
Abstract
Purkinje cell dendritic development is severely compromised after chronic activation of protein kinase C (PKC). In a recent transgenic mouse model of spinocerebellar ataxia 14, the ser361-to-gly (S361G) mutation of the protein kinase C gamma (PKCγ) gene was expressed in Purkinje cells. Purkinje cells from these mutant mice in organotypic slice cultures have the same stunted dendritic tree as Purkinje cells after pharmacological activation of PKC. Because the transgene is exclusively present in Purkinje cells, cerebellar tissue from these mice is an attractive starting material for searching genes which might be interacting with PKCγ in Purkinje cells for inducing the stunted dendritic growth. We have performed a microarray analysis and identified several candidate genes with an increased messenger RNA (mRNA) expression in the PKCγ-S361G transgenic Purkinje cells. Out of these candidates, we have further studied carbonic anhydrase 8 (CA8). We show here that CA8 mRNA and protein expression is strongly induced in PKCγ-S361G transgenic Purkinje cells. Overexpression of CA8 in Purkinje cells in dissociated cultures strongly inhibited Purkinje cell dendritic development and produced a dendritic phenotype similar to PKCγ-S361G. There was no evidence for a direct binding of CA8 to either PKCγ or the type 1 IP3 receptor. Knockdown of CA8 with miRNA did not alter Purkinje cell dendritic development and did not protect Purkinje cells in dissociated cultures from the stunted dendritic growth induced by PKCγ-S361G or by PKC activation. Our results indicate that CA8 is a novel important regulator of Purkinje cell dendritic development and that its expression is controlled by PKCγ activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The signaling molecule protein kinase C (PKC) plays an important role for long-term depression of Purkinje cells [1] and has a strong negative impact on Purkinje cell dendritic growth and development [2–4]. Increased PKC activity is likely to affect the long-term calcium equilibrium of the Purkinje cells. Blockade of voltage-gated calcium channels could partially protect Purkinje dendrite cells from the stunted growth seen with increased PKC activity [5], and Purkinje cells from Moonwalker (Mwk) mutant mice with increased calcium influx through TRPC3 channels also show a stunted dendritic morphology [6]. Similarly, inhibition of the plasma membrane calcium-ATPase 2 (PMCA2) affected Purkinje cell dendritic growth [7, 8]. These findings demonstrate the involvement of the calcium equilibrium for regulating Purkinje cell dendritic growth, but still very little is known about the downstream molecular interactions after PKC activation in Purkinje cells leading to the reduced dendritic growth. Interestingly, one form of spinocerebellar ataxias, SCA 14, is caused by point mutations in the protein kinase C gamma (PKCγ) gene [9, 10] and there is evidence that at least some of the missense mutations may confer a gain of function phenotype with a constitutive activation of PKCγ [11, 12]. We have recently created a transgenic mouse model of SCA 14 by introducing a transgene carrying the ser361-to-gly (S361G) mutation of the PKC gene specifically into Purkinje cells. This mutation is located in the catalytic domain [13] and was shown previously to confer an increased catalytic activity as tested by in vitro assays [12]. PKCγ-S361G transgenic mice have pathological changes typical for cerebellar disease and have an increased PKCγ activity in vivo reflected by the reduced dendritic development typically seen with increased PKC activity [14]. Purkinje cells from these mutant mice thus show both an increased PKCγ activity and a severe dendritic reduction. Because the transgene is exclusively activated in Purkinje cells, cerebellar tissue from these mice is an attractive starting material for searching other genes which might be interacting with PKCγ for inducing the stunted dendritic growth. We have performed a gene chip microarray analysis and identified several candidate genes with an increased messenger RNA (mRNA) expression in the PKCγ-S361G transgenic Purkinje cells. Out of these candidate genes, we have further studied carbonic anhydrase 8 (CA8) which was shown previously to be involved in Purkinje cell function and cerebellar disease [15, 16]. We show here that CA8 mRNA and protein expression is strongly induced after PKC activation and that overexpression of CA8 in cultures of dissociated Purkinje cells strongly inhibits Purkinje cell dendritic development. However, we found no evidence of a binding of CA8 to either PKCγ or the type 1 inositol 1,4,5-triphosphate (IP3) receptor. Knockdown of CA8 protein expression by microRNA (miRNA) transfection did not protect Purkinje cells in dissociated cultures from the stunted dendritic growth after increased PKCγ activity. Our results indicate that CA8 is an important regulator of Purkinje cell dendritic development and that its expression is controlled by PKCγ activity.
Materials and Methods
Plasmid Construction
Plasmid L7-green fluorescent protein (GFP) was previously described [17]. CMV-CA8 and CMV-PKCγ plasmids were from Origene (Rockville, MD, USA). Restriction sites were added to the CA8 and PKCγ genes by polymerase chain reaction (PCR) using the following primers: forward primer for CA8, 5′CAG GAT CCA GCG GCC GCA TGG CTG ACC TGA GCT TC3′; reverse primer for CA8, 5′CCC TTG CTC ACC ATG GCC TGA AAG GCC GCT CGG A3′; forward primer for PKCγ, 5′CAG GAT CCA GCG GCC GCA TGG CTG GTC TGG GCC CC 3′; and reverse primer for PKCγ, 5′CCC TTG CTC ACC ATG GCC ATG ACG GGC ACA GGC A3′. These PCR products were incubated with the L7-GFP vector in the presence of the restriction enzymes, NotI and NcoI (New England BioLabs, Massachusetts, USA) in suitable buffer. After 30-min incubation at 37 °C, DNA fragment and linearized vector were fused using In-Fusion HD Cloning Kits (Clontech, Mountain View, CA). The constructed expression vectors were confirmed by DNA sequencing (Microsynth, Balgach, Switzerland).
Organotypic Slice Cultures
Animal experiments were carried out in accordance with the EU Directive 2010/63/EU for animal experiments and were reviewed and permitted by Swiss authorities. Cultures were prepared from B6CF1 (CB6) mice or PKCγ-S361G transgenic mice [14] as described previously [18]. Briefly, mice were decapitated at postnatal day 8 (P8), their brains were aseptically removed, and the cerebellum was dissected in ice-cold preparation medium (minimal essential medium (MEM) with 1 % glutamax (Life Technologies, Zug, Switzerland), pH 7.3). Sagittal sections, 350 μm thick, were cut on a McIlwain tissue chopper under aseptic conditions. Slices were separated, transferred onto permeable membranes (Millicell-CM, Merck-Millipore, Zug, Switzerland), and incubated on a layer of Neurobasal medium (97 % Neurobasal medium, 2 % B27 supplement, 1 % glutamax, pH 7.3) in a humidified atmosphere with 5 % CO2 at 37 °C. The medium was changed every 2–3 days for a total of 5 days for microarray analysis and of 7 days for protein analysis and immunostaining.
