
Abstract
Mitochondria are the primary source for energy generation in the cell, which manifests itself in the form of the adenosine triphosphate (ATP). Nicotinamide dinucleotide (NADH) molecules are the first to enter the so-called electron transport chain or ETC of the mitochondria. The ETC represents a chain of reducing agents organized into four major protein-metal complexes (I-IV) that utilize the flow of electrons to drive the production of ATP. An additional integral protein that is related to oxidative phosphorylation is ATP synthase, referred to as complex V. Complex V carries out ATP synthesis as a result of the electron flow through the ETC. The coupling of electron flow from NADH to molecular oxygen to the production of ATP represents a process known as oxidative phosphorylation. In this review, we describe mainly the bioenergetic properties of mitochondria, such as those found in the ETC that may be altered in Alzheimer’s disease (AD). Increasing evidence points to several mitochondrial functions that are affected in AD. Furthermore, it is becoming apparent that mitochondria are a potential target for treatment in early-stage AD. With growing interest in the mitochondria as a target for AD, it has been hypothesized that deficit in this organelle may be at the heart of the progression of AD itself. The role of mitochondria in AD may be significant and is emerging as a main area of AD research.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mitochondria are the primary source for biological energy generation in the cell, which manifests itself in the form of the coenzyme adenosine triphosphate (ATP) [1–3]. Considering the inherent complexity of biological systems, there exist numerous variations in the properties of the mitochondria that are found in certain cell types, but not others (e.g., increased levels of mitochondrial cristae folding are found primarily only in liver cells [4]). In addition to cell type variation is the existence of mitochondrial structure and/or function alterations due to genetic mutations or disease [2], such as in Alzheimer’s disease (AD).
In this review, to describe the properties of mitochondria that may be altered in AD, the normal properties of mitochondrial structure and function will be first briefly described, and then built upon to discuss dysfunction mechanisms that deviate from the normal state. For a full description of the biochemical processes highlighted below, the reader is encouraged to review biochemistry textbooks that cover such processes, such as Lehninger Principles of Biochemistry. Footnote 1 To pinpoint the properties of bioenergetics processes in the mitochondria, the electron transport system will be the primary focus of discussion when detailing structure and function.
General Mitochondrial Biochemistry
It has been hypothesized that mitochondria previously existed as a single prokaryotic cell, which at some point over the eons of time evolved symbiotically with a proto-eukaryotic cell resulting in uptake of the mitochondria and ultimately the creation of the first eukaryotic cell [2]. This endosymbiotic theory is widely accepted, driven by the discovery of mitochondrial DNA (mtDNA) and phylogenetic data supporting a monophyletic lineage for mitochondria. Following the uptake of mitochondria into the cell, there were many changes that now define the dynamic nature of the mitochondria.
The mitochondria contain an outer membrane, which is freely permeable to small molecules and ions. The unique nature of the mitochondria begins with the existence of an inner membrane that is impermeable to most molecules and ions. This membrane, referred to as the inner mitochondrial membrane, separates the mitochondria into two major subspaces: an inner space, the mitochondrial matrix; and a small region between membranes, the intermitochondrial space. This membrane also has a number of rib-like folds referred to as cristae, which increases its surface area to thereby maximize the function of the mitochondrial bioenergetic machinery. The inner mitochondrial membrane contains the electron transport chain (ETC), ADP-ATP translocases, ATP synthases, and numerous other membrane transport systems. Of central importance to this discussion is the ETC and the ATP synthases dispersed throughout this membrane, as these proteins are directly involved in utilizing the potential energy stored in substrates (e.g., glucose) to fuel the generation of ATP for biological activity.
The most important biologically relevant substrate for bioenergetic function in the mitochondria is glucose. Glucose molecules are transported into the cell through glucose transport systems, most commonly the GLUT or SLC2A transporter families, and are then prepared for catabolic processing in the cytosol [1]. As glucose is transported into the cell, it is catabolized in an enzymatic process known as glycolysis, where glucose is converted to two pyruvate molecules, resulting in a net production of two ATP, two H2O molecules, and two reduced nicotinamide dinucleotide (NADH) molecules. The production of NADH, the reduced form of NAD, is a key step in relating glucose catabolism to the ETC. These NAD molecules act as electron carriers that function to effectively transfer electrons from substrates such as glucose to the ETC. This occurs through reduction (i.e., gaining an electron) by sequestering hydrogen atoms in the form of a hydride ion (:H−) and a proton (H+), thus acquiring two electrons from a substrate to form NADH [2]. Once NADH molecules have been formed, they can be transported from the cytosol into the mitochondrion for oxidation through either the malate-aspartate shuttle or the glycerol 3-phosphate shuttle (see [1] or [2] for more information on these shuttle systems).
Next, the two molecules of pyruvate produced by glycolysis are transported into the mitochondrial matrix and then converted to acetyl-CoA by oxidative decarboxylation so that they can enter the citric acid cycle (a.k.a., Krebs cycle). It is important to note that this conversion results in the production of NADH for each pyruvate molecule, resulting in a total of four NADH molecules produced thus far. The Krebs cycle results in the incorporation of each acetyl-CoA into oxaloacetate, and after a series of reactions results in the total production of six NADH, two GTP (near ATP equivalents), two reduced flavin adenine dinucleotide (FADH2; discussed later), and four CO2. Importantly, the collective metabolic activities of glycolysis and the Krebs cycle have produced ten NADH molecules and a single FADH2.
The Electron Transport System
NADH molecules that have been reduced by Krebs cycle and to a lesser extent glycolysis and acetyl-CoA conversion are now free to enter the electron transport chain (ETC). This system represents a chain of reducing agents organized into four major protein-metal complexes that utilize the flow of electrons to drive the production of ATP. The coupling of electrons being carried from NADH to molecular oxygen with the production of ATP represents a process known as oxidative phosphorylation. Though the general description given above for this process seems simple, the complexity lies in the details that drive the flow of electrons through this chain and how this flow is utilized to generate ATP. A mechanistic description of this process warrants at minimum a general description of the structure of the electron transport system.
The Complexes and ATP Synthase
As seen in Fig. 1, four integral protein complexes comprise the electron transport system (the ETC.); (NADH)-coenzyme Q oxidoreductase, or complex I; succinate-coenzyme Q oxidoreductase, or complex II; Coenzyme Q-cytochrome c oxidoreductase, or complex III; and cytochrome c oxidase, or complex IV. An additional integral protein that is related to oxidative phosphorylation is ATP synthase, referred to also as complex V, which carries out ATP synthesis as a result of the electron flow through the ETC. This collection of protein complexes is located on the inner mitochondrial membrane separating the inner mitochondrial matrix from the intermitochondrial space, with complex V scattered throughout.

A depiction of the mitochondrial electron transport chain and complex V. Illustrated in this figure are each of the four protein complexes involved in electron flow starting from complexes I and II flowing toward complex IV. An expanded view of complex V illustrates its various subunits in the Fo and F1 regions, and how these subunits are involved in relating proton flow to ATP production. Further detail regarding the complexes illustrated in this figure is discussed in “The Electron Transport System” and “The Chemiosmotic Theory of Bioenergetics” sections
The nature of these complexes is similar in that electrons are passed through the complexes between metal and protein compounds by oxidation-reduction reactions or redox reactions. Redox reactions represent the transfer of an electron from one substance, a reducing agent, to another, an oxidizing agent [3]. The complexes are arranged such that electrons are channeled through a series of centers that are increasingly more potent-reducing reagents, so that each successive compound in the chain has a higher potential for accepting an electron than the previous compound. This allows the propagation of the electron through the chain from input at complexes I and II, up until the final electron acceptor, O2, in complex IV. In order to adequately follow the flow of electrons, however, it is necessary to initially outline the relevant components of each complex in this chain.
