
Abstract
Long interspersed nuclear elements-1 (LINE-1 or L1) are mobile DNA sequences that are capable of duplication and insertion (retrotransposition) within the genome. Recently, retrotransposition of L1 was shown to occur within human brain leading to somatic mosaicism in hippocampus and cerebellum. Because unregulated L1 activity can promote genomic instability and mutagenesis, multiple mechanisms including epigenetic chromatin condensation have evolved to effectively repress L1 expression. Nonetheless, L1 expression has been shown to be increased in patients with Rett syndrome and schizophrenia. Based on this evidence and our reports of oxidative stress and epigenetic dysregulation in autism cerebellum, we sought to determine whether L1 expression was increased in autism brain. The results indicated that L1 expression was significantly elevated in the autism cerebellum but not in BA9, BA22, or BA24. The binding of repressive MeCP2 and histone H3K9me3 to L1 sequences was significantly lower in autism cerebellum suggesting that relaxation of epigenetic repression may have contributed to increased expression. Further, the increase in L1 expression was inversely correlated with glutathione redox status consistent with reports indicating that L1 expression is increased under pro-oxidant conditions. Finally, the expression of transcription factor FOXO3, sensor of oxidative stress, was significantly increased and positively associated with L1 expression and negatively associated with glutathione redox status. While these novel results are an important first step, future understanding of the contribution of elevated L1 expression to neuronal CNVs and genomic instability in autism will depend on emerging cell-specific genomic technologies, a challenge that warrants future investigation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Long interspersed nuclear elements-1 (LINE-1 or L1) are repetitive “jumping” DNA retrotransposons capable of duplication and reinsertion into the genome by a copy-and-paste mechanism and comprise ∼17% of the human genome [1]. Recent studies have shown that L1 transposable elements in human and mouse genomes are capable of active transposition and insertion during neuronal differentiation [2–4]. The full-length mammalian L1 is ∼6-kb long and consists of three major components: a 5′ untranslated region (5′UTR), containing an internal promoter, two open reading frames (ORF1 and ORF 2), and a 3′ UTR with a poly (A) signal and tail. ORF1 codes for an RNA binding protein while ORF2 encodes an endonuclease and reverse transcriptase required for autonomous retrotransposition [5, 6]. Because the ORF2 reverse transcriptase often fails to reach the 5′ end, most L1 transcription results in 5′ truncated insertions incapable of retrotransposition. Further, most L1 copies are mutated such that very few are active in the human genome. Approximately 80–100 full-length human L1 elements are estimated to be retrotransposition-competent in the average human genome [3]. The frequency of L1 active transposition in neural progenitor cells and somatic cells is much greater than previously recognized and significantly contributes to neuronal genetic mosaicism [7]. Moreover, L1-mediated insertions are considered to be a type of endogenous mutagen capable of generating insertions/deletions and copy number variation that are established risk factors for neurobehavioral disorders including autism, schizophrenia, and Alzheimer’s disease [8, 9].
Until recently, mammalian L1 transposition was thought to only occur in germ cells and cancer cells. However, it is now well accepted that L1 is capable of de novo retrotransposition in neural progenitor cells (NPC) as well as later during adult neurogenesis. Recent studies have shown that an engineered LINE-1 sequence containing a retrotransposition reporter cassette can mobilize and insert into NPC from rat hippocampus [2] and also in human fetal brain and human embryonic stem cells in vitro [4]. Reporter assays have also shown that L1 mobilization and insertion can occur in vivo utilizing transgenic mice with human L1 retrotransposition-competent sequences [2]. Multiplex qPCR technology has confirmed that L1 copy number is significantly elevated in human brain compared to non-neural tissues and that the elevated copy number is neuron-specific [4]. By applying deep sequencing data and gene ontology analysis on the L1 integration sites in human postmortem brain, Baillie et al. concluded that the sites of integration were most enriched in genes involved in the neuronal synapse, axonigenesis, postsynaptic density, presynaptic membrane, neuron projection, and cell adhesion [9]. L1-induced individual neuronal DNA sequence diversity (somatic mosaicism) has been reported in both rodents and humans [7, 9]. Although neuron-specific L1 insertions are consistent with genomic instability, it remains to be determined whether or not increased L1 expression and mobilization have a functional impact on neuronal plasticity, cognition, and ultimately behavior.
Increased rates of neuronal L1 retrotransposition and copy number have been reported patients and mouse models of Rett syndrome and schizophrenia and provide supportive evidence for a possible contribution of L1 retrotransposition to the pathophysiology of these neurobehavioral disorders. Rett syndrome is a neurodevelopmental disorder caused by mutation in the MeCP2 gene that typically occurs in girls and is associated with comorbid autism [10]. MeCP2 binds to methylated DNA in the L1 promoter and represses transcription in NPCs [11]. Notably, postmortem brain tissue from patients with Rett syndrome had significantly higher number of L1 sequences compared to age- and gender-matched controls [11]. In addition, de novo L1 insertion frequency was 2.5-fold higher in NPCs induced from a patient with Rett syndrome. Further studies with MeCP2 K/O mice were consistent with epigenetic control of L1 expression through alterations in heterochromatin landscape [11]. It has also been reported that people with schizophrenia have increased L1 neuron-specific copy numbers in prefrontal cortex [12]. Whole-genome sequencing revealed that L1 insertions occurred primarily in synapse and schizophrenia-related genes. Together, these studies indicate that misregulation of neuronal L1 expression can occur in neurobehavioral disorders and that continued investigation into potential functional consequences is needed.