Microarray Analysis
Starting material for the microarray analysis were cerebellar slice cultures from PKCγ-S361G transgenic mice versus control mice. After 5 days in vitro (DIV 5), organotypic cerebellar slices were washed, harvested, and submerged in RNA later (Life Technologies, Zug, Switzerland) solution for storage. The slices from one litter deriving from three to four mouse pups were collected and used for RNA purification. Purified total RNA was amplified by in vitro transcription and converted to sense-strand complementary DNA (cDNA). Fragmented cDNA samples were then hybridized to GeneChip Mouse Gene 1.0 ST Arrays (Affymetrix, Santa Clara, CA, USA). Procedures were carried out as described by the manufacturers, and the analysis was done in the Life Science Training Facility (LSTF) core facility of the University of Basel by Dr. Philippe Demougin. Raw data were processed and analyzed using Partek Genomics Suite (version 6.12.0907). Transcripts were considered to be changed if the p value was smaller than 0.05. After filtering, 1045 genes were retained. We then set the threshold at a fold change exceeding 1.25 for being specifically expressed, and 98 genes were retained.
Reverse Transcriptase-Quantitative PCR
Total RNA was extracted from organotypic cerebellar slices harvested at DIV 5–7, and cDNA was synthesized with reverse transcriptase-quantitative PCR (RT-qPCR) using oligo(dT) primers (Applied Biosystems, Foster City, CA). For gene expression analysis, RT-qPCRs were conducted in a total volume of 20 μl comprising 10 μl of Mastermix with SYBR Green (Applied Biosystems, Foster City, CA), 0.5 μl of each primer (1.0 μM), 0.3 μl of sample cDNA, and 8.5 μl ultrapure water. Real-time PCRs were run on a Real-Time PCR System (Applied Biosystems, Foster City, CA) under the following reaction conditions: 1 cycle of [95 °C for 10 min], 40 cycles of [94 °C for 15 s → 65 °C for 60 s], and 1 cycle of [95 °C for 15 s → 72 °C for 30 s → 95 °C for 15 s]. Oligonucleotide primers were designed using the Primer3 software (http://bioinfo.ut.ee/primer3/). The primer sequences are given in supplementary methods. Reactions were quantified by relative standard curve system and the cycle threshold (Ct) method using the SDS2.2 software (Applied Biosystems, Foster City, CA). A relative quantitation value (RQ) for each sample from the triplicates of that sample was calculated for each gene. The data were analyzed as RQ for the gene of PKCγ transgenic mice/RQ for control mice. For each comparison, a minimum of three independent experiments were done with unrelated RNA samples.
Western Blot Analysis
Tissue slices were harvested with a cell scraper. Samples were then homogenized in radioimmunoprecipitation assay (RIPA) buffer with phosphatase inhibitor (phosphor STOP, Roche, Basel, Switzerland) and protease inhibitor (cOmplete Protease Inhibitor Cocktail Tablets, Roche, Basel, Switzerland). Homogenates were sonicated for 5 s a total of five times and cleared by centrifugation at 14,000×g for 15 min at 4 °C. Protein concentrations were determined with NanoDrop (Thermo Scientific, Reinach, Switzerland). Western blotting analysis was performed by standard protocols. Membranes were blocked with 5 % BSA in tris-buffered saline (TBS) for 1 h and incubated with appropriate primary antibodies. Rabbit anti-inositol 1,4,5-trisphosphate receptor 1 (ITPR1) (Cell Signaling, Massachusetts, USA, 1:1000), rabbit anti-CA8 (Novus Biologicals, Colorado, USA, 1:2000), rabbit anti-PKCγ (Santa Cruz Biotechnology, Santa Cruz, CA, 1:1000), rabbit anti-family with sequence similarity 40, member B (Fam40b) (AVIVA, San Diego, USA, 1:1000), rabbit anti-pleckstrin homology domain containing, family D (Plekhd1) (Thermo Scientific, Reinach, Switzerland, 1:1000), rabbit anti-Homer3 (Abgent Inc., San Diego, CA, USA, 1:1000), mouse anti-metabotropic glutamate receptor 1 (mGluR1) (BD Biosciences, Franklin Lakes, NJ, USA, 1:1000), or anti-β-actin (Millipore, Zug, Switzerland, 1:1000) were used. After washing with TBS-T, membranes were incubated with HRP-labeled secondary antibodies for 1 h. After washing, bound antibodies were visualized with ECL2 Western blotting substrate (Pierce, Thermo Scientific, Reinach, Switzerland) and quantified using a C-DiGit Western blot scanner and software (LI-COR Biosciences, Bad Homburg, Germany). For each comparison, a minimum of three independent experiments were done with unrelated protein samples.
Immunoprecipitation
Cerebellar slice cultures at DIV 7 were used as starting material. Slice cultures were harvested with a cell scraper. Samples were then homogenized in RIPA buffer with phosphatase inhibitor and protease inhibitor as described in Western blot analysis. Homogenates were sonicated for 5 s a total of five times and cleared by centrifugation at 14,000×g for 15 min at 4 °C, and protein concentrations were determined and 100 μg of each sample was used for immunoprecipitation. Before using, Protein G was washed twice at 12,000g for 10 s with PBS. Lysate was incubated with 50 µl Protein G for 30 min at 4 °C, and lysate was washed twice at 12,000g for 10 s with PBS. One microgram antibody, 50 µl pre-washed Protein G, and 50 µl PBS were added to the lysate and rotated at 4 °C overnight. Tubes were then centrifuged at 4 °C for 2 min at 3000g and washed with 1 ml PBS 4 °C for 2 min at 3000g four times. SDS-PAGE and Western blot were done as described. A minimum of three independent immunoprecipitation experiments with unrelated protein samples were done per condition.
