 0.001. b Rat primary astrocytes were pre-incubated in reduced serum medium and then transfected with PPARγ-siRNA or NT-siRNA. The cells were treated with Trog (10 μM) for 18 h and double stained with annexin V-FITC and PI. Data shown in graph (LHS) and bar diagram (RHS) demonstrate a reduced percentage of annexin V-FITC/PI staining following PPARγ-siRNA treatment compared to NT-siRNA in Trog-treated astrocytes. Graph is representative of three independent experiments and bar diagram represent means ± SE. *** P < 0.001. C-D. Rat primary astrocytes were pre-incubated in reduced serum medium and PPARγ antagonists, GW9662 (10 μM). The cells were then co-treated with vehicle (water, V), As, Cd, Pb, As + Cd, Cd + Pb, or As + Pb at LC-15 doses of the metals for 18 h. The cells were double stained with annexin V-FITC and PI followed by flow cytometry assay. Data shown in graph (LHS) and bar diagram (RHS) represent GW9662-mediated reduction in the annexin V-FITC/PI staining of individual metals (c) and binary metal mixture (d)-treated astrocytes. Graph is representative of three independent experiments and bar diagram represent means ± SE. *** P < 0.001
We then examined whether activated PPARγ was responsible for the apoptosis induced by the individual metals and their binary combinations. We detected that apoptosis induced by individual As, Cd, and Pb (Fig. 4c) and their binary combinations (Fig. 4d) could as well be blocked by GW9662.
PPARγ Binds to PPRE Sequences on PARP Gene
Recognizing the increased As + Cd + Pb-mediated PPARγ activation in astrocytes, we performed an in silico screening using the NUBIScan software [35] to identify the possible target genes of PPARγ in the apoptotic pathway. We spotted that the PARP gene contained potential PPREs (Table 4), direct repeat-5 (DR5) (Fig. 5a), and inverted repeat-3 (IR3) sequences (Fig. 5b), indicating that PARP may be a target of activated PPARγ (PPRE sequences of other apoptotic genes (caspases, BAX, BCl2, Bcl-Xl, etc.) demonstrated very low significance for PPARγ binding (data not shown)). To assess whether PPARγ binds to the PARP gene as its target, we carried out gel electrophoretic mobility shift assays of astrocyte nuclear lysates using biotin-labeled DR5 and IR3 probes (Table 5). We found that As + Cd + Pb and troglitazone triggered a shift in DNA bands for both DR5 (Fig. 5c) and IR3 (Fig. 5d), suggesting increased DNA-protein complex formation. Additionally, incubating the astrocyte nuclear lysates with PPARγ-antibody (Ab) caused a super-shift (ss), and the competition experiment (CE) using excess unlabeled probes exhibited an elimination of the shifted band (Fig. 5c, d). Thus, our data prove that like troglitazone, As, Cd, and Pb induce a PPARγ binding to DR5 and IR3-PPRE sequences on PARP gene. To understand the comparative participation of individual metals and binary and tertiary mixtures in this PPARγ-PARP-PPRE binding, we performed ChIP followed by qPCR assay using DR5 and IR3 primers and acetyl-histone H3 as a positive control (Table 6). We observed that all three single metals, with an intensity in order of Cd > As > Pb, showed an increased fold enrichment for DR5 (Fig. 5e) and IR3 ChIP-qPCR products (Fig. 5f) (calculated against an IgG negative control and input; refer to “Materials and Methods”). Between binary combinations, the effects were as As + Cd > Cd + Pb > As + Pb, and tertiary As + Cd + Pb and troglitazone demonstrated a comparable fold enrichment for ChIP-qPCR (Fig. 5e, f). Through luciferase reporter assay, by transfecting the astrocytes with pGL3-promoter vector-based constructs of DR5 and IR3-PPRE (Table 7), we verified metal(s)-mediated enhanced PARP-PPRE-driven luciferase activity (Fig. 5g, h). Corroborating the ChIP-qPCR data, we found that luciferase reporter activity was in order of Cd > As > Pb and As + Cd > Cd + Pb > As + Pb, resulting in As + Cd + Pb-mediated increased luciferase activity that was comparable with troglitazone (Fig. 5g, h).

As, Cd, and Pb, like troglitazone, induce PPARγ binding to DR5 and IR3-PPRE sequences on PARP gene, causing enhanced PARP expression in astrocytes. a, b PARP gene sequence was analyzed through NUBIscan for identifying potential PPREs. Data show potential DR5 (a) and IR3 (b) PPRE sequences. c, d Rat primary astrocytes were pre-incubated in reduced serum medium and then co-treated with vehicle (water, V) or As + Cd + Pb at LC-15 doses of the metals and troglitazone (Trog, 10 μM) for 18 h. Nuclear protein lysates from the astrocytes were isolated and incubated with DR5 or IR3 oligonucleotide probes for 30 min. DNA-protein complexes were then analyzed by EMSA. Representative blots show increased shift of DR5 (c) and IR3 (d) DNA bands relative to unbound DNA upon As + Cd + Pb (left panel) and troglitazone (right panel) treatments, respectively. Note the DNA-protein complex supershift (ss) upon anti-PPARγ antibody addition, and elimination of the band for competition experiment (CE). Data are representative of three independent experiments. e, f Rat primary astrocytes were pre-incubated in reduced serum medium and then treated with vehicle (water, V), As, Cd, Pb, or their binary and tertiary metal mixtures at LC-15 doses of metals or troglitazone (Trog, 10 μM) for 18 h. Cells were fixed and ChIP assay carried out with anti-PPARγ, anti acetyl-Histone H3 (positive control), and normal IgG (negative control) antibodies. Precipitated DNA was then amplified by qPCR using DR5 or IR3 primers. Representative data show increased fold enrichment of DR5 (E) and IR3 (F) ChIP-qPCR products in metal(s), metal mixture, and troglitazone-treated astrocytes compared to V. Data represent means ± SE of four independent experiments in triplicate. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to V. $$ P < 0.01 and $ P < 0.05. # P < 0.001 indicates significant difference compared to all other treatments. g, h Rat primary astrocytes were transfected with pGL3, pGL3-DR5, or pGL3-IR3 constructs. Transfected cells were then treated with vehicle (water, V), As, Cd, Pb, their binary and tertiary metal mixtures at LC-15 doses of metals and troglitazone (Trog, 10 μM) for 18 h. Luciferase activity of the cells was measured and expressed as percentage relative to V. Data show unaltered luciferase activity in pGL3-transfected cells and increased luciferase activity in pGL3-DR5 (g) and pGL3-IR3 (h) transfected cells following metal(s) or troglitazone treatments. Data represent means ± SE of four independent experiments in triplicate. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to V. $$ P < 0.01 and $ P < 0.05. # P < 0.001indicates significant difference compared to all other treatments
Activated PPARγ Enhances PARP Expression
We examined the effects of As, Cd, and Pb on PARP mRNA expression in the astrocytes through qPCR. We detected that the metals enhanced the PARP mRNA levels in astrocytes (Fig. 6a). Analyzing the data revealed that the effect on PARP mRNA was dose-dependent for individual, dual, and tertiary metal treatments. Moreover, the effects on PARP mRNA were in order of Cd > As > Pb, and the impact on metal mixture and troglitazone was higher. We further observed that GW9662 treatment attenuated PARP mRNA levels, verifying activated PPARγ-mediated PARP expression (Fig. 6b). We finally validated enhanced PARP levels through Western blotting that demonstrated significant contribution by Cd and its combinations, and the effect of metal mixture and troglitazone was greater (Fig. 6c). Reduction in PARP protein levels following GW9662 treatment corroborated the activated PPARγ-mediated PARP expression (Fig. 6d).