Because L1 retrotransposition is regulated by epigenetic mechanisms that are environmentally responsive, recent evidence has confirmed that L1 expression is activated by exposure to environmental stressors. For example, L1 retrotransposon expression is increased in the alcoholic human brain and associated with decreased epigenetic repression [13]. In human neuronal cell lines, methamphetamine and cocaine exposure-induced L1 mobilization that was dependent on CREB activation [14, 15]. In cultured L1-stable human NIH3T3 cells, L1 activity was significantly increased following low-level exposure to heavy metals [16] and also in human neuroblastoma cells exposed to oxidative stress [17]. Increased L1 copy number was found after immune activation in the maternal poly/IC mouse model of schizophrenia and autism and after prolonged exercise [12, 18]. Collectively, these studies are consistent with the hypothesis that neurons can epigenetically increase neuronal L1 activity to modify their genomes in response to environmental stressors.
Based on evidence of environmentally responsive regulation of L1 activity in the brain [18] and our previous demonstration of oxidative stress/damage and epigenetic dysregulation of the Engrailed-2 (EN-2) gene in autism postmortem cerebellum [19], the present study was undertaken to determine (1 ) whether L1 expression is increased in autism brain; (2) whether epigenetic alterations and glutathione redox status influence L1 expression; and (3) whether the expression of Forkhead Box O3 (FOXO3) transcription factor, a sensor of oxidative stress, influences L1 expression. The results show for the first time that (1) L1 expression is significantly increased in autism postmortem cerebellum but not in BA9, BA22 or BA24; (2) increased expression of L1 is associated with evidence of oxidative stress and reduced binding of MeCP2 and histone H3K9me3; and (3) FOXO3 expression is highly correlated with L1 expression and glutathione redox status.
Results
Level of L1 ORF1 and ORF2 Transcripts in Four Brain Regions
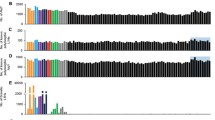
Based on evidence that L1 retrotransposition is elevated in postmortem brain from individuals with Rett syndrome and schizophrenia [11, 12], our initial aim was to explore this possibility in autism postmortem brain. To this end, we measured the levels of L1 ORF1 and ORF2 transcripts in four brain regions: BA9, BA24, BA22, and cerebellum. Expression of ORF1 and ORF2 is initiated by the promoter in the 5′UTR and both are required for retrotransposition. There was wide variation and no significant difference in transcript levels in any of the cerebral cortical BA9, BA24, and BA22 regions examined (Supplementary Fig. 1). However, as shown in Fig. 1a, b, there was a highly significant increase in total RNA and mRNA in both ORF1 and ORF2 in the autism cerebellum although there was no significant difference in overall L1 copy number (data not shown). Initial studies demonstrated that upregulation of L1 activity is primarily neuron-specific and occurs in early brain development during NPC differentiation and also during adult neurogenesis [7, 18, 20]. Interestingly, upregulation of L1 expression also occurs as an adaptive response to environmental stressors [14, 16–18]. While our data cannot define the temporal aspects of expression, we clearly show for the first time that L1 ORF1 and 2 mRNA transcripts are significantly elevated in the autism cerebellum relative to carefully matched control samples. Figure 1c shows the remarkably high correlation (r = 0.95; p = 0.0001) between the expression of ORF1 and ORF2. For full-length insertion to occur, both ORF1 and ORF2 must be expressed. Thus, the coexpression of both ORF1 and ORF2 strongly suggests that the 5′UTR promoter was fully functional since 5′-truncated L1 insertions are transcriptionally incompetent.

