
Abstract
IgA protease is secreted by various mucosal pathogenic bacteria which can cleave human immunoglobulin A1 (IgA1) in its hinge region. In addition to be considered as a virulence factor, it's reported that IgA protease can also be used for IgA nephropathy (IgAN) treatment. Our previous study identified bacteria H. influenzae 49247 expressed high activity of IgA protease with promised application in IgAN therapy. In this study, we cloned the IgA protease gene of H. influenzae 49247 with degenerate primers. Alignment analysis indicated that H. influenzae 49247 IgA protease showed unique DNA and amino acid sequence but with typical endopeptidase domain and beta transporter domain compared with known IgA proteases from the same species. To facilitate expression and purification, the H. influenzae 49247 IgA protease gene was sub-cloned into the pET28-A(+) vector with insertion of a 6xHis tag downstream of the endopeptidase domain and upstream of the potential autocleavage site. The recombined IgA protease can be constitutively expressed in E. coli and secreted into the culture medium. With a simple nickel affinity binding, the secreted IgA protease can be purified with high purity (95%) and a molecular weight of about 130 kDa. The identity of the IgA protease was validated by the presence of 6xHis tag in the purified protein by western blotting and its ability to cleave human IgA1 molecule. Collectively, the successful cloning, expression and purification of H. influenzae 49247 IgA protease will augment its therapeutic study in IgAN treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
IgA proteases are a group of endopeptidases secreted by many pathogenic bacteria and can cleave the hinge region of human IgA1 molecule [1]. Furthermore, beside IgA1, bacterial IgA protease can also impair lysosome function through cleaving LAMP1 to facilitate intracellular bacterial infection [2, 3]. Therefore, IgA protease is considered to be a virulence factor and plays important roles in pathogen invasion and colonization in the mucosal surface or even in the host cell [1, 3]. Mucosal colonized pathogenic bacteria like H. influenzae, N. gonorrhoeae, N. meningitidis and S. pneumonia are rich in IgA protease expression. However, the IgA proteases also show activity diversity caused by their highly genetic heterogeneity among different species and strains [4,5,6].
In addition to be a pathogenic factor, IgA protease was also proven to serve as a potential therapeutic agent in the treatment of IgA nephropathy (IgAN). IgAN is characterized by mesangial precipitation of IgA1-containing immune complex leading to abnormal renal function. It has been proven that IgA protease can degrade exogenous and endogenous IgA1-containing immune complex deposited in glomeruli in mouse model [6, 7]. And the impaired renal function, like proteinuria and hematuria, can also be ameliorated by IgA protease injection [8]. Furthermore, study in mouse suggested that purified IgA protease can also be employed as a vaccine to protect from the infection of corresponding pathogenic bacteria [9].
In our previous study aimed to identifying IgA protease for IgAN treatment, we screened IgA protease activity of 14 bacterial strains from 6 species. Among these bacteria, we finally identified IgA protease from H. influenzae 49247 with the highest activity and the ability to degrade in vivo deposited aberrantly glycosylated IgA1-containing immune complex [6]. Here, we reported the cloning of the H. influenzae 49247 IgA protease gene and its exogenous expression and purification in the E. coli system.
Materials and Methods
Cloning of H. influenzae 49247 IgA Gene
For designation of degenerate primers for H. influenzae 49247 IgA protease gene amplification, 16 different IgA protease gene sequences from H. influenzae of different strains were used for alignment analysis by DNAssist software to identify the conserved sequence region. The accession numbers of the 16 sequences are indicated in Fig. 2b. The degenerate primers used for the final successful amplification were: F1, 5′-ATGCTAAATAAAAAATTCAAACTCAATTTTATTGCD CTTACTGTCGCCTACGCAT-3′, R1, 5′-CCYGCAATATCTCTTGCGTG-3′, F2, 5′-CACGCAA GAGATATTGCRGG-3′ and R2, 5′-TTAAAAACTAAAACTTAGTTTYAMTTCYGCWGTT-3′. H. influenzae 49247 bacteria grown on Columbia chocolate agar plate were collected and rinsed with sterile distilled H2O. Proper amount of bacteria were directly used for PCR amplification. The PCR product was analyzed with agarose gel and sequenced by Sangon Biotech Co., Ltd (Shanghai China).