As, Cd, and Pb, like troglitazone, induce activated PPARγ-dependent PARP expression in astrocytes. a qPCR data demonstrate dose-dependent increase in PARP mRNA levels normalized against housekeeping gene GAPDH, following astrocyte treatment with As, Cd, Pb, or their binary and tertiary mixtures at LC-5, LC-10, and LC-15 doses, or troglitazone (Trog, 10 μM). Data represent means ± SE of three independent experiments in triplicate. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to vehicle (V). b Rat primary astrocytes were pre-incubated in reduced serum medium and PPARγ antagonist (GW9662, 10 μM), and then co-treated with vehicle (water, V), Cd, As + Cd, Cd + Pb, or As + Cd + Pb at LC-15 doses of the metals or troglitazone (Trog, 10 μM) for 18 h, followed by qPCR analysis. qPCR data represent GW9662-mediated reduction in PARP mRNA, normalized against GAPDH housekeeping gene in the metal(s) and troglitazone-treated astrocytes. Data represent means ± SE of three independent experiments in triplicate. *** P < 0.001. c Rat primary astrocytes were pre-incubated in reduced serum medium and then treated with vehicle (water, V), As, Cd, Pb, their binary and tertiary mixtures at LC-15 doses, or troglitazone (Trog, 10 μM) for 18 h. Cell lysates were immunoblotted for PARP and β-actin. Representative Western blot and densitometric analysis show PARP expression normalized with β-actin. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to V. $$ P < 0.01 and $ P < 0.05. # P < 0.001 indicates significant difference compared to all other treatments. d Rat primary astrocytes were pre-incubated in reduced serum medium and PPARγ antagonist (GW9662, 10 μM), and then co-treated with vehicle (water, V), Cd, As + Cd, Cd + Pb, or As + Cd + Pb at LC-15 doses of the metals or troglitazone (Trog, 10 μM) for 18 h. Cell lysates were immunoblotted for PARP and β-actin. Representative Western blot and densitometric analysis show GW9662-mediated reduction in PARP expression normalized with β-actin in the metal(s) and troglitazone-treated astrocytes. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001. e Rat primary astrocytes were pre-incubated in reduced serum medium and then transfected with NT-siRNA or PPARγ-siRNA. The cells were then treated with vehicle (water, V) or As + Cd + Pb at LC-15 doses of metals for 18 h, followed by Western blot analysis with PARP and β-actin antibodies. Representative Western blot and densitometric analysis show reduction in PARP levels normalized with β-actin for PPARγ-siRNA compared to NT-siRNA transfections in As + Cd + Pb-treated astrocytes. PPARγ-siRNA and NT-siRNA transfections in vehicle-treated astrocytes showed no change. Data represent means ± SE of the three independent experiments. *** P < 0.001. f Rat primary astrocytes were pre-incubated in reduced serum medium and a PARP inhibitor (3-aminobenzamide, 3-ABA), and then co-treated with vehicle (water, V) or As + Cd + Pb at LC-15 doses for 18 h. Cell lysates were immunoblotted for PPARγ and β-actin. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001
We observed (in Fig. 2) that the metal(s) enhanced PPARγ expression levels, promoting astrocyte apoptosis. We examined whether increased PPARγ levels participated in PARP expression. We found that co-treating with PPARγ-siRNA suppressed the metal(s)-induced PARP, indicating an increased PPARγ-dependent PARP expression by the heavy metals (Fig. 6e). Alternatively, we found that PARP inhibition failed to effect the As + Cd + Pb-induced PPARγ levels (Fig. 6f), indicating a PARP-independent PPARγ expression in the astrocytes. Thus, our data indicate that As, Cd, and Pb trigger PPARγ expression levels and activation, resulting in enhanced PARP expression in astrocytes.
As, Cd, and Pb Induce PARP-Dependent Cleaved PARP
PARP undergoes cleavage, resulting in increased cleaved PARP that marks apoptosis [44]. Therefore, we assessed the effect of metal mixture on the cleaved PARP/PARP ratio. We detected that the metal mixture stimulated a dose-dependent increase in cleaved PARP levels (Fig. 7a), resulting in enhanced cleaved PARP/PARP ratio (Fig. 7b). Troglitazone also enhanced the cleaved PARP/PARP ratio, verifying participation of troglitazone in astrocyte apoptosis (Fig. 7a, b). We, therefore, inferred that the metals enhanced PPARγ-dependent PARP, which underwent cleavage with increasing doses of the metals, resulting in augmented cleaved PARP and thus higher cleaved PARP/PARP ratio. We compared the cleaved PARP/PARP levels, and corroborating apoptosis data detected the order as Cd > As > Pb, As + Cd > Cd + Pb > As + Pb, resulting in a higher effect by As + Cd + Pb (Fig. 7c, d). To verify whether the cleaved PARP was truly dependent on increased PARP, we co-treated the metals with a PARP inhibitor, 3-amino benzamide (3-ABA) [45]. We detected that suppressing PARP levels using 3-ABA (Suppl. 3) reduced the cleaved PARP (Fig. 7e), proving cleaved PARP to be dependent on increased PARP. Supporting this PARP-dependent increased PARP cleavage, we detected that As, Cd, and Pb stimulated PARP-dependent expression of activated caspases (Suppl. 4A–B) that are known to cleave PARP [44]. Additionally, inactivating the caspases using their inhibitor, Z-VAD-FMK, attenuated cleaved PARP levels (Suppl. 4C). We finally co-related cleaved PARP and PPARγ and detected that GW9662 (Fig. 7f) and PPARγ-siRNA (Fig. 7g) reduced the metal(s)-induced cleaved PARP levels. We further established the link between PPARγ and caspases and detected an enhanced PPARγ-mediated activated caspase expression (Suppl. 4D–E). Thus, overall, our data indicate that As, Cd, and Pb enhanced the expression and transactivation function of PPARγ, thereby inducing PPARγ-dependent PARP. The increased PARP stimulated caspase activation and cleaved-PARP levels, resulting in upregulated cleaved PARP/PARP ratio and hence astrocyte apoptosis.