There are a number of elements that carry out the electron transfers within the complexes in the ETC. As detailed in the discussion of glucose catabolism above, NADH is a central electron carrier that sequesters electrons from a substrate to be delivered to the ETC. Another method of electron delivery occurs through flavoproteins, proteins that contain a flavin nucleotide, in the form of flavin mononucleotide (FMN) in complex I and flavin adenine dinucleotide (FAD) in complex II. Similar to NAD, these molecules act to accept electrons to form FMNH2 or FADH2, but operate to deliver electrons through different means since FMN acts as a relay station for electrons in complex I, and FAD transfers electrons in the reaction catalyzed by succinate dehydrogenase (SDH) in the Krebs cycle.
In addition to substrate-level electron carriers, there also exist membrane-bound electron carriers: a hydrophobic quinone (ubiquinone (Q or coenzyme Q)), cytochromes, and iron-sulfur clusters (Fe-S). Q can accept a single electron to become the semiquinone radical (:QH) or two electrons to form ubiquinol (QH2) [2]. In either form, Q can freely diffuse through the membrane since it is small and hydrophobic, allowing them to shuttle electrons between (QH2) or within complexes (:QH) [2]. The next form of electron carriers, the cytochromes, are proteins bound to an iron-containing heme group, classified as either cytochromes a, b, or c based on their light-absorption spectra, with cytochrome a having the longest wavelength band near 600 nm, b near 560 nm, and c near 550 nm [2, 3]. The iron heme groups of cytochromes operate as reducing agents and thus as carriers for electrons through the ETC. Cytochromes a and b are anchored to the membrane as integral proteins within the complexes, and cytochrome c moves through the membrane as a loosely bound soluble protein that interacts electrostatically with the inner membrane [2]. Finally, iron-sulfur clusters are groupings of iron atoms coordinated to either inorganic sulfur atoms or with the sulfur atoms of Cys residues in proteins. These iron-sulfur clusters can exist in either a [2Fe2S], [4Fe4S], or [3Fe4S] organization [2]. Rieske Fe-S clusters are similar in function to Fe-S clusters, but differ structurally in that iron atoms coordinate to His residues rather than Cys residues [2, 3]. Regardless of this structural difference, all iron-sulfur clusters function as reducing agents for passing electrons though the ETC.
Complex I contains 45 protein subunits that are segregated into a short, hydrophilic segment that slightly extends into the matrix and a long, hydrophobic segment that resides entirely in the inner membrane [2]. The short segment of complex I initiates the process of electron flow in the electron transport system, as this segment contains NADH dehydrogenase, the FMN cofactor, and the binding site for NADH, all of which are required for NADH oxidation and thus electron delivery to the system. NADH dehydrogenase oxidizes NADH by accepting two electrons from NADH, while the FMN cofactor operates as a relay center by splitting the two electrons between a series of seven iron-sulfur clusters for propagation and a holding iron-sulfur cluster [2]. This splitting process results in the propagation of one electron through the complex at a time. Through this propagation, the short subunit of complex I passes electrons from NADH through the complex to the membrane-bound electron carrier ubiquinone (Q) reducing it to ubiquinol (QH2). While this occurs, the long subunit of complex I carries out proton translocation, although this mechanism is not exactly known, it is believed that two protons are pumped by a redox-driven Q-cycle related mechanism, while two protons are pumped across by conformational transitions [2, 3].
Complex II is a small protein complex that contains four protein subunits comprised of integral anchoring proteins and succinate dehydrogenase, making it the only complex in the electron transport system with a direct enzymatic role in Krebs cycle. The two large peptides that constitute succinate dehydrogenase are separated into the Fp and Ip subunits, which are peptides with a flavin covalently attached or three Fe-S clusters attached, respectively.
This complex requires the covalently bound FAD cofactor, which operates to shuttle electrons through the complex one at a time much like FMN accomplishes in complex I [2]. In addition to the FAD cofactor, this complex also contains multiple iron-sulfur clusters and a heme group, heme b. Complex II accepts electrons directly from the Krebs cycle through succinate dehydrogenase, and much like the Fe-S chain in complex I, then carries electrons through these iron-sulfur clusters to reduce Q to QH2. Thus, both complex I and complex II operate as entry points for electrons to be passed to complex III using the carrier QH2.
The structure of complex III is unique compared to complexes I and II in that it is dimeric, composed of two identical protein chains. Each protein set contains an identical pairing of 11 protein subunits and 4 metal redox centers: cyt b562, cyt b566, a Rieske Fe-S cluster, and cyt c1 [2]. Complex III binds QH2 generated in complexes I and II at a binding site known as the Qo site, which resides near the intermembrane space [2]. Q can also bind to complex III at the Qi site, located near the mitochondrial matrix. Cytochrome c can also bind to complex III near the intermembrane space on the opposite side of the Qo site, where it acts as the final electron acceptor in the redox reactions that takes place in complex III [2].
Complex IV is the final complex in the ETC directly involved in electron flow. Complex IV is a large integral protein that much like complex III is dimeric in nature, with each half consisting of 13 protein chains and multiple metal reduction centers [2]. These metal centers consist primarily of iron and copper, organized into a bimetallic CuA site, a monometallic cytochrome a, and a bimetallic cytochrome a3-CuB site. CuA sits just outside the membrane, with the copper atoms bound by NHis, SMet, SCys, and Glu residues in protein subunits within each monomer [2]. Cytochrome a is bound to the protein subunits by two NHis groups, while cytochrome a3-CuB is bound to four NHis groups, with a very small binding site for O2 just between these atoms. Molecular oxygen is the final site for electron flow in complex IV, and although the exact mechanism of O2 reduction is not wholly understood, the theorized process is discussed below.
ATP synthase is an integral protein scattered throughout the inner mitochondrial membrane that is directly responsible for ATP synthesis. The structure of ATP synthase, or complex V, is shown in Fig. 1. It consists of two major components, the Fo portion, found within the membrane, and the F1 portion, found above the membrane within the mitochondrial matrix. The Fo region contains the subunits a, b, and c, in the ratio of ab 2 c 10–12 [2]. This region also forms a proton pore for H+ to flow through during ATP synthesis. The subunits of the Fo region are arranged into a ring of c subunits, with each c subunit orienting its amino-terminal helix within the inside of the ring. Attached to this c-ring is the a subunit, which anchors the ring to the membrane. Attached to this anchoring subunit is a dimer of b subunits, which project into the mitochondrial matrix and bind to the F1 region. The F1 region is composed of α, β, γ, ε, and δ subunits in the ratio of α3β3γδε [2]. The F1 region is anchored to both the stalk formed by the b subunits of Fo by a single δ subunit, and a central shaft made of a δ subunit, anchored to Fo by an ε subunit. This shaft extends into the F1 region and is bound to large ring of alternating α and β subunits. Conformational changes within these subunits result in ATP synthesis, brought about by rotational energy from proton flow through Fo. The process by which this occurs was proposed by Paul Boyer in the 1970s, which was referred to as the rotational catalysis mechanism, will be discussed in The Chemiosmotic Theory of Bioenergetics Section.