L1 ORF1 and ORF2 expression in autism and control cerebellar samples. Quantitative expression of ORF1 and ORF2 transcripts was measured by qRT-PCR using cDNA from total RNA and polyA-tailed mRNA (mRNA). a ORF1 total RNA and mRNA transcript levels (fold change) with significant differences at p = 0.02 and 0.04 respectively (mean ± SEM). b ORF2 total and mRNA transcript levels (fold change) with significant differences at p = 0.03 (mean ± SEM). Asterisk denotes a significant difference from the control group. c Linear regression was performed to evaluate associations between expression of ORF1- and ORF2-containing poly(A) + mRNA transcripts in autism group. The highly significant correlation between ORF1 and ORF2 expression (r value of 0.95, p = 0.0001) is consistent with coexpression that is required for L1 insertion. Each symbol represents an individual case
Reduced MeCP2 Binding to L1 5′UTR Associated with Increased ORF1 Expression
The 5′UTR promoter sequence of L1 is usually heavily methylated at multiple CpG sites as a mechanism for repressing its expression. Patients with Rett syndrome have a mutation in MeCP2, a methyl-binding protein involved in gene silencing, and these patients develop symptoms of autism. Muotri et al. [11] were the first to show that L1 transcriptional activity and retrotransposition can be modulated by MeCP2. In MeCP2 knockout mice, L1 promoter activity was about four times higher than in wild-type mice [11]. In humans, neural progenitor cells and postmortem brain derived from patients with Rett syndrome had significantly higher frequency of L1 retrotransposition compared to control cells [11]. To determine whether a similar mechanism may be involved in overexpression of L1 in autism cerebellum, we analyzed MeCP2 binding to the 5′ promoter region using ChIP technology as described in the “Materials and Methods” section. There was large variation and no significant difference between case and control samples in the overall mean binding of MeCP2 to the L1 5′UTR (data not shown). However, as shown in Fig. 2, correlation analysis between MeCP2 binding to the 5′UTR and ORF1 expression resulted in a significant negative association in the autism that was not present in matched control samples (p ≤ 0.05). These data suggest that in some cases, reduced MeCP2 binding in the L1 promoter may contribute to the increase in ORF1 expression in the autism cerebellum.

Negative correlation between MeCP2 binding to L1 5′UTR and ORF1 expression in autism and control cerebellar samples. ChIP analysis of MeCP2 binding to the 5′UTR was conducted using primers spanning two regions of L1 5′UTR (nt 24–322 and nt 390–597) in autism and control samples. There was wide variation and no significant difference in mean MeCP2 binding to the 5′UTR or to ORF1 and ORF2. However, regression analysis revealed a significant negative correlation between ORF1 expression and MeCP2 binding at both 5′UTR regions (a, b) such that higher MeCP2 binding to the 5′UTR was associated with lower ORF1 expression in autism but not in control samples. The X-axis represents the MeCP2 ChIP data expressed as the percent of input DNA after adjusting for total input DNA for each individual case
Reduced Binding of Repressive Histone H3K9me3 to ORF1 and ORF2 Sequences
Because of the mutagenic potential of L1 insertions, multiple epigenetic mechanisms, including cytosine CpG methylation and posttranslational modifications of histone proteins, have evolved to inhibit L1 expression and activity [21]. We first examined whether loss of repressive DNA methylation of CpG sites within the L1 5′UTR could be associated with increased ORF1 and ORF2 expression. Using MeDIP methodology, the results indicated that there were no significant differences in the DNA methylation density in the 5′UTR, ORF1 or ORF2 sequences comparing the case and control samples (Supplementary Fig. 2). Trimethylation of histone H3 lysine 9 is a major epigenetic mechanism responsible for the formation of condensed heterochromatin and prevention/inhibition of LINE-1 activation [22, 23]. Using ChIP methodology, we found a significant reduction in level of histone H3K9me3 at both ORF1 and ORF2 sequences but not in the L1 5′UTR as presented in Fig. 3. These data are consistent with the hypothesis that a local reduction in H3K9me3 level and associated increase in chromatin accessibility may be contributed to the overexpression of ORF1 and ORF2 in the autism cerebellum.

ChIP analysis of trimethylated histone 3 lysine 9 (H3K9me3) binding to L1 in the cerebellum of autism and control groups. The binding of H3K9me3 to both 5′UTR regions was not different between case and control samples. However, there was a significant decrease in H3K9me3 binding to both L1 ORF1 and L1 ORF2 sequences. Values are presented as mean percent of input DNA ± SEM. Asterisk denotes a significant difference from the control group
ORF1 and ORF2 Expression Negatively Correlated with Glutathione Redox Status
We previously reported that glutathione redox status, the ratio of active reduced to inactive oxidized glutathione disulfide (GSH/GSSG), is significantly decreased in the autism cerebellar cortex [24] as well as in plasma [25], lymphoblastoid cell lines, and mitochondria derived from individuals with autism [26]. Glutathione is the primary intracellular antioxidant and redox buffer that is responsible for maintaining the reduced intracellular microenvironment that is essential for normal cell function and viability. Because L1 retrotransposition has been previously associated with environmental exposures related to oxidative stress, we assessed the correlation between ORF1 and ORF2 expression and GSH/GSSG redox status in the autism cerebellar samples relative to controls. As shown in Fig. 4, there is a highly significant negative correlation between ORF1 (p = 0.01) and ORF2 (p = 0.005) expression and GSH/GSSG redox status that is not present in matched control samples. These results suggest the intriguing possibility that L1 expression may be activated under conditions of redox imbalance and oxidative stress.