As, Cd, and Pb increase PPARγ-dependent PARP that stimulates cleaved PARP levels in rat astrocytes. a, b Rat primary astrocytes were pre-incubated in reduced serum medium and then treated with vehicle (water, V) or As + Cd + Pb at LC-5, LC-10, and LC-15 doses of the metals or troglitazone (Trog, 10 μM) for 18 h. The astrocyte cell lysates were immunoblotted for PARP and β-actin. Representative Western blot shows PARP and cleaved PARP expressions normalized with β-actin (a), and bar diagram indicates increased cleaved PARP/PARP ratio (b) for As + Cd + Pb and troglitazone. c, d Rat primary astrocytes were pre-incubated in reduced serum medium and then treated with vehicle (water, V), As, Cd, Pb, or their binary and tertiary mixtures at LC-15 doses of metals for 18 h. The astrocyte cell lysates were immunoblotted for PARP and β-actin. Representative Western blot shows PARP and cleaved PARP expressions normalized with β-actin (c) and bar diagram indicate increased cleaved PARP/PARP ratio (d) for the treatments. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001, ** P < 0.01, and * P < 0.05 indicate significant difference compared to V. e Rat primary astrocytes were pre-incubated with reduced serum medium and PARP inhibitor (3-aminobenzamide, 3-ABA), and then co-treated with vehicle (water, V), As, Cd, Pb, or As + Cd + Pb at LC-15 doses of the metals for 18 h. The cell lysates were immunoblotted for cleaved PARP and β-actin. Representative Western blot and densitometric analysis show 3-ABA-mediated reduction in cleaved PARP expressions normalized with β-actin. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001. f Rat primary astrocytes were pre-incubated in reduced serum medium and PPARγ antagonist (GW9662, 10 μM), and then co-treated with vehicle (water, V), As, Cd, Pb, or As + Cd + Pb at LC-15 doses of the metals for 18 h. The cell lysates were immunoblotted for cleaved PARP and β-actin. Representative Western blot and densitometric analysis show GW9662-mediated reduction in cleaved PARP expression normalized with β-actin. g Rat primary astrocytes were pre-incubated in reduced serum medium and then transfected with NT-siRNA or PPARγ-siRNA. The cells were then treated with vehicle (water, V), As, Cd, Pb, or As + Cd + Pb at LC-15 doses of the metals for 18 h, followed by Western blot analysis with cleaved PARP and β-actin antibodies. Representative Western blot and densitometric analysis show reduction in cleaved PARP levels normalized with β-actin for PPARγ-siRNA compared to NT-siRNA transfections in the metal(s)-treated astrocytes. PPARγ-siRNA and NT-siRNA transfections in vehicle-treated astrocytes showed no change in cleaved PARP levels. Data represent means ± SE of the three independent experiments. *** P < 0.001
As, Cd, and Pb Stimulate JNK and CDK5-Dependent PPARγ and PARP
PPAR expression has an intricate connection with the P38 MAPK and JNK activities within astrocytes [23]. Hence, we examined whether P38 MAPK and JNK had any regulatory effects on As, Cd, and Pb-induced PPARγ. We observed that inhibiting P38 with SB203580 failed to affect (Fig. 8a), while the JNK inhibitor, SP600125, blocked As + Cd + Pb-induced PPARγ (Fig. 8b), proving a JNK-dependent PPARγ expression in the astrocytes.

As, Cd, and Pb induce JNK and CDK5-dependent PPARγ and PARP in astrocytes. a–c Rat primary astrocytes were pre-incubated in reduced serum medium and a P38 inhibitor (SB203580, 10 μM), JNK inhibitor (SP600125, 10 μM), or CDK5 inhibitor (R-CR8, 10 μM) and then co-treated with vehicle (water, V) or As + Cd + Pb mixture for 18 h. Cell lysates were immunoblotted for PPARγ and β-actin. Representative Western blot and densitometric analysis show the effects of SB203580 (a), SP600125 (b), and R-CR8 (c) on PPARγ expression levels normalized with β-actin in As + Cd + Pb-treated astrocytes. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001. d, e Rat primary astrocytes were pre-incubated in reduced serum medium and JNK inhibitor (SP600125, 10 μM) or CDK5 inhibitor (R-CR8, 10 μM) and then co-treated with vehicle (water, V) or As + Cd + Pb mixture for 18 h. Cell lysates were immunoblotted for PARP and β-actin. Representative Western blot and densitometric analysis show SP600125 or R-CR8-mediated reduction in PARP expression normalized with β-actin in As + Cd + Pb-treated astrocytes. Western blot is representative of three independent experiments and bar diagram data represent means ± SE. *** P < 0.001
In our earlier study, we proved that As + Cd + Pb stimulated a JNK-dependent CDK5 that promoted phospho-PPARγ levels within the astrocytes [12]. Here, we examined whether the increased PPARγ expression could be linked to CDK5. We found that inhibiting CDK5 activation using (R)-CR8 blocked As + Cd + Pb-induced PPARγ (Fig. 8c), proving a CDK5-dependent PPARγ expression in the astrocytes.
We further examined whether the JNK and CDK5 pathways participated in increased PARP expression. We observed that SP600125 (Fig. 8d) and (R)-CR8 (Fig. 8e) inhibited As + Cd + Pb-induced PARP expression in the As + Cd + Pb-treated astrocytes. Thus, our data indicate that an activated JNK and CDK5 stimulate PPARγ, and thereby PARP expression in astrocytes.
In Vivo Validation of PPARγ-Dependent Astrocyte Apoptosis by As, Cd, and Pb in Rat Brain
For in vivo verification of our in vitro findings, we exposed developing rats to environmentally relevant doses of As, Cd, and Pb (Table 3). Corroborating the in vitro observation, we found that the metal mixture caused enhanced PARP expression in astrocytes of the rat brain (Fig. 9a). We further found that co-treatment with GW9662 suppressed PARP levels (Fig. 9a), verifying activated PPARγ-dependent PARP expression. We also observed enhanced cleaved PARP expression in astrocytes, and matching in vitro data, we found that the effect of Cd was highest and that of As + Cd + Pb was comparable with troglitazone (Fig. 9b). Ultimately, an enhanced number of TUNEL-positive apoptotic astrocytes in the metal(s)-exposed rat brain verified astrocyte apoptosis, where again Cd contributed most and As + Cd + Pb and troglitazone had comparable effects (Fig. 9c).