As has been discussed, the complexes within the ETC contain components that functionally drive the production of ATP simply through continuous transfer of electrons within these complexes [2, 3]. The structural description of these complexes has detailed, but the functions they carry out remain to be discussed. With a structural description of the ETC in place, we can now turn to the discussion of how electrons flow through this transport system and how this is coupled to the generation of a proton gradient for use in ATP synthesis.
Electron Flow Through the Complexes
As electrons are passed through each complex, they are sequentially transferred from one redox center to another, at each point generating a small amount of energy through the process of oxidation-reduction [2, 3]. This energy is effectively used to then transport protons across the mitochondrial membrane, creating an electrochemical gradient across the membrane that favors H+ flow back through ATP synthase. Though the mechanisms for proton pumping have been uncovered in complexes III and IV, the mechanism by which protons are pumped in complex I is not fully understood, and thus only the hypothesized mechanism will be discussed in this case. Electrons are initially extracted from substrates by the electron carrier NAD, and to a lesser extent FAD. As previously discussed, these electron carriers can sequester electrons from catabolic activity in the cytosol and mitochondrial matrix, and subsequently deliver these electrons to the ETC at either complex I (NADH) or complex II (FADH2). Electrons flow from this point on through the complexes in sequential order, at several points coupling to the pumping of protons across the membrane. The protons are pumped from the mitochondrial matrix out to the intermembrane space, resulting in a net pH and positive charge in the intermembrane space. This forms the electrochemical gradient, henceforth known as a proton-motive force (pmf), which is the force that drives the flux of protons inward through ATP synthase, toward the matrix, where ATP is then released by ATP synthase and transported out using the ADP-ATP translocase [2, 3]. This pmf has two functional components, the chemical potential energy due to a difference in H+ concentration and electrical potential energy due to the charge differential when protons are segregated across an impermeable membrane. Since this pmf is associated with a net positive charge in the intermembrane space, this region is referred to as the positive or P side, whereas the mitochondrial matrix becomes the negative or N side [2, 3]. The overall reaction for the activity of the ETC is shown in Eq. (1).
This equation shows that a single NADH delivering two electrons reduces a half mole equivalent of O2, producing a single molecule of H2O and pumping ten protons across the membrane. The activity of each complex is discussed separately below in relation to the delivery of a single set of electrons from one NADH. In the discussion of electron flow in complex IV, however, the inclusion of a second set of electrons delivered to the chain will be discussed to fully detail the mechanism that operates in this complex.
As detailed above, the short segment of complex I binds NADH that is generated from the metabolic processes ending in the mitochondrial matrix [2]. As electrons are delivered to the complex, they are initially transferred from NADH to the flavin-containing cofactor FMN. This transfer occurs through the transfer of a single electron at a time, whereby FMN accepts an electron from NADH and immediately transfers this to the iron-sulfur cluster N1a for holding, freeing the FMN to then accept another electron to send it along the chain of iron-sulfur clusters in complex I [2]. Electrons pass from FMN to iron-sulfur clusters in the following order: FMN, N3, N1b, N4, N5, N6a, N6b, and finally N2 [2]. N2, the final iron-sulfur cluster in this chain, is directly involved in the reduction of ubiquinone firstly into semiquinone, and then when the remaining electron is funneled through this pathway the reduction of semiquinone to ubiquinol QH2 [2]. The flow of electrons in complex I is summarized in Eq. (2):
In addition to the generation of QH2, four protons from the mitochondrial matrix are pumped across the inner mitochondrial membrane, which is theorized to be driven by the coupling of this endergonic (energy requiring) pumping process to the exergonic (energy releasing) transfer of a hydride ion from NADH to QH2. The pumping of hydrogen is the first step in generating a pmf. Thus, the flow of electrons in complex I results in a net pumping of four protons to the intermitochondrial space [2, 3].
Electrons can also enter the ETC through complex II, although delivery is accomplished through the FADH2 electron carrier instead of NADH [2, 3]. As stated earlier, this complex is a unique protein complex in the ETC in that it explicitly catalyzes a reaction in the Krebs cycle, namely the oxidation of succinate to fumarate as in Eq. (3):
Electrons are directly sequestered from succinate by the covalently bound FAD molecule, which operates much like FMN in complex I in that it allows the splitting of the initial electron transfer to be carried out through complex II one electron at a time [2]. For each electron that is channeled through complex II, it is transferred through a series of iron-sulfur clusters in the following order: a [2Fe2S] cluster, a [4Fe4S] cluster, and finally a [3Fe4S] cluster [2]. These clusters carry electrons from the hydrophilic region at which succinate binds to the hydrophobic Q-binding site found within the membrane. Electrons are thus passed from [3Fe4S] to Q, forming QH2 similar to complex I. The heme b found in complex II is a metal center capable of transferring electrons; however, it is not known to be directly involved in electron transfer but instead acting as a safeguard against electrons “leaking” out of the ETC to generate reactive oxygen species (ROS) damaging radical compounds such as hydrogen peroxide (H2O2) and the superoxide radical (:O− 2) (see [1] or [2] for more information on this topic). Although this complex has some protective roles in the functioning of the mitochondria, from a bioenergetics perspective, this complex does not contribute to the generation of a pmf since no protons are directly translocated through the actions of this complex [2, 3]. The product of complexes I and II is the reduced form of the membrane-bound electron carrier QH2, which can transfer electrons to complex III by binding to the Qo binding site. The flow of electrons through complex III is carried out in a process known as the Q cycle, which involves a semi-cyclic process of serially oxidizing QH2 at the two Q binding sites [2]. The overall reaction that is carried out in the Q cycle is:
The process of the Q cycle not only utilizes the electrons taken up and delivered to complex III by QH2 but also carries out proton pumping across the inner mitochondrial membrane. Following binding of QH2 to the Qo site, ubiquinone binds to the Qi site. At the Qo site, QH2 transfers electrons divergently, sending one electron to a Rieske Fe-S protein that is attached to this binding site and the other to cytochrome b562 [2]. At this point, QH2 is oxidized to Q and is thus released from the Qo site, allowing the Rieske Fe-S protein within this Qo site to migrate to cytochrome c1 which then carries the electron from the Rieske Fe-S to an externally bound cytochrome c [2]. The heme of cytochrome c can take in a single electron, resulting in the Fe in this heme to reduce from Fe3 + to Fe2 +, and thus after this first electron is transferred, this cytochrome c can move along the ETC and carry on the process at complex IV [2, 3]. This cycle is not complete, however, as the remaining electron that was divergently supplied to cytochrome b562 is now carried through another cytochrome b566 and finally the Qi site, where Q takes up this electron and produces the radical semiquinone .Q. This necessitates induction of the Q cycle once more, this time with second QH2 binding to the Qo site while Q remains on the Qi site. The divergent electron flow described above occurs once again, resulting in the reduction of another cytochrome c and now the reduction of Q into another QH2 [2]. The fact that this process creates an additional QH2 explains how the process can continue when a single QH2 is generated at either complex I or complex II; the Q cycle requires two QH2 molecules to fully operate, but since it generates a QH2 that remains in the membrane, this can be continually used to further fuel Q cycle operations as complexes I and II operate [2].