Immunohistochemistry
At DIV 7, slices were fixed in 4 % paraformaldehyde overnight at 4 °C. All reagents were diluted in 100 mM phosphate buffer (PB), pH 7.3. Slices were incubated in blocking solution (0.5 % Triton X-100, 3 % normal goat serum) for 1 h at room temperature. Two different primary antibodies were simultaneously added to the slices in fresh blocking solution and incubated overnight at 4 °C. After washing in PB, secondary antibodies were added to the slices in PB containing 0.1 % Triton X-100 for 2 h at room temperature. For the analysis of protein expression in Purkinje cells, mouse anti-Calbindin D-28K (Swant, Marly, Switzerland, 1:1000), polyclonal rabbit anti-GFP (Abcam, Cambridge, UK, 1:1000), rabbit anti-CA8 (Novus Biologicals, Colorado, USA, 1:2000), mouse anti-ITPR1 (Abcam, Cambridge, UK, 1:100), rabbit anti-ITPR1 (Millipore, Zug, Switzerland, 1:2000), or rabbit anti-PKCγ (Santa Cruz Biotechnology, Santa Cruz, CA, 1:1000) were used as primary antibodies, and goat anti-mouse Alexa 568 (Molecular Probes, Eugene, OR, 1:1000) and goat anti-rabbit Alexa 488 (Molecular Probes, Eugene, OR, 1:1000) were used as secondary antibodies to visualize Purkinje cells. Stained slices were mounted on cover slips with Mowiol (Sigma-Aldrich, Buchs, Switzerland). Cultures were viewed on an Olympus AX-70 microscope equipped with a Spot digital camera. Recorded images were adjusted for brightness and contrast with Photoshop image processing software.
Histology
Mice were deeply anesthetized, perfused through the aorta with 4 % paraformaldehyde, and cryoprotected with 30 % sucrose overnight. Brains were embedded in tissue freezing medium (OCT), frozen, and sectioned using a Leica cryostat CM1900. Sagittal sections (25 μm) were examined by immunohistochemical staining.
Plasmid Transfection of Purkinje Cells in Dissociated Cerebellar Cultures
Dissociated cerebellar cultures were prepared from mice essentially as described [17] except that the cells were obtained from postnatal day 0 (P0) mice. The transfection of the cells was performed using a Nepa21 electroporator (NEPAGENE, Japan) according to manufacturer’s instructions. One hundred microliters of the Opti-MEM (Life Technologies, Zug, Switzerland) solution was mixed with the plasmid DNA to be transfected. This mixture was then used to suspend the cerebellar cell pellet. The cell suspension was transferred into one of the cuvettes provided in the kit and subjected to the optimized electroporation program.
After transfection, cells were plated in glass chambers that had been coated with poly-L-lysine containing 90 % v/v Dulbecco’s modified Eagle medium/F-12 nutrient-based medium (DFM), 1 % N-2 supplement, 1 % glutamax, and 10 % v/v FBS (all from Life Technologies, Zug, Switzerland); pH was adjusted to 7.2–7.4. Two hours after transfection, 0.8-ml DFM, containing 1 % N-2 supplement and 1 % glutamax at 37 °C, was added to each dish. After that, half of medium was changed once or twice a week. After 14–18 days, cells were fixed in 4 % paraformaldehyde for 1 h at 4 °C and processed for immunohistochemistry as described. A minimum of three independent transfection experiments using different dissociated cultures were done for each plasmid.
Knockdown of CA8 Expression
miRNAs were designed and generated by Life Technologies, Zug, Switzerland to target CA8 (for details see supplementary methods). Each miRNA was cloned into the BLOCK-iT™ Pol II miR RNAi expression vector according to the instructions of the manufacturer and then sequenced (Microsynth, Balgach, Switzerland). In order to test the knockdown efficacy, the different miRNA vectors together with a CA8 expression vector were transfected into HeLa cells using Lipofectamin 2000 (Life Technologies, Zug, Switzerland). The most efficient pcDNA6.2-GW/miRNA expression vectors containing CA8-specific miRNAs were subcloned to pL7 in order to transfect miRNA to Purkinje cells. In order to monitor miRNA transfection, a GFP reporter was included in the miRNA vectors. Dissociated cerebellar cultures cells were electroporated using Nepa21 (NEPAGENE, Japan) according to manufacturer’s instructions and cultured as described. A minimum of three independent transfection experiments using different dissociated cultures were done for each plasmid.
Statistical Analysis of Purkinje Cell Dendritic Tree Size
The quantification of Purkinje cell dendritic tree size and counting of branch points from organotypic slice cultures or dissociated Purkinje cell cultures were done as previously described [5]. Purkinje cells which had a dendritic tree isolated from its surroundings were selected for analysis. An image analysis program (Image Pro Plus or ImageJ) was used to trace the outline of the Purkinje cell dendritic trees yielding the area covered by the dendritic tree. Over 20 cells per experimental condition originating from at least three independent experiments were acquired and analyzed using GraphPad Prism software (San Diego, USA). The statistical significance of differences in parameters was assessed by a non-parametric Mann-Whitney’s test. Confidence intervals were 95 %; statistical significance was assumed when p < 0.05.
Results
Upregulated Genes in PKCγ-S361G Transgenic Mice Identified by Gene Chip Analysis
We have previously reported that PKCγ-S361G mice carrying a human SCA 14 allele with Purkinje cell-specific expression showed an increased PKCγ activity in Purkinje cells leading to abnormal morphology of Purkinje cells, a phenotype which is very similar to the one found after pharmacological treatment of PKC stimulator [14]. In contrast to pharmacological stimulation, the increased PKC activity in PKCγ transgenic mice is Purkinje cell specific, offering the possibility to search for downstream targets of PKCγ-S361G signaling which might be involved in the inhibition of Purkinje cell dendritic growth.
In order to search for transcripts with altered expression in Purkinje cells of PKCγ-S361G transgenic mice, we used the Affymetrix GeneChip Mouse Gene 1.0 ST Array comparing samples of cerebellar slice cultures from PKCγ transgenic mice to control mice. The microarray analysis was performed twice using two independent samples from different culture experiments. We could identify 98 genes which were upregulated and 17 genes which were downregulated in PKCγ-S361G transgenic mice based on a change of more than 1.25-fold (the complete microarray data sets are available from the authors on request). The rather moderate changes in expression of most transcripts could be explained by the fact that the Purkinje cell population is very small and only comprises around 0.1 % of the total cells of the cerebellum of adult rodents [19]. We focused on upregulated genes because our previous studies had shown that PKCγ-S361G transgenic mice had an increased activity of PKC, and we searched for candidates which might be involved in PKC signaling.
Out of the 98 upregulated genes, we selected eight genes for further analysis based on their expression level in Purkinje cells in situ and their presumed function (Table 1). A brief description of the main features of the eight candidate genes is given in the Supplementary Table 1. The expression of the mRNA of the eight candidate genes was verified using the Allen Brain Atlas. The in situ hybridization data from the Allen Brain Atlas (http://mouse.brain-map.org/) confirm that all of the eight selected candidate genes are strongly expressed in Purkinje cells. For several of these genes, an important role for Purkinje cell differentiation and function has already been suggested (Supplementary Table 1).