Bioinformatics Analysis of H. influenzae 49247 IgA Protease
The Simple Modular Architecture Research Tool (SMART) web software was used to predict the domain structure of H. influenzae 49247 IgA protease (http://smart.embl-heidelberg.de/). The NCBI web BLAST tool was applied to analyze sequence similarity between different bacterial IgA proteases. Phylogenetic tree was generated with Mega 5 software. The IgA proteases used for sequence similarity analysis and phylogenetic tree construction were retrieved from their genomic DNA sequence whose NCBI accession No. is indicated in Fig. 2b.
Exogenous Expression and Purification of H. influenzae 49247 IgA Protease
The H. influenzae 49247 IgA protease open reading frame (ORF) was codon-optimized to adapt to E. coli codon bias and cloned into pET28-A(+) vector. A 6xHis tag was introduced into the N terminus of the protein or the downstream of protease domain (adjacent to the autocleavage site). The expression vector was transformed into E. coli BL21 bacteria and grown on agar plate with 25 μg/ml kanamycin sulfate. For expression assay, 4 bacterial clones were inoculated into 4 ml of LB broth and cultured overnight at 37 °C with rotation at 160 rpm. Then, 1 mM IPTG was supplemented or not for further 6 h. The bacteria and culture supernatant were separated by centrifugation for IgA protease activity checking. The bacteria pellet was washed with PBS and lysed by ultrasonication in PBS. The cell debris was removed by centrifugation. For IgA protease activity assay, 1 μl of culture supernatant or equivalent bacterial lysate was incubated with 0.5 μg of human myeloma-derived IgA1 (Merck KGaA, Germany) overnight and subjected to SDS–PAGE gel electrophoresis followed by silver staining.
For large-scale nickel (Ni) affinity purification, a 500 ml of overnight culture was centrifuged at 4 °C at 10,000 rpm to isolate the culture supernatant followed by filtration through filter paper and 0.22-μm filter. (NH4)2SO4 was added to the cleared supernatant to 80% saturation to precipitate the protein overnight at 4 °C with gentle stirring. Next day, the precipitated protein was spun down and re-dissolved in 10 ml of PBS followed by dialysis against PBS to remove residual (NH4)2SO4. Then, imidazole was added into the raw protein preparation to 10 mM and bond with 5 ml of Ni–NTA sefinose resin (Sangon Biotech, Shanghai, China). After washing with PBS containing 10 mM imidazole, the bond protein was eluted with 10 ml of PBS containing 400 mM imidazole. The elute was dialyzed and concentrated by an Amicon Ultra-15 Centrifugal Filter Unit (Millipore, USA). IgA protease activity of each purification fraction was monitored as described above. Protein concentration of purified IgA protease was determined by the Coomassie brilliant blue method. 0.5 μg of purified IgA protease was resolved in 12% SDS–PAGE gel followed by silver staining. Gel-Pro Analyzer software was used to quantify the purity by ratio of integrated gray value of target IgA protease to that of total protein bands in the electrophoresis lane.
Western Blot
Western blot was performed as previously described [6]. In Fig. 3c, 20 μl of culture supernatant or equivalent bacteria were loaded for each lane. In Fig. 4c, 0.5 μg purified IgA protease protein was used for western blot analysis. Antibody used in this study is mouse anti-6xHis monoclonal antibody (Proteintech, USA) and peroxidase-conjugated goat anti-mouse IgG (ZSGB-BIO, China).