As noted, the Q cycle also results in the pumping of protons across the inner mitochondrial membrane. As the Q cycle operates, the QH2 that initially enters the Qo site releases the electrons into the complex, protons are also released into the intermembrane space given the close proximity of the Qo site to this side of the membrane [2]. This accounts for the translocation of two protons across the membrane into the P side. After one Q cycle operation, the Qi site contains a semiquinone, which as it has taken up an electron will also take up a proton, but this will come from the mitochondrial matrix, or N side. Similarly, after an additional turn of the Q cycle, there is an uptake of another proton from the mitochondrial matrix at the Qi to produce QH2, which after then reacting with the Qo site results in the release of these protons into the P side [2]. Protons that have been transferred to QH2 from the N side upon reduction at complexes I, II, and the Qi site are ultimately released at the Qo site and thus into the P side. This process accounts for the four protons that are translocated into the P side due to the activity of complex III [2].
Thus far the flow of electrons in complex I through complex III has produced two reduced molecules of cytochrome c, harboring the electrons that were initially introduced to the system by NADH, and eight protons translocated into the P side from the N side. This contributes to the generation of a pmf across the inner mitochondrial membrane, while continually shuttling electrons through the system without any additional energy input [2, 3]. Electrons can now enter complex IV through the carrier cytochrome c to the ETC’s final electron acceptor, oxygen. Just as in complex III, the reaction that proceeds through this complex occurs in many steps and is not a linear transfer of electrons. The overall reaction for this complex is:
In this reaction, four reduced cytochrome c’s are required for the operation of complex IV; however, progress through the system so far has followed the flow of two electrons from NADH. As mentioned in the introduction to Eq. (1), complex IV requires the activity of two sets of electrons delivered by NADH (or FAD). The reason for this becomes evident as the mechanism for electron flow is detailed. A simplified discussion of the proposed mechanism of electron flow in complex IV is given below, for a more comprehensive look at oxygen processing in this complex, see [1].
As electrons are delivered to CuA by the two cytochrome c molecules produced by complex III, each electron is passed to each copper atom of this bimetallic center one at a time. Electrons are then passed along the redox centers in the complex one at a time, first by passing from CuA to cytochrome a, and then finally to the cytochrome a3-CuB bimetallic center [2]. Each electron reduces a different component of this center, with the iron in the cytochrome a3 accepting one electron and the copper in CuB accepting the other. The reduction of these two metals simultaneously results in a slight separation between the two atoms in this center, creating a gap for O2 to bind. The oxygen atom proximal to the iron atom becomes reduced, creating a ferryl oxo molecule (Fe4 +=O), which quickly results in the remaining oxygen atom becoming similarly reduced by CuB, receiving an additional electron and a proton from the hydroxyl group of a proximal tyrosine (Tyr) residue. This results in the production of a hydroxide ion from this second oxygen atom and a tyrosyl radical left in this center. At this point, the cytochrome c molecules from complex III have been transferred to molecular oxygen to produce a ferryl oxo molecule and a hydroxide ion. The additional input of two more cytochrome c molecules are now required, and thus the processing of an additional NADH delivery must be incorporated to further reduce the oxygen. This begins by the induction of the delivery of a third electron by cytochrome c, which upon passing through the redox centers once again to the cyt a3-CuB center is taken along with two protons from the matrix to convert the tyrosyl radical back into Tyr and the hydroxide ion bound to copper into a water molecule. A final electron is again delivered by another cytochrome c to reduce the ferryl oxo molecule into oxidized iron (Fe3 +) reducing the bound oxygen into a hydroxide ion. Uptake of an additional proton from the matrix results in release of this hydroxide ion, creating the second water to be produced by complex IV. Although it may be difficult to detail precisely how this operates, the serial uptake of protons from the matrix to transfer through the cytochrome a3-CuB center results in translocation of four protons to the intermitochondrial space when they are released from the creation and reaction of the radical and intermediate formations described above.
As shown, electrons flow from the electron carriers NADH, and to a lesser extent FAD, through the ETC until they are taken up by the final electron acceptor O2 [2, 3]. This flow generates energy as electrons are passed between redox centers, which are thereby harnessed to pump protons across the membrane and establish a pmf. As outlined above, this pmf is generated by the pumping of protons into the intermembrane space, or P side, through either coupling to the energy generated by electron transfer or mechanisms specific to the operation of complexes III and IV [2, 3]. Armed with the process of generating a pmf, the mechanism by which this proton flow can be utilized to produce ATP will be discussed below.
The Chemiosmotic Theory of Bioenergetics
The pmf that is coupled to the flow of electrons through the ETC drives protons due to both a concentration gradient and a gradient driven by the separation of charge across the membranes. This pmf conserves in excess of 200 kJ per mol of free energy, which is more than sufficient to generate a mole of ATP since this requires 50 kJ of energy [1–3]. The mechanism underlying how this energy is efficiently utilized to do this was first proposed by Peter Mitchell in 1961, and it is known as the chemiosmotic model. The description of the chemiosmotic model begins with the general observation that protons driven by the pmf flow passively back into the mitochondrial matrix through a proton pore, which generates energy that is captured by an attached ATP synthase to drive ATP production. The accepted equation for this relationship is [2, 3]:
Here, we see that this is a general equation that relates the production of ATP to the generation of a pmf due to the translocation of n protons.
Confirmation for the chemiosmotic theory was derived from numerous experimental observations. Firstly, Mitchell observed that isolated mitochondria suspended in a buffer with ADP, inorganic phosphate (Pi), and an oxidizable substrate that may provide electrons to the ETC (e.g., succinate) resulted in the consumption of succinate, showing induction of Krebs cycle; the concentration of molecular oxygen decreasing; and ATP production [2]. This is no surprise given the nature of the ETC in mitochondria, as described above, although this initiated investigation into the connection between the oxidative processes in the mitochondria to ATP synthesis. The coupled nature between these processes strengthened when follow-up experiments indicated that blocking of electron flow in ATP synthesis reduced both consumption of O2 and ATP synthesis and that inhibition of ATP synthesis resulted in reduced O2 consumption and thus electron flow to O2 did not occur. The fact that the pumping of protons from electron flow in the ETC may be coupled to ATP synthesis was continually supported experimentally, by demonstrating that an artificial pmf may drive ATP synthesis in the absence of a substrate for oxidation. Again utilizing isolated mitochondria, but instead of supplying succinate, controlling cation leak (K+ ions), and increasing pH (representing proton concentration) in the buffer resulted in ATP production driven by the separation charge due to K+ and the pH difference [1]. This proceeded with or without the introduction of an oxidizable substrate.
With the establishment of the coupling between a pmf and ATP synthesis, it remained to posit the method of driving ATP synthesis through passive proton flow. The inner mitochondrial membrane is impermeable to protons; thus, pores are the only method of diffusion for protons. The chemiosmotic theory stated that proton diffusion through these pores were somehow generating energy to be used in ATP production, and this was determined to be the case after numerous structural (e.g., crystallography) and functional experiments elucidating the role of ATP synthase [1, 2].