As, Cd, and Pb increase PPARγ-dependent PARP expression, and apoptosis and cleaved PARP levels in astrocytes of developing rat brain. a Five-micron-thick cryostat sections of cerebral cortex from vehicle (water), As + Cd + Pb (at environmentally relevant doses), or GW9662 (1 mg/kg) + As + Cd + Pb-treated PND16 rats were fluorescent co-immunolabeled for PARP (red fluorescence) and GFAP (green fluorescence), and co-stained with nuclear Hoechst (blue fluorescence). Representative fluorescent photomicrograph (60× magnification) shows As + Cd + Pb-mediated increased PARP expression in the GFAP-expressing astrocytes against nuclear-Hoechst, and restoration by GW9662. Bar diagram represents PARP expression in GFAP-expressing astrocytes, normalized with nuclear Hoechst for the specidied treatments. Sections are representatives of three rats from three different litters. ***P < 0.001. b Five-micron-thick cryostat sections of cerebral cortex from vehicle, As, Cd, Pb, As + Cd + Pb, or troglitazone (Trog, 2.5 mg/kg)-treated PND16 rats were fluorescent co-immunolabeled for cleaved PARP and GFAP (green fluorescence), and co-stained with nuclear Hoechst (blue fluorescence). Representative fluorescent photomicrographs (60× magnification) show metal(s)-mediated increased cleaved PARP expression in the GFAP-expressing astrocytes against nuclear-Hoechst. Sections are representatives of three rat brains from three different litters. Bar diagram represents c-PARP expressions in GFAP-expressing astrocytes, normalized with nuclear Hoechst. Sections are representatives of three rats from three different litters. ***P < 0.001 and *P < 0.05 w.r.t. to vehicle (V) or as indicated. c Five-micron-thick cryostat sections of cerebral cortex from vehicle, As, Cd, Pd, As + Cd + Pb, and troglitazone (Trog)-treated PND16 rats were fluorescence stained for TUNEL and immunolabeled with GFAP, followed by nuclear Hoechst co-staining. Representative photomicrographs (20× magnification) show enhanced TUNEL-positive astrocytes against nuclear Hoechst in the same field. Arrows indicate the TUNEL-positive cells. Bar diagram represents apoptotic index expressed as the number of TUNEL-positive cells/100 nuclei (Hoechst stained) in GFAP-expressing astrocytes from five randomly selected fields. Sections are representatives of three rats from three different litters. ***P < 0.001, **P < 0.01, and *P < 0.05 w.r.t. to vehicle (V) or as indicated
Discussion
Our results present the first evidence of the contributions in order of Cd > As > Pb in As + Cd + Pb-induced astrocyte apoptosis. Additionally, our findings prove upregulation and activation of PPARγ as a novel pathway generating PARP and then cleaved PARP, resulting in astrocyte apoptosis (Fig. 10).

As, Cd, and Pb increase PPARγ-mediated PARP expression leading to apoptosis in astrocytes. As, Cd, and Pb induce JNK and CDK5-dependent PPARγ expression and transactivation in the rat brain astrocytes. Activated PPARγ binds to DR5 and IR3-PPRE sequences within PARP gene, resulting in enhanced PARP. Increased PARP level activates caspases and cleaved-PARP expression, resulting in astrocyte apoptosis
In terms of environmental contaminants, there are a relatively higher number of studies on astrogliosis and astroglial inflammation compared to astrocyte loss [31, 46]. Again, studies focused more on toxicant-mediated degeneration of neurons rather than astrocytes [47–49]. Through the present study, we reveal a novel mechanism of astrocyte damage upon exposure to the three key ground water contaminants, As, Cd, and Pb, at their environmentally relevant doses [11]. Damage to the CNS has been well reported to have a distinct link with astrogliosis, increased astrocyte expression, and an ultimate glial scar formation [50, 51]. Astroglial activation has also been proved to be a major reason for death and degeneration of the neighboring cell population, particularly neurons [52, 53]. Moreover, we reported that long-term treatment with As, Cd, and Pb ultimately led to astroglial hypertrophy and neurodegeneration [7]. Hence, it may be presumed that astrocyte apoptosis and damage observed in the current study at early stages, and at relatively lower doses of the heavy metals are adaptive responses towards saving the other cell population from subsequent damage. We previously proved that As + Cd + Pb induces synergistic astrocyte loss via enhanced apoptosis [11]. In the present study, we add to the observations by establishing Cd to be the most apoptotic, followed by As and Pb. Interestingly, this opposes one of our earlier reports on heavy metals, depicting the maximum impact of Pb, however, towards promoting AD pathogenesis [7]. In the AD study, an enhanced interleukin-1 (IL-1) expression appeared mainly responsible for Alzheimer’s Aβ expression, where the contribution by Pb was significantly high and Cd low [7]. Probably, a difference in the two mechanisms, IL-1 increase for AD [7] versus PPARγ activation for astrocyte apoptosis (as detected in the current study), rationalizes this disparate impact of the heavy metals. Although less compared to Cd, our findings for As-mediated astrocyte apoptosis in the current investigation also challenge a previous report demonstrating astrocytes to be relatively resistant to As administration [54]. Nonetheless, unlike ours, the earlier study mainly focused upon the tumor necrosis factor-mediated effects of arsenic [54], justifying the differential responses. Taken together, our data that clearly identify contributions of Cd, As, and Pb in astrocyte apoptosis confirmed these heavy metals as risk factors for CNS damage. Our findings also hint at the necessity of adopting relatively greater precautions against Cd-induced astrocyte apoptosis to protect the developing brain.
Astrocytes play an important role in regulating lipid and glucose metabolism within the brain [55, 56]. Likewise, PPARγ contributes to the brain energy metabolism [42, 57] and undergoes activation by prostaglandins that are vital astrocyte components [58]. Additionally, because we had earlier found that PPARγ participated in modulating the GFAP levels [12], we were especially interested to understand the role of the nuclear receptor in astrocytes for the present investigation. PPARγ is known to play an important role in inflammatory brain pathologies [59] where astrogliosis plays a prominent role [60, 61]. PPARγ activation has also been reported in astroglioma or glioblastoma cells [62, 63]. Several studies have as well shown ligands of PPARγ to cause apoptosis in astrocytoma and glioma cells [26, 63–65]. However, there are relatively very few reports correlating PPARγ and apoptosis in the primary astrocytes. Chattopadhyay et al. found modulation of the apoptotic process as a vital function of PPARγ in the human astrocytes [15]. Perez-Ortiz et al. proved PPARγ-mediated rat astrocyte apoptosis, however, using a higher dose of PPARγ agonist, ciglitazone [19]. Our findings not only established that PPARγ participates in primary astrocyte apoptosis, but also validated the event in both heavy metal and troglitazone-treated rat astrocytes. Interestingly, unlike the data of Spagnolo et al. that showed primary astrocytes to be selectively resistant to the cytotoxic effects of troglitazone [17], we detected that the PPARγ agonist stimulated rat astrocyte apoptosis at a low to moderate dose of troglitazone. We also validated troglitazone-mediated apoptosis through pertinent apoptotic assay methods, such as annexin V/PI and cleaved PARP/PARP levels, and supportively found increased activated caspases as well (Suppl. 5). Unlike our study that specifically focused on the apoptosis aspect, Spagnolo et al. dealt more on the effects of troglitazone on cellular energetics and astrocyte metabolic rates [17], probably justifying the difference in opinions. Additionally, while Spagnolo et al. focused primarily on the lactate dehydrogenase release assay and on necrosis and autophagy-mediated death in astrocytes by troglitazone, the current study emphasized upon apoptosis. Thus, disparities in study designs may have also contributed to the differential effects of Spagnolo et al. compared to the present one.