RT-qPCR Analysis and Western Blotting Confirm the Upregulation of Candidate Genes
In order to confirm the GeneChip microarray results (Fig. 1a), we performed RT-qPCR assays using cerebellar slice cultures. As expected, all of the candidate genes were upregulated in the cerebellar slice cultures from PKCγ-S361G transgenic mice (Fig. 1b). In order to confirm the increased expression of candidates on the protein level, we performed Western blot analysis. After 7 days of culture (DIV 7), cerebellar slice cultures of control mice and PKCγ-S361G transgenic mice were collected and candidate proteins were detected with Western blotting. At DIV 7, there is a strong reduction of the dendritic expansion of Purkinje cells in slice cultures from PKCγ-S361G transgenic mice [14]. In PKCγ-S361G transgenic mice, the PKCγ protein expression level is higher than that of control (Fig. 2a). Notably, fold change of PRKCG gene in GeneChip microarray was around 1.6 (data not shown). Within the candidates, the protein expression of CA8, Fam40b, and Plekhd1 was induced in PKCγ-S361G transgenic mice (Fig. 2b, c). CA8 was more than five times upregulated, while Fam40b and Plekhd1 were only slightly up at around 1.3 of control levels. As for the other candidates, either we could not see a strong difference in protein expression or no antibody suitable for Western blot analysis is available so far. Therefore, further analysis was restricted to CA8, Fam40b, and Plekhd1.

Gene chip and quantitative RT-PCR data for cerebellum of PKCγ-S361G transgenic mice and control mice (a). Induction of the expression of the eight selected candidate genes as found in the gene chip assay (b). Quantitative RT-PCR confirmed that all candidates are upregulated in PKCγ-S361G transgenic mice

Western blot analysis of cerebellum of PKCγ-S361G transgenic mice and control mice (a). Protein expression of PKCγ is increased in the cerebellum of PKCγ-S361G transgenic mice. β-Actin was used as a loading control. b Western blot results of CA8, Fam40b, and Plekhd1. β-Actin as loading control. c Quantitation of Western blot data. Signal intensity of each candidate was normalized with β-actin. Data were from three independent experiments, and signal intensity of samples from control mice was set to one
CA8 Is Specifically and Strongly Expressed in Purkinje Cells
To determine the expression pattern of CA8 in brain, we performed immunohistochemical analysis on mid-sagittal brain sections of postnatal day 14 (P14) wild-type mice and PKCγ-S361G transgenic mice. Purkinje cells were identified by staining for Calbindin-D28K together with CA8. We found that CA8 was specifically expressed in Purkinje cells and that the staining intensity was higher in PKCγ-S361G transgenic mice (Fig. 3a, b). Further, we performed immunostaining in organotypic slice cultures. CA8 was again most strongly expressed in cultures derived from PKCγ-S361G transgenic mice (Fig. 3c, d). These results suggest that CA8 is specifically expressed in Purkinje cells and that the expression level is induced by the PKCγ-S361G transgene.

Immunofluorescence-staining analysis of cerebellum of PKCγ-S361G transgenic mice and control mice. a–f Immunofluorescence staining of frozen section from P14 control mice in a–c and from PKCγ-S361G mice in d–f with anti-Calbindin-D28K (a, d) and with anti-CA8 (b, e). Merged images in (c, f). g–l Immunofluorescence staining of cerebellar slice cultures after 1 week in vitro. Cultures from control mice in g–i and from PKCγ-S361G mice in j–l stained with anti-Calbindin-D28K (g, j) and with anti-CA8 (h, k). Merged images in (i, l). Note the strong induction of anti-CA8 immunostaining in PKCγ-S361G cerebellum. Scale bar in H = 50 μm
Immunostaining for Fam40b and Plekhd1 with commercially available antibodies did not give satisfactory results and showed either no staining or only poor staining intensity on sections or organotypic cerebellar slice cultures. Therefore, we did not further analyze these candidates and focused on CA8.
The Inositol 1,4,5-Trisphosphate Receptor 1 (ITPR1) Is Also Induced in PKCγ-S361G Transgenic Mice
CA8, CA10, and CA11 are called carbonic anhydrase-related proteins (CARPs) because they lack classical carbonic anhydrase enzymatic activity due to the missing catalytic zinc-coordinating residues [20]. Thus, their biological functions are still unclear. Hirota et al. [21] showed that CARPs bind to ITPR1 using a yeast hybrid system and also found abundant co-expression and co-localization of CARP and ITPR1 in Purkinje cells. The ITPR1 functions as a ligand-gated ion channel that releases Ca2+ from intracellular stores [22]. We analyzed the ITPR1 expression level in PKCγ-S361G transgenic mice. Although we had not identified ITPR1 as a candidate in our GeneChip microarray because the p value was above 0.05 (p value was 0.052 and fold change was 1.42), ITPR1 mRNA was induced in PKCγ-S361G transgenic mice when tested with RT-qPCR (Fig. 4a). Western blotting analysis confirmed that ITPR1 protein is strongly upregulated in PKCγ-S361G transgenic mice (Fig. 4b, c). Immunofluorescence-staining results showed specific expression in Purkinje cells which was increased in PKCγ-S361G transgenic mice (Fig. 4d, e).

ITPR1 is induced in PKCγ-S361G transgenic mice (a). Quantitative RT-PCR showing upregulation of ITPR1 but not ITPR2 and ITPR3 in PKCγ-S361G. b Western blot results of ITPR1 from cerebellum of control and PKCγ-S361G transgenic mice. β-Actin as loading control. c Quantitation of Western blot data. Signal intensity was normalized with β-actin. d, e Anti-ITPR1 immunostaining of cerebellar slice cultures after 1 week in vitro. Note the strong induction of anti-ITPR1 immunostaining in PKCγ-S361G slices (e) versus control slices (d). Scale bar in E = 50 μm
No Evidence for a Binding Between CA8 and ITPR1 in Cerebellar Purkinje Cells
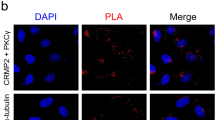
The co-expression of ITPR1 and CA8 in Purkinje cells and the induction of both proteins by increased PKCγ activity raise the question about a direct interaction of ITPR1 and CA8 in Purkinje cells. Binding of CA8 to ITPR1 has been previously shown using a glutathione S-transferase (GST) pull-down assay with recombinant proteins, but it is not known whether such an interaction also occurs endogenously in Purkinje cells. In order to identify an interaction in Purkinje cells, we have carried out co-immunoprecipitation using lysates from cerebellar slice cultures. Using this assay, we were unable to detect an interaction between ITPR1 and CA8 (Fig. 5) in cerebellar tissue. Furthermore, no interaction of CA8 and ITPR1 was detected using the Duolink proximity ligation assay (data not shown). Our data do not support the idea of a direct binding of CA8 and ITPR1 in cerebellar Purkinje cells.