Results
Amplification and Sequencing of IgA Gene Loci of H. influenzae 49247
Bacterial species enjoy highly genetic variation even among the different strains from the same species. And this also happens for IgA protease gene in H. influenzae [4, 5]. To clone the sequence of IgA protease gene of H. influenzae 49247, we designed the degenerate primers based on the alignment analysis of 16 known IgA protease gene sequences of different H. influenzae strains. As shown in Fig. 1, the whole coding region of IgA protease gene of H. influenzae 49247 was successfully amplified in two continuous fragments. The length of upstream fragment is about 2 kbp (kilo base pairs) and downstream fragment 3 kbp. The two PCR fragments were sequenced and the intact coding sequence was assembled. The IgA protease gene of H. influenzae 49247 is 5238 bp in size.

PCR amplification of IgA gene with H. influenzae 49247 bacteria as template. The PCR was conducted with a range of anneal temperature as indicated up the lanes
Bioinformatic Analysis of Cloned IgA Protease of H. influenzae 49247
Nucleotide acid sequence alignment indicated that both H. influenzae 49247 and the above 16 bacterial stains’ IgA protease genes from the same species presented unique sequence (data not shown), in line with the fact that IgA protease gene showed highly genetic heterogeneity [4]. Theoretically, the H. influenzae 49247 IgA protease gene encodes a polypeptide of 1745 amino acids. Amino acid sequence analysis with the SMART web software showed that the deduced protein contained an autotransporter beta domain and a serine protease domain of peptidase_S6 superfamily (Fig. 2a). Both of the two domains are typically shared by serine-type bacterial IgA protease [1]. Furthermore, the deduced H. influenzae 49247 IgA protease also contained a conserved serine-type IgA protease enzymatic active motif “VLGDSGSPLF” and a similar potential autocleavage site (compared with the reported H. influenzae Rd IgA protease autocleavage site) (Fig. 2a) [1, 10]. The cloned H. influenzae 49247 IgA protease shared 63 to 80% of sequence similarity compared with IgA protease proteins from different strains of the same species. However, lower than 51% of sequence similarity can be found between H. influenzae 49247 IgA protease with those from N. meningitidis and N. gonorrhoeae although they all belong to serine-type IgA protease (Fig. 2b) [1]. Phylogenetic tree analysis also indicated that the cloned H. influenzae 49247 IgA protease can be classified into the IgA protease subgroup of H. influenzae (Fig. 2b). These data confirmed the successful cloning of H. influenzae 49247 IgA protease gene and also its unique sequence identity.

Bioinformatic analysis of H. influenzae 49247 IgA protease. a The domain distribution predicted by SMART software. The enzyme active site and autocleavage site are also indicated. The conserved active site motif is underlined, and potential autocleavage proline residual is labeled with asterisk. b Phylogenetic tree generated with MEGA 5 software. The amino acid sequence similarity in comparison with H. influenzae 49247 IgA protease is indicated on the left. Note that some bacteria without a strain code were labeled as UN (unknown). For N. meningitidis 510612 and N. gonorrhoeae DGI18 who have two IgA proteases gene loci, the two IgA proteases are discriminated as No. 1 and No. 2
Prokaryotic Expression and Purification of H. influenzae 49247 IgA Protease
Next, we tried to express the cloned H. influenzae 49247 IgA protease gene (thereafter indicated as IgA protease for short) in E. coli which is more convenient and safer for exogenous protein preparation. The H. influenzae 49247 IgA protease gene ORF was subjected to codon optimization to facilitate E. coli expression and further cloned into prokaryotic expression vector pET28-A(+). To facilitate protein purification, we firstly fused a 6xHis tag on the N terminus of IgA protease ORF. E. coli transformed with the vector expressed IgA protease activity (cleavage of human IgA1) mainly in the culture supernatant without the induction by IPTG (data not shown), suggesting constitutive expression and secretion of IgA protease. However, no 6xHis tag can be detected in the bacterial body or supernatant by western blot (data not shown). We suspected that the 6xHis tag must be removed along with the signal peptide during the secretion of de novo synthesized IgA protease. We next moved the 6xHis tag to the site at the N terminus of the potential autocleavage site of the IgA protease and downstream of the protease domain (Fig. 3a). As shown in Fig. 3b, c, introduction of the 6xHis tag in site adjacent to the autocleavage site preserved its constitutive expression and autosecretion feature of the exogenously expressed IgA protease. Western blot analysis detected weak band with molecular weight of about 130 kDa in the bacterial body but not in the supernatant (arrow in Fig. 3c). This may be caused by low concentration of secreted IgA protease protein in the supernatant. Interestingly, there was also a more intensive band with molecular weight similar to EGFP protein to be observed.