As evident in Fig. 1, the rotational catalysis mechanism involves the structural features of the Fo and F1 regions. Fo has a ring of c subunits, or a c-ring, that is bound to the F1 region through two stalks: a stalk made of β subunits that are a rigid anchor between the regions, and a γ shaft that combines rotation of the c-ring to the rotation of the α and β ring that comprises region F1 [1, 2]. The process of rotational catalysis begins with the rotation of the c-ring. The c-ring of Fo begins rotating due to electrostatic interactions within the ring brought about by the flow of H+ through a proton pore within Fo. As the c-ring is anchored to the shaft, this also results in rotation of this shaft. The α and β subunits that compose a ring surrounding the outermost section of this shaft itself does not rotate, however, as it is anchored to the membrane by the b stalk previously mentioned. Thus, the subunit rotates within this ring, making contact to only a single β subunit at a time. At any given time, these β subunits are in one of three states: β-ADP, β-ATP, and β-empty [1]. Contact between the γ shaft and a β subunit results in a conformational change that forces release of ATP from the contacted β and thus converts β-ATP to β-empty, and when this occurs, the farthest neighbor β-ADP can now tightly bind a phosphate group since is there is now no hindrance from a neighboring β-ATP [1]. Thus, as the subunit rotates within this ring, each 120° rotation results in contact between the stalk and a different β-ATP, forcing this ATP to release an adjacent β to bind a Pi with ADP to tightly bind ATP, until it too comes into contact with the shaft [1]. This repeats as long as H+ flows through the Fo region, and thus maximizing the flux of H+ through this pore will maximize the activity of ATP synthase through continued rotation of this γ shaft. It is clear that the pmf generated by the ETC results in ATP synthesis through this rotational catalysis mechanism, and this process is coupled as well to the consumption of oxygen.
As with many biological processes, the efficiency of functioning for any part of this coupled system can be manipulated chemically, and so it may be helpful to detail the reagents that may be applied to alter the function the components of this system. Manipulation of the system after all affords the potential to determine how dysfunction may arise, and as we shall see in the discussion of AD, this opens a new approach for determining the underlying functional deficits of AD in the mitochondria.
Altering Oxidative Phosphorylation
The previous sections illustrate the deep connection between the structure of the ETC and the role of said structures in incorporating electron flow, oxygen consumption, and proton pumping in ATP synthesis. Thus, it follows that chemical agents may influence either the structure or functioning of any component in the ETC to exert an effect on oxidative phosphorylation. For NADH to deliver electrons to the ETC, it must bind to complex I near the mitochondrial matrix side of the inner mitochondrial matrix, but the binding site may be blocked by the compound adenosine diphosphate ribose. This reagent competitively binds to the NADH binding site, resulting in the complete halting of complex I induction [5]. In a similar fashion, the best known inhibitor of complex I function, rotenone, and the lesser known inhibitors myxothiazol, amytal, and piericidin A, all competitively bind to complex I at the Q binding site by attaching to the N2 Fe-S clusters and disallow transfer of electrons from this Fe-S cluster to Q [1, 6]. In similar fashion to complex I, inhibitors for complex II operate through competitive inhibition at both the input (succinate) and output (Q) binding sites. Interestingly, the inhibitors for the succinate binding site are themselves intermediates of the Krebs cycle: malate and oxaloacetate, which may be the a control system in place to disallow reverse flow of electrons in the Krebs cycle and ETC [7]. Turning to complex III, there are a few well-known functional inhibitors that are worth discussing. As with the aforementioned complexes, both input and output sites can be blocked to reduce or cease functioning of the complex. At the input site, the Qo site stigmatellin binds to inhibit the transfer of electrons from QH2 to the Rieske Fe-S proteins [1]. At the output site of complex IV, antimycin A binds to the Qi site and disrupts electron transfer from cytochrome b to cytochrome c1, which results in the inhibition of QH2 oxidation and thus proton pumping across the inner mitochondrial membrane [8]. As antimycin A interrupts complex IV activity following partial electron flow in the complex, and also disrupts proton pumping late in the ETC, this inhibition has been known to result in increased ROS generation [8]. Complex IV inhibitors operate much like inhibitors for erythrocytes in that reagents that have a higher binding affinity for an exposed heme group than oxygen may competitively, and in some cases permanently, bind to the cytochrome a3-CuB site upon opening. These reagents, namely cyanide, sulfide, azides, and carbon monoxide, all result in the inhibition of oxygen binding, which halts electron flow in the ETC altogether. Inhibition of complex IV may also proceed allosterically, through a feedback mechanism whereby high levels of ATP can result in inhibition, thus slowing down ATP production [1]. In ATP synthase, as there is no input binding site, there is no competitive inhibition per se, but a potent and well-known inhibitor of ATP synthase, oligomycin A, has been used to block the functioning of the proton channel in the Fo subunit [9]. ATP synthesis is inhibited by oligomycin A, but the electron flow through the ETC still occurs due to proton leak, which involves rarely occurring proton movement through channels other than the proton pore of ATP synthase (e.g., thermogenin) [10].
In addition to inhibiting the structure and/or function of the complexes themselves, there exists an additional class of inhibitors known as uncouplers, which detach the functioning of the ETC from ATP synthase. This class of inhibitors nullifies the coupling that underlies oxidative phosphorylation by providing an additional route for the generated pmf to be relieved. The most commonly known uncoupling agents are 2,4-dinitrophenol (DNP) and carbonylcyanide-p-triuoromethoxyphenylhydrazone (FCCP), which result in inhibited ATP synthesis without disruption to the structure of the mitochondria. These compounds operate as proton carriers through the membrane, since they move through the membrane as a hydrophobic weak acid in the protonated form, releasing a proton in the mitochondrial matrix and returning to the intermitochondrial space in the unprotonated form. This reverse translocation of protons bypasses the ATP synthase, and thus uncouples proton flow from ATP synthesis, and also reduces the pmf generated by the activity of the ETC [1].
Many chemical reagents impair and influence the bioenergetic processes of cellular mitochondria, which then may provide insight into how these impairments may mechanistically correlate to the dysfunction witnessed in a diseased state. Furthermore, experimental paradigms may utilize these chemical reagents to manipulate mitochondrial functioning and determine how different mechanisms of influencing this functioning may be affected in diseased states. Along with direct experimental observations, these approaches have been used to further provide insight into the dysfunction associated with Alzheimer’s disease, which will now be discussed.