Co-immunoprecipitation for CA8, ITPR1, PKCγ, and Homer3. At DIV 7, cerebellar slices were lysed (see “Materials and Methods”) and subjected to co-immunoprecipitation analysis. Western blots with the antibodies indicated at the left margin of co-immonoprecipitates from the antibodies indicated in the top line. Immunoprecipitation was only observed for the protein of the antibody used, and no co-immunoprecipitation of other proteins was found
CA8 Overexpression in Purkinje Cells Inhibits Purkinje Cell Dendritic Development
In order to characterize the functions of CA8 on dendritic development of Purkinje cells, we performed transfection experiments in dissociated cerebellar cell cultures with a Purkinje cell-specific expression vector based on the L7 promoter [17]. The L7 promoter plasmid was transfected by electroporation and efficiently and selectively mediated expression of the transfected genes in Purkinje cells at a high rate and with a continued expression for more than 3 weeks. We first transfected a plasmid containing the mutated PKCγ-S361G construct that was used for the generation of the PKCγ-S361G transgenic mice [14]. Control Purkinje cells in these cultures transfected with a GFP plasmid showed a well-developed branched dendritic tree (Fig. 6c) which was indistinguishable from that of non-transfected Purkinje cells (Fig. 6a). In contrast, Purkinje cells transfected with the PKCγ-S361G plasmid developed only very small dendritic trees with little branching (Fig. 6d). This morphology was indistinguishable from that of Purkinje cells derived from PKCγ transgenic mice (Fig. 6b). This finding confirms that the dissociated cultures are useful for studying some aspects of Purkinje cell dendritic development and that the PKCγ-mediated inhibition of Purkinje cell dendritic growth is present in the dissociated cultures. We then transfected a plasmid containing the CA8 gene inducing an overexpression of CA8 in the transfected Purkinje cells. CA8 overexpression resulted in a strong inhibition of Purkinje cell dendritic development compared to control Purkinje cells (Fig. 6e) with a morphology of the dendritic tree in transfected cells (Fig. 6f) very similar to that after transfection of the PKCγ-S361G containing plasmid (Fig. 6d). Quantitative measurements of the size of the Purkinje cell dendritic tree with the different transfections confirm that both PKCγ-S361G and overexpression of CA8 lead to a severe and similar reduction of the size of Purkinje cell dendritic trees (Fig. 6g–i). Our findings show that CA8 overexpression has similar effects on Purkinje cell dendritic development as a strong activation of PKCγ and suggest that PKCγ and CA8 may be acting in the same or related signaling pathways within Purkinje cells.

Morphology of Purkinje cells after PKCγ-S361G and CA8 transfection after 2 weeks in vitro. Immunostaining with anti-Calbindin-D28K (a, b) or anti-GFP (c–f). a Purkinje cells from control mice in dissociated culture with expanded dendritic trees. b Purkinje cell from PKCγ-S361G transgenic mice with very small dendritic trees. c Purkinje cells transfected with a control GFP vector with normal dendritic expansion. d Purkinje cells transfected with a PKCγ-S361G-GFP vector with small dendritic trees similar to those of PKCγ-S361G transgenic mice. e Purkinje cells transfected with a control GFP with normal dendritic expansion. f Purkinje cells transfected with a CA8-GFP vector also with small dendritic trees. Scale bar in H = 50 μm. g–i Quantification of the total dendritic area of Purkinje cells in the transfection experiments. The analysis shows that Purkinje cells transfected with PKCγ-S361G (h) or CA8 (i) had very small dendritic trees similar to those of Purkinje cells from PKCγ-S361G transgenic mice (g). The number of measured cells (n) was 20 for all experiments, and the results were statistically significant with p < 0.0001 in all conditions. Error bars represent the SEM
miRNA-Mediated Suppression of CA8 Protein Does Not Protect From Reduction of Dendritic Growth by Increased Activity of PKC
Our finding that overexpression of CA8 reduces dendritic growth raises the question whether CA8 might be part of a signaling complex which converts increased PKC activity into reduced dendritic growth. We have therefore aimed at suppressing the expression of CA8 protein in Purkinje cells by miRNA transfection. In order to knock down CA8 protein expression in Purkinje cells, we used two different miRNAs, mi327 and mi616, which were selected based on their knockdown efficiency in HeLa cells and which were cloned into the L7 promoter plasmid together with a GFP reporter (for details, see supplementary methods). mi327- and mi616-transfected Purkinje cells showed a greatly reduced CA8 expression as revealed by immunohistochemistry (Fig. 7d, f, h) compared to control miRNA-transfected (Fig. 7a) or miRNA-non-transfected cells. Purkinje cells with a CA8 knockdown displayed a rather normal morphology with no significant reduction of the size of the dendritic tree (Fig. 7c). When CA8 knockdown was combined with stimulation of PKC activity with phorbol-12-myristate-13-acetate (PMA), the CA8 knockdown did not provide any protection from the reduction of the dendritic tree seen under these conditions (Fig. 7e, f). A similar result was obtained when CA8 knockdown was done in Purkinje cells from PKCγ-S361G transgenic mice. The CA8 knockdown did not rescue the dendritic tree from the severe reduction typical for this condition (Fig. 7g, h). A quantitative evaluation of the dendritic tree sizes is shown in Fig. 7i confirming the qualitative findings. These data indicate that the signaling from PKCγ to the control of dendritic growth in Purkinje cells does not require a strong contribution of CA8 protein function.