Expression of the cloned H. influenzae 49247 IgA protease in E. coli. a Schematic graph shows the insertion position of 6xHis tag. b IgA protease activity of bacterial body lysate and culture supernatant is assayed by digestion incubation with human IgA1 substrate followed by SDS–PAGE electrophoresis and silver staining. M protein marker. N negative control. B bacteria. S culture supernatant. H and L are heavy and light chain of IgA1, respectively. Fc and Fd are the cleavage product of heavy chain of IgA1. c Western blot analysis to check the 6xHis tag in the bacteria and culture supernatant. Bacteria expressing 6xHis-tagged EGFP is used as a positive control. Arrow indicates the potential IgA protease band
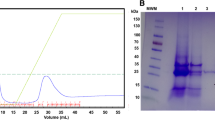
As the majority of the protease activity was distributed in the culture medium, we next performed the Ni affinity chromatography with a larger batch of culture (500 ml) as described in the methods section. As shown in Fig. 4a, the IgA protease activity can be fully recovered by Ni affinity chromatography. After elution and concentration of bound protein, a single protein band of about 130 kDa can be observed by silver staining with a purity of about 95% (Fig. 4b). Western blot confirmed the existence of the 6xHis tag in the purified protein (Fig. 4c). The final yield of this purification procedure is about 2 mg from 1 l of initial culture medium. Although ITPG is not essential for the expression of recombined IgA protease, addition of IPTG (1 mM) can indeed slightly increase the final yields (to about 2.47 mg from 1 l of initial culture). Furthermore, the purified IgA protease showed highly increased enzymatic activity compared with the crude culture medium from both transformed E. coli and its natural host bacteria H. influenzae 49247 (Fig. 4d). These data suggested the successful purification of the H. influenzae 49247 IgA protease.

Purification of H. influenzae 49247 IgA protease by Ni chromatography. IgA protease activity monitoring of purification fraction of Ni chromatography. 1, negative control with only IgA1. 2, raw protein solution before Ni column binding. 3, protein solution after Ni column binding. 4, washing solution. 5, IgA protease eluted with 400 mM imidazole. b Silver staining of 0.5 μg of purified IgA protease. c Western blot analysis to check the 6xHis tag in purified IgA protease. d IgA protease activity comparison between purified recombined IgA protease (lane 4) and culture supernatant of natural host bacteria (H. influenzae 49247, lane 2) and transformed E. coli (lane 3). Lane 1 is control blank IgA1 substrate. In a, b, c and d, M means protein marker. In a and b, H and L are heavy and light chain of IgA1. Fc and Fd are the cleavage product of IgA1 heavy chain
Discussion
In this study, we reported the cloning of IgA protease gene with degenerate primers from a novel H. influenzae strain and engineered expression in E. coli with a fused 6xHis tag. We showed that the engineered IgA protease can constitutively be expressed and secreted into the medium with preserved activity. Finally, the exogenously expressed IgA protease can be easily purified with high purity.
The successful expression of secreted H. influenzae IgA protease in E. coli also indicates that the beta transporter domain of IgA protease is functional compatible in different gram-negative bacteria, which keeps in line with the previous reports [11, 12].