Bioenergetic Dysfunction in Alzheimer’s Disease
Introduction to Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common form of dementia, showing an increasing rate of morbidity in the elderly population over the past few decades. AD is a devastating neurodegenerative disorder that is characterized symptomatically by cognitive and motor decline, progressing through a stage of mild cognitive impairment (MCI) affecting memory, attention, speech patterns, and as AD worsens, late-stage cognitive impairment affecting all aspects of behavior [11, 12]. Despite the rapid rise in cases of AD, there remains little in the form of treatment to slow or stop the progression of AD. This is not helped by the fact that AD is largely idiopathic, with various theories attributing the cause of AD to many different physiological factors. The leading hypotheses regarding the etiology of AD revolve around either genetic heritability [13], reduced synthesis of acetylcholine [14], the accumulation of neurotoxic protein plaques of amyloid β peptide (Aβ) [15], and fibrillary tangles of hyperphosphorylated tau-protein [16] or irregular function and dynamics of cellular mitochondria (the mitochondrial cascade hypothesis). In all experimental endeavors, the neuropathological findings of late-stage deposition of tau-tangles and accumulation of Aβ have been a consistent finding for AD pathology, although it remains unclear whether these accumulations are a consequence or causative factor for AD. Our focus in this review is directed toward the relationship of protein interactions with oxidiative phosphorylation. As the manner by which the protein plaques and neurofibrillary tangles result in neurotoxicity are yet to be established, discussion with regard to this relationship will also be restricted to neurotoxic effects on cellular mitochondria with particular emphasis on dysfunction in oxidative phosphorylation processes. Although it is not entirely known how Aβ is translocated into the mitochondrial matrix from extracellular plaques [17], if in fact this is the case, there have nonetheless been numerous reports of Aβ exerting an effect on mitochondrial function [17, 18]. Furthermore, there have been numerous reports indicating that mitochondrial function is disrupted in early-stage AD [19, 20], providing promise for investigating mitochondrial dysfunction as an early therapeutic target for AD treatment. Beginning with the Aβ, plaques and tau tangles are an inherent pathological finding for AD, with these accumulated proteins becoming more prominent as AD progresses. Aβ plaques arise from the cleaving of Aβ precursor protein (APP) by β-secretase [21], which studies indicate, results in aggregation of the Aβ sections of APP into plaques that begin to accumulate extracellularly [21]. In AD, tau protein is enzymatically phosphorylated three to four times higher than under normal physiological conditions, resulting in aggregation of this protein into paired helical filaments (PHF) intracellularly [21]. With the findings supporting consistent aggregation of these proteins in AD, whether due to genetic, environmental, or biochemical factors, this lent early support for their role as the potential primary biochemical basis for AD development. It remains unclear, however, whether the accumulation of these proteins in fact lead to the development of AD or whether these proteins are pathological markers that have developed as a product of AD. The latter notion became evident as more evidence surfaced that indicated that plaque development alone did not result in neurotoxicity, and that other biochemical processes were showing dysfunction, such as dysfunction in the regulation and dynamics of mitochondria [22, 23], regardless of plaque and tangle deposition [19, 20]. This motivated the formation of the mitochondrial cascade hypothesis, which posits that mitochondria are the inherent driving force for the development of AD [24, 25], is based on mounting evidence of mitochondrial dysfunction in early-stage AD. This hypothesis will be detailed below following a discussion of the mitochondrial dysfunction often seen in AD.
Mitochondrial Dysfunction in AD
The Mitochondrial Cascade Hypothesis
In 2004, Swerdlow and Khan hypothesized that the regulation over mitochondrial baseline activity is inherent genetically but can be influenced by other factors, and along with this influence, mitochondria may accumulate damage resulting in both the symptoms and neuropathology found in AD [24, 25]. This theory states that given the accumulating evidence for early-stage mitochondrial dysfunction in AD potentially producing other molecular phenomena in AD, even in the absence of Aβ plaques and tau tangles, mitochondrial dysfunction may be at the heart of AD development. In other words, inherent to each individual is a baseline level of mitochondrial functioning, largely determined by genetic predisposition and also influenced by environmental factors, combining to influence how baseline functioning may change over time. Aβ in this model is seen as a marker of brain aging, but not as the cause of AD. While mitochondrial functioning may be influenced by the accumulation and deposition of Aβ, this model posits that the process of accumulation and deposition of Aβ is initiated by mitochondrial dysfunction. Thus, it stands that manipulating mitochondrial function and understanding the deficits that result in mitochondrial bioenergetics may help to suspend the development of AD.
Specific Mitochondrial Dysfunctions Associated with AD
Overview
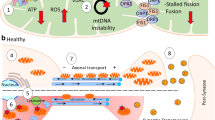
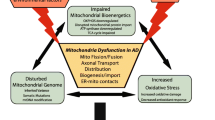
Several mitochondrial functions have been shown to be affected in AD. These include diminished glucose metabolism, mitochondrial enzymatic failure, and increased ROS production (before Aβ and tau tangles have begun forming [26]). Several facets of mitochondrial dynamics are also dysfunctional in early-stage AD [27], including a disruption in the balance between mitochondrial fusion and fission, representing mitochondria dividing and binding with one another, decreased axonal transport of mitochondria, a lower percentage of mitochondria within a cell, and a change in size, with mitochondria in AD maintaining much shorter, wider shapes [26, 28] as depicted in Fig. 2a. Importantly, there is also a disruption to glycolytic processes, impairments to the enzymatic activity of the protein complexes of the ETC, and negative alterations in antioxidant enzymatic activity [26]. Much research has also been devoted to relating dysfunction in AD to complex IV activity, as it is often shown to be defective in early stages of the disease [26, 29, 30]. Many of the dysfunctions discussed below are illustrated in Fig. 2.

a An illustration of some mitochondrial dysfunctions found in Alzheimer’s disease. Explicitly shown is altered morphology of the mitochondria and synaptic space, as well as the reduced level of mitochondrial axonal transport. b A closer inspection of the mitochondria in a diseased state showing translocation of Aβ into the mitochondria through the TIM and TOM complexes. Once Aβ is transported into the mitochondrial matrix, Aβ is shown to affect complex IV activity, Ca2+ efflux and result in mtDNA mutation
Catabolic Processes
Additional effects on energetics in AD center around decreased activity in the catabolic processes in glycolysis and Krebs cycle, particularly the pyruvate dehydrogenase complex (PDHC), the α-ketoglutarate dehydrogenase complex (KDHC), and enzymes in the ETC such as succinate dehydrogenase (in complex II) [31]. It has been shown, however, that these deficits are not specific to AD but are also common across other neurodegenerative disorders [23].
The deficit found in complex IV activity (discussed in Complex IV), was also shown to likely be a consequence of abnormal substrate binding kinetics (e.g., cytochrome c binding), showing that mutations in oxidative phosphorylation subunit genes may be driving the defects found in complex IV activity [31]. Furthermore, while many studies produced defects in complex IV activity, some reported no significant decrease in the activity of this complex in certain brain regions (temporal lobe) and additionally reported that localization of decreased activity in complexes I–III were specific to certain cortex locations as well [31]. Following the discussion of decreased activity in complex IV due to Aβ accumulation, it has also been shown to suppress activity of complex II, though it is not entirely sure if this is due to the binding of Aβ to redox centers or not [31]. Although there is evidence to show decreases in functionality of complex II, there is primarily only evidence showing that levels of complex IV decrease as AD progresses, not complexes I–III [31]. Complex organization is also disrupted in AD, as it has been shown that the structural organization of the complexes in the ETC may result in changes in conformation and complex interaction in the formation of supercomplexes [32].
Complex IV
Evidence exists to show that impaired activity of complex IV may be accompanied by reserved levels of complex IV subunit levels, indicating that some facets of mitochondrial dysfunction, particularly deficit in complex IV function, are inherent to AD and not related to structural degradation and induction of apoptosis due to plaque formation [33]. Impairment of function in complex IV was also detected in mice that were genetically altered to develop the neuropathology of AD. For example, the 3xTg-AD mouse model [34] is widely used, where these studies showed that oxygen consumption at complex IV in mitochondria were deficient in AD [23, 26]. Bioenergetic deficits have been shown to precede formation of Aβ and tau aggregation in several studies but have also been shown to become exacerbated by the presence of these accumulated proteins [26]. Aβ-induced mitochondrial dysfunction has been demonstrated on several occasions [26]. PC12 cell from rat adrenal medulla cells treated with varied concentrations of Aβ peptides showed improper function in electron transport through complexes in the ETC [35]. Additionally, these cells showed an impairment in glycolysis and thus a reduction in ATP generation [35].