Morphology of Purkinje cells after CA8 miRNA-mediated knockdown after 2 weeks in vitro. Immunostaining with anti-GFP (a, c, e, g, i, l) or anti-CA8 (b, d, f, h, k, m). a, b Control miRNA-GFP-transfected Purkinje cells express the GFP reporter (a) and show normal CA8 immunostaining (b) and a normal morphology. c, d A Purkinje cell transfected with CA8 miRNA-GFP expresses GFP and shows a normal morphology (c). A non-transfected Purkinje cell is well stained in the anti-CA8 immunostaining, whereas the CA8 miRNA-GFP-transfected Purkinje cell (arrow) is almost not visible. e, f Treatment with the PKC stimulator PMA dramatically reduced dendritic growth. e Anti-GFP immunostaining and f anti-CA8 immunostaining. Transfection with CA8 miRNA-GFP strongly reduced the CA8 immunostaining but did not protect from the PMA-induced dendritic reduction. g, h Purkinje cell from a PKCγ-S361G transgenic mouse with strongly reduced dendrites. g Anti-GFP immunostaining and h anti-CA8 immunostaining. Transfection with CA8 miRNA-GFP strongly reduced the CA8 immunostaining but did not protect from the dendritic reduction. Scale bar in H = 50 μm. i Quantification of the dendritic area of Purkinje cells in the CA8 miRNA-GFP transfection experiments. The analysis shows that transfection with CA8 miRNA-GFP could not rescue the dendritic tree size after stimulation with PMA (labeled WT + PMA) or in PKCγ-S361G transgenic Purkinje cells (labeled Tg). The data include control experiments with transfections of a control miRNA-GFP plasmid for which no images are shown in (a–h). The number of measured cells (n) was 20 for all experiments, and the reduction of the dendritic tree in the experiments with either PMA treatment or PKCγ-S361G transgenic mice was statistically significant with p < 0.0001 in all conditions irrespective of the presence of miRNA. Error bars represent the SEM
Discussion
Using a transgenic mouse model expressing a constitutive active form of PKC specifically in Purkinje cells, we have identified several candidate genes which might be involved in the stunted dendritic growth of Purkinje cells seen after PKC activation. One of these candidate genes, CA8, has a greatly increased mRNA and protein expression after PKC activation. Overexpression of CA8 in cultures of dissociated Purkinje cells strongly inhibited Purkinje cell dendritic development. Knockdown of CA8 protein expression by miRNA transfection did not protect Purkinje cells in dissociated cultures from the stunted dendritic growth after increased PKCγ activity. Our results show that CA8 is an important regulator of Purkinje cell dendritic development and that its expression is controlled by PKCγ activity.
Genes with Increased mRNA Expression in Purkinje Cells with Activated PKCγ
Increased activity of PKC in Purkinje cells results in a dramatic reduction of dendritic outgrowth and in the formation of a greatly reduced dendritic tree. The downstream signaling events converting the activation of PKC to reduced dendritic growth are still very unclear. We have used a transgenic mouse model expressing a mutant PKCγ with increased catalytic activity specifically in Purkinje cells resulting in stunted dendritic growth [14] as starting point for searching for genes with an increased expression in such Purkinje cells using a gene chip approach. We have identified many candidate molecules, although the observed upregulation was typically only in the range of 1.5–1.2× the original expression. The rather moderate changes in expression of most transcripts could be explained by the fact that the Purkinje cell population is very small and only comprises around 0.1 % of the total cells of the cerebellum of adult rodents [19]. When we confirmed the increased expression of some of the candidates by RT-qPCR, we actually found factors of 2 and more (Fig. 1). All of the selected candidate molecules are expressed in Purkinje cells and have the potential to be involved in PKC signaling and Purkinje cell dendritic development (see Supplementary Table 1). We decided to focus the further analysis on CA8 because it was the only candidate molecule for which commercially available antibodies gave satisfactory results in Western blotting and immunohistochemistry, both of which were required for further analysis of a potential function in Purkinje cell dendritic development.
CA8 is a carbonic anhydrase lacking enzymatic activity [20]. In the wdl mouse mutant with a 19-base-pair deletion in the CA8 gene, the loss of function of CA8 results in ataxia [15] and synaptic changes in the cerebellum [16] which are associated with alterations in calcium signaling [23]. CA8 knockdown in zebrafish resulted in abnormal cerebellar development and ataxia [24]. Mutations in the human CA8 gene were identified which are associated with cerebellar ataxia and mild cognitive retardation [25, 26], and antibodies directed against CA8 have been identified as a cause of Purkinje cell degeneration and cerebellar ataxia in paraneoplastic syndromes [27]. While through these studies there is a strong association of CA8 mutations with Purkinje cell death and cerebellar ataxia, only little is known about CA8 function in Purkinje cells. Our findings that CA8 mRNA is approximately 2-fold increased and that its protein shows a 6-fold increase in Purkinje cells of PKCγ-S361G transgenic mice suggest that it may be involved in the signaling events downstream of PKC activation. In view of the reported interaction between CA8 and ITPR1 [21], we have now shown that ITPR1 is strongly induced in Purkinje cells from PKCγ-S361G transgenic mice. Mutations in ITPR1 are causing spinocerebellar ataxia [28, 29], linking the final outcome of disturbances of PKCγ, CA8, and ITPR1 and suggesting that these proteins may interact functionally for the control of Purkinje cell synaptic function and dendritic development.
PKCγ, CA8, and ITPR1 Do Not Co-immunoprecipitate
CA8 was previously identified as a protein binding to ITPR1 in a yeast two hybrid screen, and the interaction was confirmed with a GST pull-down assay [21]. In a recent report, CA8 was shown to modulate pain sensation in sensory neurons by inhibiting ITPR1-mediated signaling and calcium release [30]. Although the ITPR1 was not originally identified as being upregulated in our gene chip screening (because the p value was slightly above 0.05), we have confirmed that it is also increased with an almost 5-fold-increased protein expression in Purkinje cells with activated PKC. In the classical signaling pathway, both PKCγ and the ITPR1 are activated in parallel via diacylglycerol (DAG) and IP3. It is an interesting finding that a chronic activation of PKC induces a strong induction of the protein expression of ITPR1 and the presumed interacting protein CA8. The activation of the PKC pathway thus induces an induction of its parallel pathway which is activated by IP3. This could mean that these pathways are not only linked by the common activation by DAG and IP3 but that PKC activation by itself can recruit the parallel IP3 pathway. The exact mechanism of this interaction still needs to be determined. It is well known that the IP3 receptor is a target of PKC phosphorylation and that phosphorylation changes the IP3-induced calcium release [31]. Our finding suggests that PKC-mediated phosphorylation of the ITPR1 may eventually increase the protein expression of this receptor.
Despite the parallel rise of protein expression of ITPR1 and CA8 in Purkinje cells of PKCγ-S361G transgenic mice and the finding that an interaction between these two proteins was identified in a yeast two hybrid screen and by a GST pull-down assay [21], we could not show a co-immunoprecipitation of CA8 and ITPR1 in Purkinje cells. Neither ITPR1 nor PKCγ co-immunoprecipitated with CA8. Similarly, in the Duolink proximity assay, no evidence for a close interaction was found (data not shown). Of course we cannot exclude that this failure to demonstrate a direct binding was due to technical limitations, but both proteins are solidly expressed, and we have tried different immunoprecipitation protocols, none of which was showing co-immunoprecipitation (data not shown). Therefore, the exact nature of the interaction between PKCγ, CA8, and ITPR1 in Purkinje cells still needs to be studied further.