Correlation between ORF1 and ORF2 expression and GSH/GSSG redox ratio in autism and control cerebellar samples. a Significant negative correlation between ORF1 mRNA and GSH/GSSG redox in autism samples that was not present in control samples. b Significant negative correlation between ORF2 mRNA and GSH/GSSG that was not present in control samples. Lower GSH/GSSG redox status (increased oxidative stress) was associated with increased ORF1 and ORF2 expression. Each symbol represents an individual case
FOXO3 Expression and Binding to L1 5′UTR Promoter
FOXO3 family of transcription factors are sentinels of oxidative stress that mediate multiple transcriptional targets including signals for proliferation, apoptosis, stress resistance, and mitochondrial metabolism [27, 28]. Given that the FOXO3 ortholog is predominantly expressed in the brain and our previous evidence of oxidative stress in the autism cerebellum [24], we chose to examine the expression of FOXO3 in our samples. In Fig. 5a, we show a significant increase in FOXO3 expression in the autism cerebellum relative to the control samples (p = 0.01) that is negatively associated with the glutathione redox ratio (Fig. 5b). Thus, the lower the GSH/GSSG ratio, the higher the FOXO3 expression (r = − 0.65; p = 0.01). FOXO3 expression is also highly correlated with ORF1 expression (Fig. 5c; r = 0.81, p = 0.001) and also ORF2 expression (Fig. 5d; r = 0.79, p = 0.001). These data provide evidence for the first time that FOXO3 expression is (1) elevated in the autism brain, (2) may be negatively regulated by the glutathione redox ratio, and (3) strongly associated with L1 ORF1 and ORF2 expression. Finally, it was of interest to determine whether FOXO3 protein binds to the LINE-1 5′UTR promoter. Based on literature references [29, 30], we found three potential binding sites for FOXO3 in the L1 5′UTR promoter regions covered by our primers. Relative to the control samples, there was a highly significant increase in FOXO3 binding to between nt 24 and 322 (p = 0.05) and between nt 390 and 597 (p = 0.01) in the autism samples despite wide variability (Fig. 5e, f). Together, the data are consistent with the possibility that the FOXO3 response to oxidative stress may contribute to L1 activation in the autism cerebellum.

FOXO3 expression and protein binding to L1 5′UTR. a The level of FOXO3 mRNA was significantly elevated in autism compared to control samples. Values are presented as mean ± SEM. Asterisk denotes a significant difference from the control group. b Highly significant negative correlation between FOXO3 expression and GSH/GSSG redox status in autism group is consistent with previous reports that FOXO3 expression is upregulated under conditions of oxidative stress. Each symbol represents an individual case. c, d Highly significant positive correlation in the autism group between FOXO3 expression and ORF1 and ORF2 expression, respectively. e ChIP analysis of FOXO3 protein binding to two regions of the L1 5′UTR in the cerebellum of autism and control patients. Values are presented as mean percent of input DNA ± SEM. Asterisk denotes a significant difference from the control group. FOXO3 protein binding is shown to be significantly elevated in both primer regions of the L1 5′UTR
Discussion
The long interspersed nuclear elements are the only autonomous mobile elements that are active in humans. Although L1 expression is suppressed in most peripheral somatic cells, recent evidence has convincingly shown that neuronal progenitor cells (NPCs) are a notable exception. NPC in humans and mice are capable of high levels of L1 expression and insertion during neurogenesis that can lead to subsequent somatic mosaicism, a de novo form of mutagenesis that can promote neuronal CNVs and genomic instability [2–4, 9, 31]. Interestingly, in postmortem human brain, L1 retrotransposition has been shown to be active in the subgranular zone of the hippocampus which is a major site of adult neurogenesis [4, 9, 18]. Deep-sequencing techniques further demonstrated that L1 integrates predominantly in intragenic sites compared to exonic sites and is enriched in protein-coding regions. Subsequent gene ontology analysis revealed enrichment for genes involved in neurogenesis and synaptic function although these results have not yet been replicated [9]. In a recent ground-breaking study, Erwin et al. [31] demonstrated that L1 insertions occur in hippocampal and cerebellar DNA and that approximately 0.5% of cells from both tissues contained the L1 sequence suggesting a heritable L1 component that originally derived from a progenitor cell insertion. Using sophisticated single nuclei sequencing technology, they confirmed that L1 retrotransposon insertions occur in NPCs and also in neurons and glia. Their data further indicate that L1 sequences are prone to double-stranded DNA damage preferentially at germline L1 loci mediated by the upregulation of ORF2 endonuclease during neural differentiation. In previous work, this group demonstrated that retrotransposition of an engineered L1 in mice resulted in overexpression of the DLG2 gene which altered the differentiation pattern of NPCs [2]. They now report a somatic L1 variant in the same gene in differentiated human hippocampal progenitor cells. Of interest, mutations in DLG2 have been reported in schizophrenia [32, 33] and a copy number variation in DLG2 has been reported in autism [34]. These results suggest that genomic variation in this gene, including L1 somatic variants, may contribute to neurobehavioral disorders.
Our results are limited in that they are global in scope and not cell specific in terms of the observed increase in ORF1 and ORF2 expression. It is curious that L1 copy number was not increased despite significant increase in the expression of both ORF elements. This suggests that the increased expression did not occur during heritable NPC lineage differentiation but more likely occurred as a de novo adaptive response that was not reflected in global copy number. Further, the highly significant correlation between ORF1 and ORF2 expression (Fig. 1c) is consistent with competent full-length L1 insertion potential that could impact neuronal gene expression and predisposition to disease as recently reviewed by Elbarbary et al. [35]. Nonetheless, the functional consequence of increased L1 expression, if any, cannot be ascertained until identification of specific transcripts disrupted by somatic retrotransposition is technically possible and shown to negatively affect neuroplasticity, cognition, or behavior.