The present investigation proved that our heavy metals enhanced PPARγ levels, and the siRNA data showed that the increased PPARγ participated in astrocyte apoptosis, where Cd contributed most. PPARγ is a positive regulator of astrocyte mitochondriogenesis and energy homeostasis and metabolism [42]. On the other hand, heavy metals, particularly Cd, are known to hamper the normal processes of lipid and glucose metabolism in astrocytes and disturb the mitochondrial functions [66–69]. Likewise, we reported that As, Cd, and Pb damaged the antioxidative defense machinery of astrocytes and induced oxidative stress [11]. Thus, as observed earlier for the Cd-treated kidney cells [70], it may be stated that the increase in PPARγ for astrocytes may have resulted from heavy metal-mediated loss in mitochondrial bioenergetics. The study had also shown that for acute responses to Cd exposure, a markedly increased PPARγ, along with oxidative stress and mitochondrial destabilization, generated aberrant PPARγ activation and, thereby, enhanced the expression of pro-apoptotic proteins [70]. Thus, an analogous situation may have led to an activated PPARγ and ultimate astrocyte apoptosis here following metals, predominantly Cd treatment, in astrocytes.
Via computer-assisted screening tools, as well as gel shift, ChIP, and luciferase reporter assays, we identified two functional PPREs within the PARP gene. Several novel facts emerge from this finding. Firstly, although PARP modulation by PPAR agonists has been casually reported in the glioblastoma cells [65], there have been no studies correlating the two in primary brain cells. We not only unveiled PPARγ and PARP linkage in primary astrocytes, but also proved a direct dependence of PARP expression on PPARγ via genomic interactions. Therefore, our data unveil PPARγ-PARP link as a new mechanism that deserves significant consideration in astrocyte damage. Hence, the present study in conjunction with our previous report demonstrating phospho-PPARγ-mediated suppression of the astrocyte marker, GFAP [12], affirms the prominent role of PPARγ in astrocyte degeneration and damage.
The current investigation reveals that As, Pb, and Cd, individually and in mixtures, upregulate cleaved PARP expression, known to contribute in apoptosis [71]. An unchecked PARP expression may either lead to necrotic cell death or apoptosis via an increased cleavage [72]. Our findings that show PARP cleavage, supported by enhanced caspase-3 and -7 that are mainly responsible for the cleavage [44], suggest a preference of the heavy metals for apoptotic pathway. On the whole, notably and very interestingly, our findings detected a consistent trend in Cd > As > Pb-induced toxicity, starting from PPARγ expression, PPARγ and PARP-PPRE binding, and eventually PARP cleavage and thereby apoptosis. Thus, as an extrapolation of our earlier study that showed increased apoptosis by the As + Cd + Pb-mixture [11], our current study reveals the complex mechanism involved and contribution of individual metals and binary mixtures towards the tertiary mixture-induced astrocyte damage.
The current study proves that increased PPARγ and hence an ultimate PARP induction was JNK-dependent in the As-, Cd-, and Pb-treated astrocytes. The PPAR triad is known to have intricate connections with MAPKs and displays sensitivity to P38 and JNK inhibitions at variable degrees [23, 73]. Interestingly, quite like the current study, PPARγ regulation in astrocytes appeared to be under greater control of JNK activity compared to P38 [23]. Nonetheless, opposing the earlier concept, where JNK inhibition could stimulate PPARγ activity [23], the current study reveals an increased JNK-dependent PPARγ expression within the astrocytes. The differences in the stimuli, viz. toll-like receptor agonists for the previous study [23] and heavy metals for the current one, may well explain the discrepant behavior. However, the two studies certainly provide convincing evidence on participation of MAPKs in controlling PPAR levels and thereby cellular responses within astrocytes. An earlier report from our group also provided a supportive information showing As + Cd + Pb-mediated JNK activation, which led to an increased phospho-PPARγ expression [12]. This increased phosphorylation occurred at an earlier time point, around 10–15 min following the heavy metals(s) exposure [12]. Thus, it remains exploring whether this early phospho-PPARγ could have any role in regulating PPARγ expression at a later time point, overnight as observed presently. Another intriguing link lies in an As-, Cd-, and Pb-mediated increased CDK5 activity in the astrocytes. While an enhanced CDK5-dependent phospho-PPARγ expression appeared as the key regulator maintaining astrocyte integrity in the previous study [12], the current study depicts a CDK5-mediated PPARγ expression. Thus, our two studies emphasize the prominent participation of JNK and CDK5 activities in controlling heavy metal-induced responses on phospho-PPARγ and PPARγ. Additionally, the current study also portrays the marked involvement of these kinases in regulating PPARγ transcriptional activity, resulting in increased PARP expression. Thus, largely, it appears that the extracellular signals activating intracellular kinase pathways may play a significant influential role in maintaining PPARγ expression and activity, contributing towards vital cellular responses within the astrocytes.
Conclusion
Altogether, our study enlightens a novel mechanism of Cd > As > Pb-induced primary astrocyte apoptosis, where the enhanced and activated PPARγ played key roles. Two intricately associated crucial aspects in terms of astrocyte toxicity emerge from these observations: (1) predominance of Cd and less of As and Pb and (2) participation of PPARγ. Consequently, our findings emphasize the need for precautionary measures against heavy metals, particularly Cd-induced apoptosis and toxicity, in relation to CNS disorders like Alexander disease, schizophrenia, bipolar disorder, etc. characterized by severe astrocyte loss [74, 75]. Most importantly, our data point towards the PPARγ antagonists, GW9662, as a therapeutic against astrocyte death, especially in situations of heavy metal toxicity.