CA8 and Purkinje Cell Dendritic Development
The strong upregulation of CA8 with activation of PKCγ raises the possibility that CA8 is directly or indirectly involved in the stunted growth of the Purkinje cell dendritic tree seen with constitutive activation of PKCγ in PKCγ-S361G transgenic mice. We have addressed this issue by overexpressing CA8 in dissociated Purkinje cell cultures. The dissociated culture system is of course less complex and more artificial compared to organotypic slice cultures, but it allows to reliably transfect Purkinje cells by electroporation and the use of an L7-based Purkinje cell-specific expression vector [17]. Purkinje cells derived from PKCγ-S361G transgenic mice show a much reduced dendritic development in such cultures, and also, transfection of a PKCγ-S361G plasmid resulted in a strong reduction of dendritic outgrowth (Fig. 6), indicating that the dissociated culture does indeed reflect the changes of dendritic outgrowth originally seen in organotypic slice cultures. When Purkinje cells were transfected with a CA8 plasmid resulting in CA8 overexpression, a similar stunted dendritic growth was observed as seen after overexpression of PKCγ or of the mutated PKCγ-S361G (Fig. 6). This finding shows that CA8 function does have an inhibitory effect on Purkinje cell dendritic growth and that it is likely that PKCγ and CA8 are both part of a common or related signaling pathway eventually controlling dendritic outgrowth and retraction. If CA8 was a downstream effector of PKC activation, a loss of function of CA8 should reduce the effects of PKCγ on the Purkinje cell dendrites. To this end, we have used transfection of miRNA in order to suppress CA8 expression. After transfection with miRNA, protein expression of CA8 was reduced to a degree that cells were only weakly detected by immunohistochemistry (Fig. 7), confirming an efficient suppression of CA8 protein expression by the miRNA. Despite this marked reduction of CA8 expression, activation of PKC with PMA still showed the typical stunted dendritic growth indicating that CA8 is not required for this effect. There was also no rescue of the stunted dendritic tree by CA8 knockdown in Purkinje cells derived from PKCγ-S361G transgenic mice. While these findings leave open the possibility that the small residual amount of CA8 protein present in Purkinje cells with CA8 knockdown is sufficient for mediating the effects of PKCγ activation, they make it very unlikely that CA8 is the dominant mediator of PKCγ activation. They leave open the possibility that CA8 may be more loosely associated with PKCγ signaling or that its function might be redundant with another yet to be identified mediator of PKCγ activation.
Conclusions
In this study we have identified CA8 and the ITPR1 as two genes with strongly increased expression in Purkinje cells with chronic activation of PKCγ. We could not find evidence for a direct interaction of CA8 with the ITPR1 but show that overexpression of CA8 in Purkinje cells results in a phenotype with stunted dendritic growth similar to that seen after activation or overexpression of PKCγ. Although CA8 is probably not a direct mediator of PKCγ for the effects on dendritic growth, it is likely to be part of a larger signaling network including PKCγ and the ITPR1 which controls synaptic function and dendritic growth of Purkinje cells.
References
Ito M, Yamaguchi K, Nagao S, Yamazaki T (2014) Long-term depression as a model of cerebellar plasticity. Prog Brain Res 210:1–30. doi:10.1016/B978-0-444-63356-9.00001-7
Metzger F, Kapfhammer JP (2000) Protein kinase C activity modulates dendritic differentiation of rat Purkinje cells in cerebellar slice cultures. Eur J Neurosci 12:1993–2005. doi:10.1046/j.1460-9568.2000.00086.x
Schrenk K, Kapfhammer JP, Metzger F (2003) Altered dendritic development of cerebellar Purkinje cells in slice cultures from protein kinase C gamma-deficient mice. Neuroscience 110:675–689. doi:10.1016/S0306-4522(01)00559-0
Kapfhammer JP (2004) Cellular and molecular control of dendritic growth and development of cerebellar Purkinje cells. Prog Histochem Cytochem 39:131–182
Gugger OS, Hartmann J, Birnbaumer L, Kapfhammer JP (2012) P/Q-type and T-type calcium channels, but not type 3 transient receptor potential cation channels, are involved in inhibition of dendritic growth after chronic metabotropic glutamate receptor type 1 and protein kinase C activation in cerebellar Purkinje cells. Eur J Neurosci 35:20–33. doi:10.1111/j.1460-9568.2011.07942.x
Becker EB, Oliver PL, Glitsch MD, Banks GT, Achilli F, Hardy A, Nolan PM, Fisher EM, Davies KE (2009) A point mutation in TRPC3 causes abnormal Purkinje cell development and cerebellar ataxia in Moonwalker mice. Proc Natl Acad Sci U S A 106:6706–6711. doi:10.1073/pnas.0810599106
Huang H, Nagaraja RY, Garside ML, Akemann W, Knöpfel T, Empson RM (2010) Contribution of plasma membrane Ca ATPase to cerebellar synapse function. World J Biol Chem 1:95–102. doi:10.4331/wjbc.v1.i5.95
Sherkhane P, Kapfhammer JP (2013) The plasma membrane Ca2 + -ATPase2 (PMCA2) is involved in the regulation of Purkinje cell dendritic growth in cerebellar organotypic slice cultures. Neural Plast 2013:321685. doi:10.1155/2013/321685
Pandolfo M, van de Warrenburg BP (2005) Spinocerebellar ataxia type 14: opening a new door in dominant ataxia research? Neurology 64:1113–1114. doi:10.1212/01.WNL.0000160013.10806.53
Chen DH, Raskind WH, Bird TD (2012) Spinocerebellar ataxia type 14. Handb Clin Neurol 103:555–559. doi:10.1016/B978-0-444-51892-7.00036-X
Verbeek DS, Knight MA, Harmison GG, Fischbeck KH, Howell BW (2005) Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain 128:436–442. doi:10.1093/brain/awh378