The redox state of the cell is determined by the relative balance between oxygen/nitrogen free radical accumulation and the cellular antioxidant defense potential. Although a more oxidized microenvironment is often considered to be negative and damaging to cellular structure and function, it is now well accepted that transient low-level reactive oxygen species (ROS) such as superoxide and hydrogen peroxide provide critical signaling molecules that ultimately regulate cell fate decisions to proliferate or differentiate during neurogenesis [36]. The redox poise during neurogenesis is dynamic and varies with the homeostatic fluctuation of multiple reduced and oxidized redox couples in response to internal and environmental signals [36]. The intracellular redox homeostasis and antioxidant defense is primarily maintained by the GSH/GSSG redox couple which is especially important in tissues like the brain where 20% of total body oxygen is consumed for oxidative phosphorylation and energy production. An unexpected finding was the region-specific increase in L1 expression in the cerebellar cortex that was not present in frontal cortical regions. Interestingly, several other investigators have found a cerebellum-specific increase in oxidative stress that theoretically could underlie the increase in L1 expression.
In a previous publication, we reported a significant decrease in glutathione GSH/GSSG redox ratio in the same cerebellar samples utilized in the present study [24]. Combining data, we found a highly significant correlation between the decrease in GSH/GSSG redox potential and both ORF1 and ORF2 expression in autism samples not present in matched control samples (Fig. 2). An increase in L1 expression under oxidizing conditions has been previously reported by several investigators [16, 17, 37, 38]. Taken together, these data support the possibility that an increase in LINE-1 expression in the autism cerebellum may be upregulated under prooxidant conditions.
Moutri et al. have provided convincing evidence that the relative MeCP2 binding to the L1 promoter can regulate their expression in the mouse brain and that L1 retrotransposition frequency is higher in postmortem brain samples from people with Rett syndrome relative to controls [11]. Similar to our results, Muotri et al. found no difference in copy number in postmortem brain samples from patients with Rett syndrome although an increase in L1 copy number has been reported in patients with schizophrenia [12]. Although we found no significant difference in global DNA methylation density or in global MeCP2 binding to the L1 5′UTR promoter region in the autism cerebellar samples, we did find a significant negative correlation between MeCP2 binding to the L1 promoter region and the expression of ORF1 that was not present in control samples (Fig. 3). The possibility that a decrease in repressive MeCP2 binding could result in an upregulation of ORF1 expression is supported by similar results in MeCP2-deficient mice as reported by Moutri et al. [11]. Using ChIP qPCR methodology, we also explored whether reduced level of chromatin repressive H3K9Me3 could contribute to the increased expression of ORF1 and ORF2. The significant decrease in H3K9me3 binding to both ORF1 and ORF2 sequences (Fig. 4) suggests the possibility that a more accessible local chromatin structure may also have contributed to the observed increase in ORF1 and ORF2 expression. Together, the reduction in both MeCP2 and H3K9me3 binding in the autism cerebellum are consistent with recent evidence suggesting that epigenetic mechanisms influence L1 expression and retrotransposition potential [39].
Members of the forkhead box protein O (FOXO) family of transcription factors are largely regulated by two conserved pathways: negative regulation occurs in response to insulin and growth factor signaling whereas positive regulation occurs in the presence of oxidative stress through JNK activity [29]. FOXO3 is activated by cysteine oxidation under conditions of elevated intracellular ROS and functions to activate target genes involved in the cellular response to oxidative stress [28, 40]. Given that both L1 and FOXO3 expression can be activated by oxidative stress and our previous observation that GSH/GSSG was significantly decreased in the autism cerebellum, we investigated FOXO3 transcript levels and FOXO3 protein binding to the L1 5′UTR promoter. Not only was overall FOXO3 gene expression significantly higher in the autism samples, the expression of FOXO3 was also negatively correlated with GSH/GSSG such that lower GSH/GSSG was associated with higher FOXO3 expression (p < 0.001; Fig. 5a, b) consistent with redox regulation of FOXO3 in the autism cerebellum. A possible functional relationship between FOXO3 and L1 expression is further supported by the highly significant correlation between FOXO3 expression and the expression of both ORF1 and ORF2 (p = 0.01 and 0.001 respectively; Fig. 5c, d). Finally, ChIP analysis demonstrated in a significant increase in FOXO3 protein binding to the 5′UTR L1 promoter region in the autism relative to the control cerebellar samples (p = 0.01 and 0.05; Fig. 5e, f). An increase in FOXO3 expression protects cells from oxidative stress by binding to promoter regions and by transactivating multiple genes involved in antioxidant defense including manganese superoxide dismutase (MnSOD), catalase and peroxiredoxin 3 (Prx3), and thioredoxin 2 (Trx2) [27, 41]. Our results suggest the possibility that upregulation of FOXO3 occurred in response to a chronic reduction in GSH/GSSG antioxidant capacity and resulting oxidative stress in the autism cerebellum.