Abbreviations
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- PARP:
-
Poly (ADP-ribose) polymerase
- LC:
-
Lethal concentration
- PPRE:
-
Peroxisome proliferator-activated receptorresponse elements
- As:
-
Arsenic
- Cd:
-
Cadmium
- Pb:
-
Lead
- GFAP:
-
Glial fibrillary acidic protein
- EMSA:
-
Electrophoretic mobility shift assay
- ChIP:
-
Chromatin immunoprecipitation
- ABA:
-
3-aminobenzamide
- GFP:
-
Green fluorescence protein
- PND:
-
Postnatal day
- TUNEL:
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- TBP:
-
Tata-binding protein
- Trog:
-
Troglitazone
References
Montgomery DL (1994) Astrocytes: form, functions, and roles in disease. Vet Pathol 31(2):145–167
Allen NJ (2014) Astrocyte regulation of synaptic behavior. Annu Rev Cell Dev Biol 30:439–463. doi:10.1146/annurev-cellbio-100913-013053
Cohly HH, Panja A (2005) Immunological findings in autism. Int Rev Neurobiol 71:317–341
Schreiner B, Romanelli E, Liberski P, Ingold-Heppner B, Sobottka-Brillout B, Hartwig T, Chandrasekar V, Johannssen H et al (2015) Astrocyte depletion impairs redox homeostasis and triggers neuronal loss in the adult CNS. Cell Rep 12(9):1377–1384. doi:10.1016/j.celrep.2015.07.051
Sobieski C, Jiang X, Crawford DC, Mennerick S (2015) Loss of local astrocyte support disrupts action potential propagation and glutamate release synchrony from unmyelinated hippocampal axon terminals in vitro. J Neurosci 35(31):11105–11117. doi:10.1523/JNEUROSCI.1289-15.2015
Jadhav SH, Sarkar SN, Patil RD, Tripathi HC (2007) Effects of subchronic exposure via drinking water to a mixture of eight water-contaminating metals: a biochemical and histopathological study in male rats. Arch Environ Contam Toxicol 53(4):667–677. doi:10.1007/s00244-007-0031-0
Ashok A, Rai NK, Tripathi S, Bandyopadhyay S (2015) Exposure to As-, Cd-, and Pb-mixture induces Abeta, amyloidogenic APP processing and cognitive impairments via oxidative stress-dependent neuroinflammation in young rats. Toxicol Sci 143(1):64–80. doi:10.1093/toxsci/kfu208
Bui AT, Nguyen HT, Nguyen MN, Tran TH, Vu TV, Nguyen CH, Reynolds HL (2016) Accumulation and potential health risks of cadmium, lead and arsenic in vegetables grown near mining sites in northern Vietnam. Environ Monit Assess 188(9):525. doi:10.1007/s10661-016-5535-5
Obiri S, Yeboah PO, Osae S, Adu-Kumi S (2016) Levels of arsenic, mercury, cadmium, copper, lead, zinc and manganese in serum and whole blood of resident adults from mining and non-mining communities in Ghana. Environ Sci Pollut Res Int 23(16):16589–16597. doi:10.1007/s11356-016-6537-0
Rai NK, Ashok A, Rai A, Tripathi S, Nagar GK, Mitra K, Bandyopadhyay S (2013) Exposure to as, Cd and Pb-mixture impairs myelin and axon development in rat brain, optic nerve and retina. Toxicol Appl Pharmacol 273(2):242–258. doi:10.1016/j.taap.2013.05.003
Rai A, Maurya SK, Khare P, Srivastava A, Bandyopadhyay S (2010) Characterization of developmental neurotoxicity of as, Cd, and Pb mixture: synergistic action of metal mixture in glial and neuronal functions. Toxicol Sci 118(2):586–601. doi:10.1093/toxsci/kfq266
Rai A, Tripathi S, Kushwaha R, Singh P, Srivastava P, Sanyal S, Bandyopadhyay S (2014) CDK5-induced p-PPARgamma(Ser 112) downregulates GFAP via PPREs in developing rat brain: effect of metal mixture and troglitazone in astrocytes. Cell Death Dis 5:e1033. doi:10.1038/cddis.2013.514
Bordet R, Ouk T, Petrault O, Gele P, Gautier S, Laprais M, Deplanque D, Duriez P et al (2006) PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans 34(Pt 6):1341–1346. doi:10.1042/BST0341341
Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB (1998) Distribution of mRNAs encoding the peroxisome proliferator-activated receptor alpha, beta, and gamma and the retinoid X receptor alpha, beta, and gamma in rat central nervous system. J Neurochem 70(4):1366–1375
Chattopadhyay N, Singh DP, Heese O, Godbole MM, Sinohara T, Black PM, Brown EM (2000) Expression of peroxisome proliferator-activated receptors (PPARS) in human astrocytic cells: PPARgamma agonists as inducers of apoptosis. J Neurosci Res 61(1):67–74
Cristiano L, Bernardo A, Ceru MP (2001) Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J Neurocytol 30(8):671–683
Spagnolo A, Grant EN, Glick R, Lichtor T, Feinstein DL (2007) Differential effects of PPARgamma agonists on the metabolic properties of gliomas and astrocytes. Neurosci Lett 417(1):72–77. doi:10.1016/j.neulet.2007.02.036
Zander T, Kraus JA, Grommes C, Schlegel U, Feinstein D, Klockgether T, Landreth G, Koenigsknecht J et al (2002) Induction of apoptosis in human and rat glioma by agonists of the nuclear receptor PPARgamma. J Neurochem 81(5):1052–1060
Perez-Ortiz JM, Tranque P, Vaquero CF, Domingo B, Molina F, Calvo S, Jordan J, Cena V et al (2004) Glitazones differentially regulate primary astrocyte and glioma cell survival. Involvement of reactive oxygen species and peroxisome proliferator-activated receptor-gamma. J Biol Chem 279(10):8976–8985. doi:10.1074/jbc.M308518200
Varga T, Czimmerer Z, Nagy L (2011) PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim Biophys Acta 1812(8):1007–1022. doi:10.1016/j.bbadis.2011.02.014
Chiu SC, Lin YJ, Huang SY, Lien CF, Chen SP, Pang CY, Lin JH, Yang KT (2015) The role of intermittent hypoxia on the proliferative inhibition of rat cerebellar astrocytes. PLoS One 10(7):e0132263. doi:10.1371/journal.pone.0132263
Wang J, Deng X, Zhang F, Chen D, Ding W (2014) ZnO nanoparticle-induced oxidative stress triggers apoptosis by activating JNK signaling pathway in cultured primary astrocytes. Nanoscale Res Lett 9(1):117. doi:10.1186/1556-276X-9-117
Chistyakov DV, Aleshin SE, Astakhova AA, Sergeeva MG, Reiser G (2015) Regulation of peroxisome proliferator-activated receptors (PPAR) alpha and -gamma of rat brain astrocytes in the course of activation by toll-like receptor agonists. J Neurochem 134(1):113–124. doi:10.1111/jnc.13101
Xiao Y, Yuan T, Yao W, Liao K (2010) 3T3-L1 adipocyte apoptosis induced by thiazolidinediones is peroxisome proliferator-activated receptor-gamma-dependent and mediated by the caspase-3-dependent apoptotic pathway. FEBS J 277(3):687–696. doi:10.1111/j.1742-4658.2009.07514.x