Inflammation
Inflammation is a central process for several disorders, such as cancer, obesity, diabetes, and AD. It is also observed in normal brain aging [36]. In most situations, the inflammatory process is a natural mechanism of defense against infections and injury, which is designed to first respond to threats and then restore the body to normal homeostasis. In AD, markers of inflammation have been found in the brain, which has been demonstrated by increased levels of cytokines, chemokines, and gliosis; this includes TNF-α, IL-6, and IL-1β, to name a few [37]. Overall, in AD, Aβ is thought to induce inflammation via mechanisms that need further clarification.
Aβ
To elucidate any connection between mitochondrial dysfunction and Aβ toxicity, Ntera2 cells containing no functional ETC showed no adverse effects following application of Aβ, whereas cells that contained functional ETC prior to application of Aβ exhibited cytotoxicity through cell death and reduced energetic activity [38]. Many studies have since corroborated these results, showing that treatment with Aβ resulted in deficits in energy metabolism [26]. Studies have also shown that Aβ has the potential to permeate not only the cell membrane but also the inner and outer mitochondrial membranes as well [26]. Although it is not entirely known for certain how Aβ is transported into the membrane, it is currently proposed that it is imported through the translocase of the outer membrane (TOM) complexes Tom20, Tom70, and Tom40, then transported through the inner mitochondrial membrane by the translocase of the inner membrane (TIM) [26]. The operation of these complexes and the translocation of Aβ is shown in Fig. 2b.
It is believed that mitochondrial dysfunction precedes Aβ formation, which upon aggregation then fuels further decline in mitochondrial functioning once it has permeated the membranes. One major way that Aβ can influence mitochondrial functioning is by binding to heme groups, which reduces their functionality as redox centers in the ETC, which occurs predominantly in complex IV heme groups [23]. Aβ may also bind to Aβ-binding alcohol dehydrogenase (ABAD) resulting in an increase in ROS generation through impairment in complex IV molecular O2 processing, reduced cytochrome c release, and a decrease in ATP production [23]. Furthermore, aside from Aβ peptide interacting with certain TOM and TIM complexes to allow translocation into the mitochondria, they can also block TOM40 and TIM23 from importing subunits of complex IV into the mitochondria [23], thereby also resulting in affected complex IV activity.
Oxidative Stress
Increasing evidence suggests that oxidative stress is linked to AD [39]. However, exactly how this occurs is not known. Interestingly, some data suggest that oxidative stress is an early event in disease progression. Investigators have also hypothesized that redox balance decreases during conditions associated with neurodegeneration, which results in mitochondrial dysfunction [39]. Whether increased oxidative stress leads to mitochondrial dysfunction or if mitochondrial dysfunction promotes oxidative stress remains to be clarified.
In addition, a link between cancer and AD has been proposed that may be via a nitric oxide (NO)-induced oxidative stress pathway resulting in mitochondrial structural and changes and DAN damage [40]. Interestingly, in both cancer and AD, energy demand increases. Due to this increased demand, which is coupled to hypoxia and oxidative stress, some tumors switch their metabolic processes to glycolysis (i.e., Warburg effect; [41]), and depend less on mitochondrial ATP output and more on ATP output from glycolysis. Several studies have found similar patterns of this switching in AD models. For example, using PET scans, Vlassenko et al. [42] found significant correlations exist between brain aerobic glycolysis and Aβ deposition in human brain. The significance of aerobic glycolysis in proliferating cells has been well documented in cancer studies, but the observation that glycolysis occurs in AD pathophysiology is new and worthy of further investigation.
Mitochondrial Gene Expression
The mitochondria possess their own compact circular genome (mtDNA), which lacks histone proteins and any major repair mechanisms, making it highly vulnerable to ROS that are produced in close proximity. However, not all ROS production is detrimental, and data also suggest some ROS are important for normal cell signaling. Human mtDNA possesses 37 genes that encode for 13 polypeptides, which is remarkably similar to mouse mtDNA (see MitoCarta database, http://www.broadinstitute.org/pubs/MitoCarta/). Studies show that mtDNA genes code for many of the subunits of all 5 complexes of the ETC (but some subunits are coded by nuclear DNA), 2 rRNAs, and 22 tRNAs. In almost every case, mtDNA is inherited along maternal lineages.
Interestingly, studies suggest that mtDNA mutations play an important role in mitochondrial dysfunction in AD. For example, variations in mtDNA were found to be associated with AD pathogenesis. These included, heteroplasmic somatic mtDNA control region mutations [43] and point/missense mutations in mitochondrial encoded cytochrome c oxidase subunits I, II, and III genes [44].
Mitochondrial Dynamics
The endosymbiosis theory, proposed by Dr. Lynn Margulis, states that mitochondria were once independent entities that at one point became associated with eukaryotic cells. Given this, it should come as little surprise that mitochondria do not exist in cells in fixed positions. In fact, mitochondria have been observed to be highly dynamic organelles constantly fluxing between fusional and fissional states. Moreover, it is the alteration of mitochondrial dynamics that is now believed to be at the core of many neurodegenerative disorders including AD.
For example, proteins such as optic atrophy 1 (OPA1), mitofusin 1 (MFN1), and mitofusin 2 (MFN2) have been implicated in regulating mitochondrial morphology (i.e., balancing fusion and fission) and when mutated result in disease [45]. In addition, data suggest that tau and Aβ proteins can influence the regulation of mitochondrial dynamics and involve proteins such as dynamin-related protein 1 (DRP1), which is thought to possibly alter processes of mitochondrial fission in AD.
Conclusion
The discussion of ETC bioenergetics began with an introduction to some of the main characteristics of the mitochondria, substrate processing, and the structural components of the ETC. Understanding the involvement of these components in utilizing the electrons sequestered from relevant oxidizable substrates (e.g., glucose) provides a framework for understanding how oxidative phosphorylation occurs mechanistically. Through this framework, understanding function and dysfunction are both enabled. In reviewing the dysfunctions commonly associated with AD, it is apparent that the mitochondria are an increasingly interesting target for understanding early-stage AD and potential treatments. With growing interest in the mitochondria as a potential target for understanding AD, it has even been hypothesized that deficit in this organelle may be at the heart of the progression of AD itself. With enough evidence to support this hypothesis, it is clear that the role of mitochondria in AD is significant, but the extent of this significance still remains an interesting topic for research.
Notes
Sections Introduction, General Mitochondrial Biochemistry, The Electron Transport System, The Chemiosmotic Theory of Bioenergetics heavily reference [1] unless otherwise indicated.