Conclusions
To our knowledge, this is the first report of an increase in L1 expression in the autism cerebellum and its association with oxidative stress and FOXO3 activation. Whether or not these observations are functionally related to the etiology or pathophysiology of autism cannot be ascertained from results in postmortem brain. Although many technologic challenges remain, the use of retrotransposon inhibitors, genome editing, and recent cell-specific and site-specific technologies may serve to uncover the basis and implications of the selective increase in L1 expression in the autism brain. Nonetheless, the evidence presented in this report is novel and warrants deeper investigation into L1 expression during fetal and adult neuronal differentiation to better understand the potential relationship between L1 insertional mutagenesis, genomic instability, and the neurodevelopmental abnormalities in the autism brain.
Materials and Methods
Postmortem Cerebellar Samples
The frozen blocks of postmortem cerebellar cortex from autism individuals (n = 13) and unaffected control individuals (n = 13) were obtained from the NICHD and Tissue Bank for Developmental Disorders (University of Maryland) and from the Autism Tissue Program at the Harvard Brain Tissue Resource Center, Belmont, MA, USA. All case donors had a confirmed diagnosis of autism based on the Autism Diagnostic Observation Schedule (ADOS), the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV), and/or Autism Diagnostic Interview Revised (ADI-R). Autism and control samples were rigorously matched for postmortem interval, age, gender, and race, and cause of death was matched as closely as possible (Supplementary Table 1).
L1 Primer Design
In addition to published primers for human LINE-1 5′UTR (h5UTR-s3), LINE-1 ORF2 (hORF2-s1), and alpha satellite repetitive element internal control (SATA) as described by Coufal et al. [4], we designed several new primers following the approach described in Bundo M et al. [12]. Briefly, the full-length L1 sequences from L1Base (http://line1.bioapps.biozentrum.uni-wuerzburg.de/l1base.php) were aligned using Kalign software (EMBL-EBI) (http://www.ebi.ac.uk/Tools/msa/kalign/). The primers were designed for the conserved regions of L1 5′UTR (h5UTR-s1–2), ORF1 (h5ORF-s1), and ORF2 (hORF-s2,3) using the Real-Time qPCR Assay software (Integrated DNA Technologies; IDT), and then were tested with in-silico Blat (USCS) (https://genome.ucsc.edu/cgi-bin/hgPcr?db=hg19) and with L1 DNA sequences available in NCBI Nucleotide database (http://www.ncbi.nlm.nih.gov/nuccore). Forward (F) and reverse (R) primers for each application, as well as probe (P) sequences, are listed in Supplementary Table 2. The qPCR probes were conjugated to 6FAM (5UTR, ORF1, ORF2) or VIC (SATA). All primers and probes were synthesized at Integrated DNA Technologies (IDT, Coralville, IA) or at Applied Biosystems (Thermo Fisher Scientific, Waltham, MA). The amplification efficiency of a qPCR was calculated based on the slope of the standard curve.
L1 RNA Transcript Measurements
Total RNA was extracted from cerebellum brain samples using TRI Reagent (Ambion, Thermo Fisher Scientific, Waltham, MA), DNAse I treated and purified with RNeasy Mini Kit (Qiagen, Valencia, CA). RNA quality (RNA integrity number) was assessed with Agilent RNA 6000 Nano Kit and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). All samples had a RIN number >4 which is considered a minimum standard for postmortem RNA quality in human brain tissue [42]. PolyA-tailed mRNAs were isolated from total RNA by using mRNA Catcher™ PLUS (Invitrogen, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Total RNA or polyA-tailed mRNA was reverse transcribed using random primers and high-capacity cDNA reverse transcription kit (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol, and cDNA was analyzed in a 96-well plate assay format using the 7900HT Fast Real-Time PCR System (Life Technologies). The qPCR primers for ORF1 and ORF2 were used for L1 transcript level estimation (Supplementary Table 2). The cycle threshold (Ct) for each sample was determined from the linear region of the amplification plot. The ΔCt values for all genes were determined relative to the endogenous control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and succinate dehydrogenase subunit A (SDHA). The L1 transcript level was presented as fold change. All qRT-PCR reactions were conducted in triplicate and repeated twice.
LINE-1 Copy Number Profiling Using Real-Time Quantitative PCR
DNA was extracted and purified with DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. DNA quality was confirmed by agarose electrophoresis and DNA concentration was strictly adjusted using the Nanodrop 2000c/2000 UV-Vis spectrophotometer (Thermo Fisher Scientific, Waltham, MA) and Quant-iT™ PicoGreen ® dsDNA Kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA). Genomic DNA aliquots used in qPCR reactions corresponded to spectrophotometric estimates of 120 pg of DNA. Each sample was routinely analyzed in triplicate. The comparative Ct method with SATA as internal control was used to calculate CNV with Copy Caller v2.0 software (Applied Biosystems,Thermo Fisher Scientific, Waltham, MA).