Kang DW, Choi CH, Park JY, Kang SK, Kim YK (2008) Ciglitazone induces caspase-independent apoptosis through down-regulation of XIAP and survivin in human glioma cells. Neurochem Res 33(3):551–561. doi:10.1007/s11064-007-9475-x
Liu DC, Zang CB, Liu HY, Possinger K, Fan SG, Elstner E (2004) A novel PPAR alpha/gamma dual agonist inhibits cell growth and induces apoptosis in human glioblastoma T98G cells. Acta Pharmacol Sin 25(10):1312–1319
Alano CC, Ying W, Swanson RA (2004) Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279(18):18895–18902. doi:10.1074/jbc.M313329200
Ying W, Chen Y, Alano CC, Swanson RA (2002) Tricarboxylic acid cycle substrates prevent PARP-mediated death of neurons and astrocytes. J Cereb Blood Flow Metab 22(7):774–779. doi:10.1097/00004647-200207000-00002
Alam S, Pal A, Kumar R, Mir SS, Ansari KM (2015) Nexrutine inhibits azoxymethane-induced colonic aberrant crypt formation in rat colon and induced apoptotic cell death in colon adenocarcinoma cells. Mol Carcinog. doi:10.1002/mc.22368
Campbell SE, Stone WL, Whaley SG, Qui M, Krishnan K (2003) Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer 3:25. doi:10.1186/1471-2407-3-25
Maurya SK, Mishra J, Abbas S, Bandyopadhyay S (2016) Cypermethrin stimulates GSK3beta-dependent Abeta and p-tau proteins and cognitive loss in young rats: reduced HB-EGF signaling and downstream neuroinflammation as critical regulators. Mol Neurobiol 53(2):968–982. doi:10.1007/s12035-014-9061-6
Lee KW, Ku YH, Kim M, Ahn BY, Chung SS, Park KS (2011) Effects of sulfonylureas on peroxisome proliferator-activated receptor gamma activity and on glucose uptake by thiazolidinediones. Diabetes Metab J 35(4):340–347. doi:10.4093/dmj.2011.35.4.340
Dwivedi SK, Singh N, Kumari R, Mishra JS, Tripathi S, Banerjee P, Shah P, Kukshal V et al (2011) Bile acid receptor agonist GW4064 regulates PPARgamma coactivator-1alpha expression through estrogen receptor-related receptor alpha. Mol Endocrinol 25(6):922–932. doi:10.1210/me.2010-0512
Yadav M, Singh AK, Kumar H, Rao G, Chakravarti B, Gurjar A, Dogra S, Kushwaha S et al (2016) Epidermal growth factor receptor inhibitor cancer drug gefitinib modulates cell growth and differentiation of acute myeloid leukemia cells via histamine receptors. Biochim Biophys Acta 1860(10):2178–2190. doi:10.1016/j.bbagen.2016.05.011
Podvinec M, Kaufmann MR, Handschin C, Meyer UA (2002) NUBIScan, an in silico approach for prediction of nuclear receptor response elements. Mol Endocrinol 16(6):1269–1279. doi:10.1210/mend.16.6.0851
Ashok A, Rai NK, Raza W, Pandey R, Bandyopadhyay S (2016) Chronic cerebral hypoperfusion-induced impairment of Abeta clearance requires HB-EGF-dependent sequential activation of HIF1alpha and MMP9. Neurobiol Dis 95:179–193. doi:10.1016/j.nbd.2016.07.013
Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE (2005) Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience 130(3):685–696. doi:10.1016/j.neuroscience.2004.10.021
Jensen EC (2013) Quantitative analysis of histological staining and fluorescence using ImageJ. Anat Rec (Hoboken) 296(3):378–381. doi:10.1002/ar.22641
Maurya SK, Rai A, Rai NK, Deshpande S, Jain R, Mudiam MK, Prabhakar YS, Bandyopadhyay S (2012) Cypermethrin induces astrocyte apoptosis by the disruption of the autocrine/paracrine mode of epidermal growth factor receptor signaling. Toxicol Sci 125(2):473–487. doi:10.1093/toxsci/kfr303
Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14(2):121–141
Gronemeyer H, Gustafsson JA, Laudet V (2004) Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov 3(11):950–964. doi:10.1038/nrd1551
Dello Russo C, Gavrilyuk V, Weinberg G, Almeida A, Bolanos JP, Palmer J, Pelligrino D, Galea E et al (2003) Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem 278(8):5828–5836. doi:10.1074/jbc.M208132200
Harada S, Hiromori Y, Nakamura S, Kawahara K, Fukakusa S, Maruno T, Noda M, Uchiyama S et al (2015) Structural basis for PPARgamma transactivation by endocrine-disrupting organotin compounds. Sci Rep 5:8520. doi:10.1038/srep08520
Soldani C, Scovassi AI (2002) Poly(ADP-ribose) polymerase-1 cleavage during apoptosis: an update. Apoptosis 7(4):321–328
Choi SK, Galan M, Kassan M, Partyka M, Trebak M, Matrougui K (2012) Poly(ADP-ribose) polymerase 1 inhibition improves coronary arteriole function in type 2 diabetes mellitus. Hypertension 59(5):1060–1068. doi:10.1161/HYPERTENSIONAHA.111.190140
Stoltenburg-Didinger G, Punder I, Peters B, Marcinkowski M, Herbst H, Winneke G, Wiegand H (1996) Glial fibrillary acidic protein and RNA expression in adult rat hippocampus following low-level lead exposure during development. Histochem Cell Biol 105(6):431–442
Cobbina SJ, Chen Y, Zhou Z, Wu X, Zhao T, Zhang Z, Feng W, Wang W et al (2015) Toxicity assessment due to sub-chronic exposure to individual and mixtures of four toxic heavy metals. J Hazard Mater 294:109–120. doi:10.1016/j.jhazmat.2015.03.057
Check L, Marteel-Parrish A (2013) The fate and behavior of persistent, bioaccumulative, and toxic (PBT) chemicals: examining lead (Pb) as a PBT metal. Rev Environ Health 28(2–3):85–96. doi:10.1515/reveh-2013-0005
Garcia-Arenas G, Ramirez-Amaya V, Balderas I, Sandoval J, Escobar ML, Rios C, Bermudez-Rattoni F (2004) Cognitive deficits in adult rats by lead intoxication are related with regional specific inhibition of cNOS. Behav Brain Res 149(1):49–59
Middeldorp J, Hol EM (2011) GFAP in health and disease. Prog Neurobiol 93(3):421–443. doi:10.1016/j.pneurobio.2011.01.005
Eddleston M, Mucke L (1993) Molecular profile of reactive astrocytes--implications for their role in neurologic disease. Neuroscience 54(1):15–36
Dahlke C, Saberi D, Ott B, Brand-Saberi B, Schmitt-John T, Theiss C (2015) Inflammation and neuronal death in the motor cortex of the wobbler mouse, an ALS animal model. J Neuroinflammation 12:215. doi:10.1186/s12974-015-0435-0