References
Nelson D, Cox M (2004) Lehninger principles of biochemistry, 4th edn. W.H. Freeman
Scheffler I (2007) Mitochondria, 2nd edn. Wiley
Nicholls D, Ferguson S (2013) Bioenergetics, 4th edn. Elsevier
Mannella CA (2006) Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta 1763(5–6):542–548
Zharova TV, Vinogradov AD (1997) A competitive inhibition of the mitochondrial NADH-ubiquinone oxidoreductase (complex I) by ADP-ribose. Biochim Biophys Acta 1320(3):256–264
Watabe M, Nakaki T (2008) Mitochondrial complex I inhibitor rotenone inhibits and redistributes vesicular monoamine transporter 2 via nitration in human dopaminergic SH-SY5Y cells. Mol Pharmacol 74(4):933–940
Muller FL, Liu Y, Abdul-Ghani MA, Lustgarten MS, Bhattacharya A, Jang YC, Van Remmen H (2008) High rates of superoxide production in skeletal-muscle mitochondria respiring on both complex I- and complex II-linked substrates. Biochem J 409(2):491–499
Dairaku N, Kato K, Honda K, Koike T, Iijima K, Imatani A, Sekine H, Ohara S et al (2004) Oligomycin and antimycin a prevent nitric oxide-induced apoptosis by blocking cytochrome C leakage. J Lab Clin Med 143(3):143–151
Nakata M, Ishiyama T, Akamatsu S, Hirose Y, Maruoka H, Suzuki R, Tatsuta K (1995) Synthetic studies on oligomycins. Synthesis of the oligomycin B spiroketal and polypropionate portions. Bull Chem Soc Jpn 68(3):967–989
Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Martin D (2011) “NIH Public Access,” 53–67
Arnáiz E, Almkvist O (2003) Neuropsychological features of mild cognitive impairment and preclinical Alzheimer’s disease. Acta Neurol Scand Suppl 179:34–41
Kurz HFA (1999) “Clinical features of Alzheimer’s disease,”. 288–290
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL (2006) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63(2):168–174
Francis PT, Palmer AM, Snape M, Wilcock GK (1999) “The cholinergic hypothesis of Alzheimer’s disease : a review of progress”.137–147
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12(10):383–388
Mudher A, Lovestone S (2002) Alzheimer’s disease—do tauists and baptists finally shake hands? Trends Neurosci 25(1):22–26
Hashimoto M, Rockenstein E, Crews L, Masliah E (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol Med 4(1–2):21–36
Hauptmann S, Keil U, Scherping I, Bonert A, Eckert A, Müller WE (2006) Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp Gerontol 41(7):668–673
Chen X, Yan SD (2006) Mitochondrial abeta: a potential cause of metabolic dysfunction in Alzheimer’s disease. IUBMB Life 58(12):686–694
Lunnon K, Ibrahima Z, Proitsi P, Lourdusamy A, Newhouse S, Sattlecker M, Furney S, Saleem M et al (2012) Mitochondrial dysfunction and immune activation are detectable in early alzheimer’s disease blood. J Alzheimers Dis 30(3):685–710
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Perspective 81(2):741–767
García-Escudero V, Martín-Maestro P, Perry G, Avila J (2013) “Deconstructing mitochondrial dysfunction in alzheimer disease”. Oxidative Med Cell Longev 2013
Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X (2010) Alzheimer’s disease: diverse aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol 3(6):570–581
Swerdlow RH, Burns JM, Khan SM (2014) The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta Mol basis Dis 1842(8):1219–1231
Swerdlow RH, Khan SM (2004) A ‘mitochondrial cascade hypothesis’ for sporadic Alzheimer’s disease. Med Hypotheses 63(1):8–20
Esteves AR, Arduino DM (2009) “Mitochondrial metabolism in age-related neurodegenerative disorders: Alzheimer’s and Parkinson’s revisited.” In: Svensson OL (ed.) Mitochondria: structure, functions and dysfunctions
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Zhu X, Perry G, Smith MA, Wang X (2013) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 33(Suppl 1):S253–S262
Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM et al (1992) Brain cytochrome oxidase in Alzheimer’s disease. J Neurochem 59(2):776–779
Mutisya EM, Bowling AC, Beal MF (1994) Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J Neurochem 63(6):2179–2184
Shoffner JM (1997) Oxidative phosphorylation defects and Alzheimer’s disease. Neurogenetics 1(1):13–19
Seelert H, Dani DN, Dante S, Hauß T, Krause F, Schäfer E, Frenzel M, Poetsch A et al (2009) From protons to OXPHOS supercomplexes and Alzheimer’s disease: structure-dynamics-function relationships of energy-transducing membranes. Biochim Biophys Acta Bioenerg 1787(6):657–671
Cardoso S, Santana I, Swerdlow R, Oliveira C (2004) Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem 89(6):1417–1426
Sterniczuk R, Dyck RH, Laferla FM, Antle MC (2010) Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: part 1. Circadian changes. Brain Res 1348:139–148
Pereira C, Santos MS, Oliveira C (1999) Involvement of oxidative stress on the impairment of energy metabolism induced by A beta peptides on PC12 cells: protection by antioxidants. Neurobiol Dis 6(3):209–219
Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, Swerdlow RH (2014) “Mitochondrial lysates induce inflammation and alzheimer’s disease-relevant changes in microglial and neuronal cells.” J Alzheimers Dis
De Felice FG, Ferreira ST (2014) Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes 63(7):2262–2272
Cardoso S, Santos S, Swerdlow RH, Oliveira CR (2001) Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J 15(8):1439–1441
Wang X, Wang W, Li L, Perry G, Lee H, Zhu X (2014) Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta Mol basis Dis 1842(8):1240–1247
Aliev G, Obrenovich ME, Tabrez S, Jabir NR, Reddy VP, Li Y, Burnstock G, Cacabelos R, Kamal MA (2013) “Link between cancer and Alzheimer disease via oxidative stress induced by nitric oxide-dependent mitochondrial DNA overproliferation and deletion.” Oxidative Med Cell Longev 2013
Ngo H, Tortorella SM, Ververis K, Karagiannis TC (2014) The Warburg effect: molecular aspects and therapeutic possibilities. Mol Biol Rep 42(4):825–834
Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME et al (2010) Spatial correlation between brain aerobic glycolysis and amyloid-β (Aβ) deposition. Proc Natl Acad Sci U S A 107(41):17763–17767
Coskun PE, Beal MF, Wallace DC (2004) Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A 101(29):10726–10731
Hamblet NS, Ragland B, Ali M, Conyers B, Castora FJ (2006) Mutations in mitochondrial-encoded cytochrome c oxidase subunits I, II, and III genes detected in Alzheimer’s disease using single-strand conformation polymorphism. Electrophoresis 27(2):398–408
Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P (2014) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11(1):11–24
Acknowledgments
This work was funded in part by the National Sciences and Engineering Research Council (to B.C.A.), the Everett Endowment Fund (to B.C.A.), the Edwards Family Endowment (to B.C.A.), the University of Manitoba (to C.C.), and the St. Boniface General Hospital Research Foundation (to B.C.A. and M. G. S.). B.C.A. is a Research Affiliate at the University of Manitoba’s Centre on Aging and holds the Honourable Douglas Everett, Patricia Everett and the Royal Canadian Properties Endowment Fund Chair and the Manitoba Dementia Research Chair.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Cadonic, C., Sabbir, M.G. & Albensi, B.C. Mechanisms of Mitochondrial Dysfunction in Alzheimer’s Disease. Mol Neurobiol 53, 6078–6090 (2016). https://doi.org/10.1007/s12035-015-9515-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9515-5