Chromatin Immunoprecipitation Assay of MeCP2, Histone H3 Lysine 9 Trimethylation (H3K9Me3) and FOXO3
Chromatin immunoprecipitation (ChIP) assays were performed using the Magna ChIP A Kit (EMD Millipore, Billerica, MA) and primary antibodies: mouse monoclonal antibodies against MeCP2 (Active Motif, Carlsbad, CA), rabbit polyclonal antibody against Histone H3 lysine 9 trimethylated (Abcam, Cambridge, MA), rabbit polyclonal antibody to FOXO3A (Abcam). Purified immunoprecipitated DNA and input DNA (pre-immunoprecipitation) were subsequently coamplified by qPCR. The ChIP primers within the L1 promoter were designed as described earlier. All assays were run in duplicate and data expressed as the mean (±S.E) percent input DNA after adjusting for total input DNA: 100 × 2(adjusted input Ct − IP Ct).
5-Methylcytosine MeDIP Quantification within the LINE-1 Promoter and Gene Body
The 5-methylcytosine MeDIP assay was carried out within L1 promoter and in the gene body as described in the protocol for the magnetic methylated DNA immunoprecipitation kit (Diagenode, Denville, NJ). (Diagenode, NJ, USA). Briefly, 1 μg of genomic DNA fragments were immunoprecipitated with 2.5 μg of mouse monoclonal 5-mC antibody or with 2.5 μg of mouse IgG per tube on the magnetic beads. DNA from the antibody-bound fractions was purified by Proteinase K in DNA Isolation Buffer (DIB) and stored at −20 °C. DNA was subjected to real-time qPCR analysis. Percent enrichment was calculated by 100 × 2 (ΔCTadjusted input − ΔCTenriched). Input DNA ΔCT value was adjusted from 10 to 100% equivalent to subtracting 3.32 ΔCts or log2 10.
Statistical Analyses
Results are presented as mean ± SD or SEM. Regression analysis was performed using Prism 6 software. P values ≤0.05 were considered significant.
References
Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr (2003) Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A 100:5280–5285. doi:10.1073/pnas.0831042100
Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435:903–910. doi:10.1038/nature03663
Thomas CA, Paquola AC, Muotri AR (2012) LINE-1 retrotransposition in the nervous system. Annu Rev Cell Dev Biol 28:555–573. doi:10.1146/annurev-cellbio-101011-155822
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131. doi:10.1038/nature08248
Feng Q, Moran JV, Kazazian HH Jr, Boeke JD (1996) Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell 87:905–916. doi:10.1016/S0092-8674(00)81997-2
Erwin JA, Marchetto MC, Gage FH (2014) Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci 15:497–506. doi:10.1038/nrn3730
Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH (2010) LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci 33:345–354. doi:10.1016/j.tins.2010.04.001
Guffanti G, Gaudi S, Fallon JH, Sobell J, Potkin SG, Pato C, Macciardi F (2014) Transposable elements and psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet 165B:201–216. doi:10.1002/ajmg.b.32225
Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479:534–537. doi:10.1038/nature10531
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23:185–188. doi:10.1038/13810
Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446. doi:10.1038/nature09544
Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, Sunaga F, Toritsuka M (2014) Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81:306–313. doi:10.1016/j.neuron.2013.10.053
Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD (2012) Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci 32:1884–1897. doi:10.1523/JNEUROSCI.3136-11.2012
Okudaira N, Ishizaka Y, Nishio H (2014) Retrotransposition of long interspersed element 1 induced by methamphetamine or cocaine. J Biol Chem 289:25476–25485. doi:10.1074/jbc.M114.559419
Moszczynska A, Flack A, Qiu P, Muotri AR, Killinger BA (2015) Neurotoxic methamphetamine doses increase LINE-1 expression in the neurogenic zones of the adult rat brain. Sci Rep 5:14356. doi:10.1038/srep14356
Kale SP, Moore L, Deininger PL, Roy-Engel AM (2005) Heavy metals stimulate human LINE-1 retrotransposition. Int J Environ Res Public Health 2:14–23
Giorgi G, Marcantonio P, Del Re B (2011) LINE-1 retrotransposition in human neuroblastoma cells is affected by oxidative stress. Cell Tissue Res 346:383–391. doi:10.1007/s00441-011-1289-0
Muotri AR, Zhao C, Marchetto MC, Gage FH (2009) Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus 19(10):1002–1007. doi:10.1002/hipo.20564
James SJ, Shpyleva S, Melnyk S, Pavliv O (2013) Pogribny IP (2013) complex epigenetic regulation of engrailed-2 (EN-2) homeobox gene in the autism cerebellum. Transl Psychiatry 3:e232. doi:10.1038/tp.2013.8
Kurnosov AA, Ustyugova SV, Nazarov VI, Minervina AA, Komkov AY, Shugay M, Pogorelyy MV, Khodosevich KV (2015) The evidence for increased L1 activity in the site of human adult brain neurogenesis. PLoS One 10:e0117854. doi:10.1371/journal.pone.0117854
Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8:272–285. doi:10.1038/nrg2072