Lee Y, Chun HJ, Lee KM, Jung YS, Lee J (2015) Silibinin suppresses astroglial activation in a mouse model of acute Parkinson's disease by modulating the ERK and JNK signaling pathways. Brain Res 1627:233–242. doi:10.1016/j.brainres.2015.09.029
Kim EH, Yoon MJ, Kim SU, Kwon TK, Sohn S, Choi KS (2008) Arsenic trioxide sensitizes human glioma cells, but not normal astrocytes, to TRAIL-induced apoptosis via CCAAT/enhancer-binding protein homologous protein-dependent DR5 up-regulation. Cancer Res 68(1):266–275. doi:10.1158/0008-5472.CAN-07-2444
Belanger M, Allaman I, Magistretti PJ (2011) Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14(6):724–738. doi:10.1016/j.cmet.2011.08.016
Yi CX, Habegger KM, Chowen JA, Stern J, Tschop MH (2011) A role for astrocytes in the central control of metabolism. Neuroendocrinology 93(3):143–149. doi:10.1159/000324888
Sarruf DA, Yu F, Nguyen HT, Williams DL, Printz RL, Niswender KD, Schwartz MW (2009) Expression of peroxisome proliferator-activated receptor-gamma in key neuronal subsets regulating glucose metabolism and energy homeostasis. Endocrinology 150(2):707–712. doi:10.1210/en.2008-0899
Dentesano G, Serratosa J, Tusell JM, Ramon P, Valente T, Saura J, Sola C (2014) CD200R1 and CD200 expression are regulated by PPAR-gamma in activated glial cells. Glia 62(6):982–998. doi:10.1002/glia.22656
Boes K, Russmann V, Ongerth T, Licko T, Salvamoser JD, Siegl C, Potschka H (2015) Expression regulation and targeting of the peroxisome proliferator-activated receptor gamma following electrically-induced status epilepticus. Neurosci Lett 604:151–156. doi:10.1016/j.neulet.2015.08.007
Barroso E, del Valle J, Porquet D, Vieira Santos AM, Salvado L, Rodriguez-Rodriguez R, Gutierrez P, Anglada-Huguet M et al (2013) Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPARbeta/delta-null mice. Biochim Biophys Acta 1832(8):1241–1248. doi:10.1016/j.bbadis.2013.03.006
Defaux A, Zurich MG, Braissant O, Honegger P, Monnet-Tschudi F (2009) Effects of the PPAR-beta agonist GW501516 in an in vitro model of brain inflammation and antibody-induced demyelination. J Neuroinflammation 6:15. doi:10.1186/1742-2094-6-15
De Rosa A, Pellegatta S, Rossi M, Tunici P, Magnoni L, Speranza MC, Malusa F, Miragliotta V et al (2012) A radial glia gene marker, fatty acid binding protein 7 (FABP7), is involved in proliferation and invasion of glioblastoma cells. PLoS One 7(12):e52113. doi:10.1371/journal.pone.0052113
Zang C, Wachter M, Liu H, Posch MG, Fenner MH, Stadelmann C, von Deimling A, Possinger K et al (2003) Ligands for PPARgamma and RAR cause induction of growth inhibition and apoptosis in human glioblastomas. J Neuro-Oncol 65(2):107–118
Benedetti E, Galzio R, Cinque B, Biordi L, D'Amico MA, D'Angelo B, Laurenti G, Ricci A et al (2008) Biomolecular characterization of human glioblastoma cells in primary cultures: differentiating and antiangiogenic effects of natural and synthetic PPARgamma agonists. J Cell Physiol 217(1):93–102. doi:10.1002/jcp.21479
Strakova N, Ehrmann J, Bartos J, Malikova J, Dolezel J, Kolar Z (2005) Peroxisome proliferator-activated receptors (PPAR) agonists affect cell viability, apoptosis and expression of cell cycle related proteins in cell lines of glial brain tumors. Neoplasma 52(2):126–136
Fern R, Black JA, Ransom BR, Waxman SG (1996) Cd(2+)-induced injury in CNS white matter. J Neurophysiol 76(5):3264–3273
Sagara J, Makino N, Bannai S (1996) Glutathione efflux from cultured astrocytes. J Neurochem 66(5):1876–1881
Dringen R, Spiller S, Neumann S, Koehler Y (2016) Uptake, metabolic effects and toxicity of arsenate and arsenite in astrocytes. Neurochem Res 41(3):465–475. doi:10.1007/s11064-015-1570-9
Zhang Y, Sun LG, Ye LP, Wang B, Li Y (2008) Lead-induced stress response in endoplasmic reticulum of astrocytes in CNS. Toxicol Mech Methods 18(9):751–757. doi:10.1080/15376510802390908
Nair AR, Lee WK, Smeets K, Swennen Q, Sanchez A, Thevenod F, Cuypers A (2015) Glutathione and mitochondria determine acute defense responses and adaptive processes in cadmium-induced oxidative stress and toxicity of the kidney. Arch Toxicol 89(12):2273–2289. doi:10.1007/s00204-014-1401-9
Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M (1999) Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem 274(33):22932–22940
Chaitanya GV, Steven AJ, Babu PP (2010) PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell communication and signaling : CCS 8:31. doi:10.1186/1478-811X-8-31
Chistyakov DV, Aleshin S, Sergeeva MG, Reiser G (2014) Regulation of peroxisome proliferator-activated receptor beta/delta expression and activity levels by toll-like receptor agonists and MAP kinase inhibitors in rat astrocytes. J Neurochem 130(4):563–574. doi:10.1111/jnc.12757
Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, Yolken RH (2000) Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley neuropathology consortium. Mol Psychiatry 5(2):142–149
Messing A, Goldman JE, Johnson AB, Brenner M (2001) Alexander disease: new insights from genetics. J Neuropathol Exp Neurol 60(6):563–573
Acknowledgement
We acknowledge Miss. Shipra Kartik and Mr. Devendra Pratap Singh for their help in Western blotting and primary rat astrocyte culture.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding Information
This work was supported by Council of Scientific and Industrial Research-network miND; Department of Biotechnology, Govt. of India [GAP285] and Science and Engineering Research Board, Govt. of India [GAP278].
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
.
Supplementary Fig S1
(DOCX 532 kb)
.
Supplementary Fig S2
(DOCX 317 kb)
.
Supplementary Fig S3
(DOCX 207 kb)
.
Supplementary Fig S4
(DOCX 671 kb)
.
Supplementary Fig S5
(DOCX 219 kb)
Rights and permissions
About this article

Cite this article
Kushwaha, R., Mishra, J., Tripathi, S. et al. Arsenic, Cadmium, and Lead Like Troglitazone Trigger PPARγ-Dependent Poly (ADP-Ribose) Polymerase Expression and Subsequent Apoptosis in Rat Brain Astrocytes. Mol Neurobiol 55, 2125–2149 (2018). https://doi.org/10.1007/s12035-017-0469-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0469-7