To defend the host mucosal IgA1, the bacteria convergently evolved the IgA proteases which fall into three groups, serine, metallo- and cysteine-type IgA protease [1]. Genetic analysis indicated highly inter- and intraspecies heterogeneity caused by gene transfer and recombination between different species and strains [13]. In line with the feature of high genetic variance, the cloned IgA protease gene of H. influenzae 49247 also showed unique nucleotide and amino acid sequence compared with the other 16 strains of the same species (Fig. 2b and data not shown). This unique sequence feature may contribute to its relative higher enzymatic activity demonstrated in our previous study [6].
Several studies have reported the purification of natural or recombined IgA protease. Purification of natural IgA protease from the native host bacteria always needed finely equipped technical platform (kinds of chromatography equipments, usually) and complex procedure with skillful manipulation. However, the yields were always extremely low (several to hundreds microgram per liter of culture supernatant depending on bacterial species, strains and purification procedure) and the purity was barely satisfactory [14,15,16]. Recently, Shinong et al. reported successful expression and purification of the 6xHis tagged-IgA protease domain (without the β transporter domain) of H. influenzae in E. coli. Although the final production was plausible, a considerable part of recombined IgA protease existed as insoluble inclusive bodies and a finely controlled induction condition was needed. Furthermore, a combination of ion-exchange chromatography and size-exclusive gel filtration was still required for high purity of IgA protease [17]. Lamm and his colleagues inserted the 6xHis tag at the C terminus of IgA protease domain in host bacterial genome (H. influenzae). The engineered H. influenzae strain can constitutively express secreted recombined IgA protease which can be further purified with Ni column [7]. However, culturing large batch of pathogenic H. influenzae for large amount of IgA protease preparation may cause a bio-safety problem. In our study, the engineered IgA protease was constitutively expressed and secreted into the culture medium in E. coli (BL21 strain) which is much safer than culturing pathogenic host bacteria. And, as inspired by Lamm’s work, we also inserted the 6xHis tag in the C terminus of IgA protease domain (also upstream of the autocleavage site) to prevent the 6xHis tag from loss during IgA protease secretion. With our procedure, high purity of IgA protease can be made from the culture medium by simple Ni affinity chromatography without other complex chromatography. Although the total production (about 2.47 mg/l with IPTG) is lower than that from Shinong’s study (20–40 mg/l), it’s much higher than the yields from natural host bacteria [11, 18]. Furthermore, higher yield may be applicable by increasing the culture time or renewing the culture medium with our system.
Collectively, we successfully cloned the IgA protease gene of H. influenzae 49247 followed by expression and purification in E. coli system. This simple and high efficient purification procedure can provide high quality of IgA protease with satisfied quantity for its therapeutic study in IgAN treatment.
References
Mistry, D., & Stockley, R. A. (2006). IgA1 protease. The International Journal of Biochemistry & Cell Biology, 38, 1244–1248.
Lin, L., Ayala, P., Larson, J., Mulks, M., Fukuda, M., Carlsson, S. R., et al. (1997). The Neisseria type 2 IgA1 protease cleaves LAMP1 and promotes survival of bacteria within epithelial cells. Molecular Microbiology, 24, 1083–1094.
Clementi, C. F., Hakansson, A. P., & Murphy, T. F. (2014). Internalization and trafficking of nontypeable Haemophilus influenzae in human respiratory epithelial cells and roles of IgA1 proteases for optimal invasion and persistence. Infection and Immunity, 82, 433–444.
Poulsen, K., Reinholdt, J., & Kilian, M. (1992). A comparative genetic study of serologically distinct Haemophilus influenzae type 1 immunoglobulin A1 proteases. Journal of Bacteriology, 174, 2913–2921.
Lomholt, H., Poulsen, K., & Kilian, M. (1995). Comparative characterization of the IgA gene encoding IgA1 protease in Neisseria meningitidis, Neisseria gonorrhoeae and Haemophilus influenzae. Molecular Microbiology, 15, 495–506.