Adachi N, Kobayashi T, Takahashi H, Kawasaki T, Shirai Y, Ueyama T, Matsuda T, Seki T, Sakai N, Saito N (2008) Enzymological analysis of mutant protein kinase Cgamma causing spinocerebellar ataxia type 14 and dysfunction in Ca2+ homeostasis. J Biol Chem 283:19854–19863. doi:10.1074/jbc.M801492200
Chen DH, Cimino PJ, Ranum LP, Zoghbi HY, Yabe I, Schut L, Margolis RL, Lipe HP, Feleke A, Matsushita M, Wolff J, Morgan C, Lau D, Fernandez M, Sasaki H, Raskind WH, Bird TD (2005) The clinical and genetic spectrum of spinocerebellar ataxia 14. Neurology 64:1258–1260. doi:10.1212/01
Ji J, Hassler ML, Shimobayashi E, Paka N, Streit R, Kapfhammer JP (2014) Increased protein kinase C gamma activity induces Purkinje cell pathology in a mouse model of spinocerebellar ataxia 14. Neurobiol Dis 70:1–11. doi:10.1016/j.nbd.2014.06.002
Jiao Y, Yan J, Zhao Y, Donahue LR, Beamer WG, Li X, Roe BA, Ledoux MS, Gu W (2005) Carbonic anhydrase-related protein VIII deficiency is associated with a distinctive lifelong gait disorder in waddles mice. Genetics 171:1239–1246. doi:10.1534/genetics.105.044487
Hirasawa M, Xu X, Trask RB, Maddatu TP, Johnson BA, Naggert JK, Nishina PM, Ikeda A (2007) Carbonic anhydrase related protein 8 mutation results in aberrant synaptic morphology and excitatory synaptic function in the cerebellum. Mol Cell Neurosci 35:161–170. doi:10.1016/j.mcn.2007.02.013
Wagner W, McCroskery S, Hammer JA 3rd (2011) An efficient method for the long-term and specific expression of exogenous cDNAs in cultured Purkinje neurons. J Neurosci Methods 200:95–105. doi:10.1016/j.jneumeth.2011.06.006
Kapfhammer JP (2010) Cerebellar slice cultures. In: Doering LC (ed) Protocols for neural cell culture, 4th edn. Humana Press, New York, pp 285–298
Tomomura M, Rice DS, Morgan JI, Yuzaki M (2001) Purification of Purkinje cells by fluorescence-activated cell sorting from transgenic mice that express green fluorescent protein. Eur J Neurosci 14:57–63. doi:10.1046/j.0953-816x.2001.01624.x
Sjöblom B, Elleby B, Wallgren K, Jonsson BH, Lindskog S (1996) Two point mutations convert a catalytically inactive carbonic anhydrase-related protein (CARP) to an active enzyme. FEBS Lett 398:322–325. doi:10.1016/S0014-5793(96)01263-X
Hirota J, Ando H, Hamada K, Mikoshiba K (2003) Carbonic anhydrase-related protein is a novel binding protein for inositol 1,4,5-trisphosphate receptor type 1. Biochem J 372:435–241. doi:10.1042/BJ20030110
Banerjee S, Hasan G (2005) The InsP3 receptor: its role in neuronal physiology and neurodegeneration. Bioessays 27:1035–1047. doi:10.1002/bies.20298
Lamont MG, Weber JT (2015) Mice deficient in carbonic anhydrase type 8 exhibit motor dysfunctions and abnormal calcium dynamics in the somatic region of cerebellar granule cells. Behav Brain Res 286:11–16. doi:10.1016/j.bbr.2015.02.035
Aspatwar A, Tolvanen ME, Ortutay C, Parkkila S (2014) Carbonic anhydrase related proteins: molecular biology and evolution. Subcell Biochem 75:135–156. doi:10.1007/978-94-007-7359-2_8
Kaya N, Aldhalaan H, Al-Younes B, Colak D, Shuaib T, Al-Mohaileb F, Al-Sugair A, Nester M, Al-Yamani S, Al-Bakheet A, Al-Hashmi N, Al-Sayed M, Meyer B, Jungbluth H, Al-Owain M (2011) Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am J Med Genet B Neuropsychiatr Genet 156B:826–834. doi:10.1002/ajmg.b.31227
Türkmen S, Guo G, Garshasbi M, Hoffmann K, Alshalah AJ, Mischung C, Kuss A, Humphrey N, Mundlos S, Robinson PN (2009) CA8 mutations cause a novel syndrome characterized by ataxia and mild mental retardation with predisposition to quadrupedal gait. PLoS Genet 2009, e1000487. doi:10.1371/journal.pgen.100048
Höftberger R, Sabater L, Velasco F, Ciordia R, Dalmau J, Graus F (2014) Carbonic anhydrase-related protein VIII antibodies and paraneoplastic cerebellar degeneration. Neuropathol Appl Neurobiol 40:650–653. doi:10.1111/nan.12118
van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung HC, Lin X, Hernandez D, Simon-Sanchez J, Wood NW, Giunti P, Rafferty I, Hardy J, Storey E, Gardner RJ, Forrest SM, Fisher EM, Russell JT, Cai H, Singleton AB (2007) Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet 3, e108. doi:10.1371/journal.pgen.0030108
Schorge S, van de Leemput J, Singleton A, Houlden H, Hardy J (2010) Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci 33:211–219. doi:10.1016/j.tins.2010.02.005
Zhuang GZ, Keeler B, Grant J, Bianchi L, Fu ES, Zhang YP, Erasso DM, Cui JG, Wiltshire T, Li Q, Hao S, Sarantopoulos KD, Candiotti K, Wishnek SM, Smith SB, Maixner W, Diatchenko L, Martin ER, Levitt RC (2015) Carbonic anhydrase-8 regulates inflammatory pain by inhibiting the ITPR1-cytosolic free calcium pathway. PLoS One 10:e0118273. doi:10.1371/journal.pone.0118273
Vermassen E, Parys JB, Mauger JP (2004) Subcellular distribution of the inositol 1,4,5-trisphosphate receptors: functional relevance and molecular determinants. Biol Cell 96:3–17. doi:10.1016/j.biolcel.2003.11.004
Acknowledgments
We thank Dr. John A. Hammer III (National Heart, Lung and Blood Institute, NIH, Bethesda, MD, USA) for providing the pL7-mGFP plasmid and Markus Saxer for technical assistance. Dr. Phillipe Demougin from the Life Science Training Facility (LSTF) of the University of Basel performed the microarray experiments. This work was supported by the University of Basel and the Swiss National Science Foundation (31003A-116624).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 70 kb)
Rights and permissions
About this article

Cite this article
Shimobayashi, E., Wagner, W. & Kapfhammer, J.P. Carbonic Anhydrase 8 Expression in Purkinje Cells Is Controlled by PKCγ Activity and Regulates Purkinje Cell Dendritic Growth. Mol Neurobiol 53, 5149–5160 (2016). https://doi.org/10.1007/s12035-015-9444-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-015-9444-3