Huda A, Marino-Ramirez L, Jordan IK (2010) Epigenetic histone modifications of human transposable elements: genome defense versus exaptation. Mob DNA 1:2. doi:10.1186/1759-8753-1-2
Bulut-Karslioglu A, De La Rosa-Velazquez IA, Ramirez F, Barenboim M, Onishi-Seebacher M, Arand J, Galan C, Winter GE (2014) Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol Cell 55:277–290. doi:10.1016/j.molcel.2014.05.029
Rose S, Melnyk S, Pavliv O, Bai S, Nick TG, Frye RE, James SJ (2012) Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry 2:e134. doi:10.1038/tp.2012.61
James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, Cutler P, Bock K (2006) Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet 141B:947–956. doi:10.1002/ajmg.b.30366
James SJ, Rose S, Melnyk S, Jernigan S, Blossom S, Pavliv O, Gaylor DW (2009) Cellular and mitochondrial glutathione redox imbalance in lymphoblastoid cells derived from children with autism. FASEB J 23:2374–2383. doi:10.1096/fj.08-128926
van der Vos KE, Coffer PJ (2011) The extending network of FOXO transcriptional target genes. Antioxid Redox Signal 14:579–592. doi:10.1089/ars.2010.3419
Storz P (2011) Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal 14:593–605. doi:10.1089/ars.2010.3405
Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A (2013) Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol 9:638. doi:10.1038/msb.2012.74
Tsai KL, Sun YJ, Huang CY, Yang JY, Hung MC, Hsiao CD (2007) Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res 35:6984–6994. doi:10.1093/nar/gkm703
Erwin JA, Paquola AC, Singer T, Gallina I, Novotny M, Quayle C, Bedrosian TA, Alves FI (2016) L1-associated genomic regions are deleted in somatic cells of the healthy human brain. Nat Neurosci 19:1583–1591. doi:10.1038/nn.4388
Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K (2012) De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry 17:142–153. doi:10.1038/mp.2011.154
Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, Georgieva L, Rees E (2014) De novo mutations in schizophrenia implicate synaptic networks. Nature 506:179–184. doi:10.1038/nature12929
Egger G, Roetzer KM, Noor A, Lionel AC, Mahmood H, Schwarzbraun T, Boright O, Mikhailov A (2014) Identification of risk genes for autism spectrum disorder through copy number variation analysis in Austrian families. Neurogenetics 15:117–127. doi:10.1007/s10048-014-0394-0
Elbarbary RA, Lucas BA, Maquat LE (2016) Retrotransposons as regulators of gene expression. Science 351:aac7247. doi:10.1126/science.aac7247
Ostrakhovitch EA, Semenikhin OA (2013) The role of redox environment in neurogenic development. Arch Biochem Biophys 534:44–54. doi:10.1016/j.abb.2012.08.002
Trivedi M, Shah J, Hodgson N, Byun HM, Deth R (2014) Morphine induces redox-based changes in global DNA methylation and retrotransposon transcription by inhibition of excitatory amino acid transporter type 3-mediated cysteine uptake. Mol Pharmacol 85:747–757. doi:10.1124/mol.114.091728
Kloypan C, Srisa-art M, Mutirangura A, Boonla C (2015) LINE-1 hypomethylation induced by reactive oxygen species is mediated via depletion of S-adenosylmethionine. Cell Biochem Funct 33:375–385. doi:10.1002/cbf.3124
Yang F, Wang PJ (2016) Multiple LINEs of retrotransposon silencing mechanisms in the mammalian germline. Semin Cell Dev Biol 59:118–125. doi:10.1016/j.semcdb.2016.03.001
Holmstrom KM, Finkel T (2014) Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15:411–421. doi:10.1038/nrm3801
Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL (2002) Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419:316–321. doi:10.1038/nature01036
Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE (2006) Critical factors in Gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry 60:650–658. doi:10.1016/j.biopsych.2006.06.019
Acknowledgements
We would like to thank the families of individuals with autism for the thoughtful donation of postmortem tissues to the Autism Tissue Program at the Harvard Brain Tissue Resource Center, Belmont, MA, and the NICHD Brain and Tissue Bank for Developmental Disorders at the University of Maryland, Baltimore, MD. This work was supported, in part, by the National Institute of Child Health and Development (RO1 HD051873 to SJJ) and the Jane Botsford Johnson Foundation.
The views expressed in this manuscript do not necessarily represent those of the US Food and Drug Administration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Grant Sponsors: National Institute of Child Health and Development; Grant number: 1RO1HD051873; Arkansas Biosciences Institute; Jane Botsford Johnson Foundation
Electronic supplementary material
Supplementary Figure 1
(PNG 99.9 kb)
Supplementary Figure 2
(PNG 95.4 kb)
Supplementary Table 1
(DOCX 16 kb)
Supplementary Table 2
(DOCX 18 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Shpyleva, S., Melnyk, S., Pavliv, O. et al. Overexpression of LINE-1 Retrotransposons in Autism Brain. Mol Neurobiol 55, 1740–1749 (2018). https://doi.org/10.1007/s12035-017-0421-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0421-x