Wang, L., Li, X., Shen, H., Mao, N., Wang, H., Cui, L., et al. (2016). Bacterial IgA protease-mediated degradation of agIgA1 and agIgA1 immune complexes as a potential therapy for IgA nephropathy. Scientific Reports, 6, 30964.
Lamm, M. E., Emancipator, S. N., Robinson, J. K., Yamashita, M., Fujioka, H., Qiu, J., et al. (2008). Microbial IgA protease removes IgA immune complexes from mouse glomeruli in vivo: Potential therapy for IgA nephropathy. The American Journal of Pathology, 172, 31–36.
Lechner, S. M., Abbad, L., Boedec, E., Papista, C., Le Stang, M. B., Moal, C., et al. (2016). IgA1 protease treatment reverses mesangial deposits and hematuria in a model of IgA nephropathy. Journal of the American Society of Nephrology, 27, 2622–2629.
Kotelnikova, O. V., Zinchenko, A. A., Vikhrov, A. A., Alliluev, A. P., Serova, O. V., Gordeeva, E. A., et al. (2016). Serological analysis of immunogenic properties of recombinant meningococcus IgA1 protease-based proteins. Bulletin of Experimental Biology and Medicine, 161, 391–394.
Plaut, A. G., Lexington, M., Qiu, J., & Westborough, M. (2009). Combination therapy with IgA protease (Vol. US 2009/0041746 A1, p. 35). Boston, MA: Tufes Medical Center.
Halter, R., Pohlner, J., & Meyer, T. F. (1984). IgA protease of Neisseria gonorrhoeae: Isolation and characterization of the gene and its extracellular product. The EMBO Journal, 3, 1595–1601.
Klauser, T., Pohlner, J., & Meyer, T. F. (1990). Extracellular transport of cholera toxin B subunit using Neisseria IgA protease beta-domain: Conformation-dependent outer membrane translocation. The EMBO Journal, 9, 1991–1999.
Poulsen, K., Reinholdt, J., Jespersgaard, C., Boye, K., Brown, T. A., Hauge, M., et al. (1998). A comprehensive genetic study of streptococcal immunoglobulin A1 proteases: Evidence for recombination within and between species. Infection and Immunity, 66, 181–190.
Iagudaeva, E., Zhigis, L. S., Razguliaeva, O. A., Zueva, V. S., Mel’nikov, E. E., Zubov, V. P., et al. (2010). Isolation and determination of activity of IgA1 protease from Neisseria meningitidis. Bioorganicheskaia Khimiia, 36, 89–97.
Mortensen, S. B., & Kilian, M. (1984). Purification and characterization of an immunoglobulin A1 protease from Bacteroides melaninogenicus. Infection and Immunity, 45, 550–557.
Simpson, D. A., Hausinger, R. P., & Mulks, M. H. (1988). Purification, characterization, and comparison of the immunoglobulin A1 proteases of Neisseria gonorrhoeae. Journal of Bacteriology, 170, 1866–1873.
Long, S., Phan, E., & Vellard, M. C. (2010). The expression of soluble and active recombinant Haemophilus influenzae IgA1 protease in E. coli. Journal of Biomedicine & Biotechnology, 2010, 253983.
Blake, M. S., & Swanson, J. (1978). Studies on gonococcus infection. XVI. Purification of Neisseria gonorrhoeae immunoglobulin A1 protease. Infection and Immunity, 22, 350–358.
Acknowledgements
This study was supported by the national nature science foundation of China (No. 81641025), the nature science foundation of Science & Technology department of Sichuan province (No. 2016JY0194), the nature science foundation of Southwest Medical University (No. 2015QN-32) and the scientific research innovation team grant of Education department of Sichuan province (17TD0046).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Wang, H., Zhong, X., Li, J. et al. Cloning and Expression of H. influenzae 49247 IgA Protease in E. coli. Mol Biotechnol 60, 134–140 (2018). https://doi.org/10.1007/s12033-017-0054-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-017-0054-3