
Abstract
Next generation sequencing is a high-throughput technique widely used for transcriptome profiling. Isolation of high quality RNA is a prerequisite for such large scale transcriptome analysis. Phyllanthus emblica is an important medicinal plant having high amount of metabolites like vitamin C, flavonoids, polyphenolic compounds, tannins, which are responsible for its wondered medicinal properties. High concentration of secondary metabolites like polysaccharides and polyphenols proved to be an obstacle in isolating RNA of good quality. Any compromise with quality of RNA affects the downstream applications and requires extra cleaning steps that further reduce RNA quantity. We have developed a protocol for isolation of high quality RNA from P. embilca. RNA was successfully assessed for downstream applications like reverse transcription polymerase chain reaction, rapid amplification of cDNA ends, mRNA library preparation, and sequencing using HiSeq™ 2000 sequencing technology. The protocol is simple and can be completed in 4–5 h.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Phyllanthus emblica (Emblica officinalis; family Euphorbiaceae) is one of the important medicinal plants possessing antioxidant [1], antiviral [2], antimicrobial [3], antitumor [4], ulcer healing [5], hepatoprotective [6], and immune-modulatory [7] properties. These properties are imparted by the presence of high amount of vitamin C, flavonoids and other polyphenolic compounds such as tannins [8]. Keeping in view the immense medicinal properties attributed to this plant, studies were initiated on understanding molecular aspects of biosynthesis of secondary metabolites imparting these properties. For such studies, nucleic acids isolation is a primary requirement. However, there are no standard methods for the isolation of nucleic acids applicable to all the plant species [9]. P. emblica is the rich source of vitamin C, carbohydrates, and polyphenolic compounds [8, 10, 11]. Compounds such as polysaccharides, polyphenols interferes in isolation of good quality RNA. Polyphenolic compounds oxidized very quickly during RNA extraction and form quinones which bind with RNA [9] and polysaccharides co-precipitate with the RNA in low ionic concentration buffer [12]. The problem increases with the tissue source such as fruits and flowers, which are rich in such compounds. We have tried previously published protocols and commercial kits available to isolate RNA from different parts of plant (13, 14, 15, 16, RNeasy® Kit (Qiagen, Germany) and Trizol® reagent (Invitrogen, USA)) but either quality or quantity or both were very poor especially in case of flowers and mature leaves, pellet was of black color. The RNA isolation protocol developed for woody plants having high level of polyphenols by Lal et al. [13], Jaakola et al. [14], and Chang et al. [15], were time consuming (about 16 h or more) as it required overnight precipitation and also the RNA yield and quality was very poor in case of P. emblica. The phenol-based isolation system developed by Ghawana et al. [16] was rapid and less cumbersome, but using this procedure for P. emblica, the yield was low and pellet was brown in color suggesting high amount of polysaccharides co-precipitated with the nucleic acids, thus makes the RNA unsuitable for further applications. Commercially available, Trizol reagent (Invitrogen, USA) and RNeasy® mini kit (Qiagen, Germany) were also used but none of these worked well with P. emblica.
Thus, we aimed to develop a method for isolation of good quality RNA from P. emblica. We standardized a protocol using two buffers, both at acidic pH. Buffer I, in the presence of high amount of polyvinylpyrrolidone (PVP) removes polysaccharides, proteins, and polyphenolics, while buffer II removes all the traces of interfering proteins and other metabolites persisted in aqueous phase after treating with buffer I. Low pH of buffers stabilize RNA rather than DNA which enhance the recovery of RNA. Use of high amount of absolute ethanol with lithium chloride (LiCl) reduces incubation time for precipitation. The specificity and applicability of the protocol was evaluated by downstream processes such as cDNA synthesis, polymerase chain reaction (PCR), rapid amplification of cDNA ends (RACE), preparation of mRNA library and sequencing using Illumina’s HiSeq 2000 sequencing technology.
Materials and Methods
Plant Materials
A healthy P. emblica plant growing under normal environmental conditions in the Botanical garden of Panjab University, Chandigarh was selected. Leaves (young and mature), flowers and fruit samples were collected, frozen immediately in liquid nitrogen and stored at −80°C till further use. Fruits were chopped off into small pieces before storage.
Reagents and Solutions
All the glassware and plasticware were treated with 0.1% diethyl pyrocarbonate (DEPC)-treated water overnight and autoclaved. All the solutions were prepared in 0.1% DEPC-treated water. Gel running apparatus was treated with 3% H2O2, rinsed and washed with DEPC-treated autoclaved water.
-
Extraction buffer I Cetyl trimethyl ammonium bromide (CTAB, 2% (w/v); Sigma, USA), 3.5 M guanidine thiocyanate (GTC) (Sigma, USA), 100 mM sodium citrate buffer (pH 4.5), 3 M Potassium acetate (Sigma, USA), 1.0% sodium dodecyl sulfate (SDS) (Sigma, USA) (added just before the start of experiment) and RNase free water for final volume makeup.
-
Extraction buffer II Acidic phenol (pH 4.5) (Sigma, USA), 2.5 M sodium chloride (Sigma, USA), 0.5% SDS.
-
Chloroform: Isoamyl alcohol (24:1)
-
12 M LiCl
-
Ethanol (absolute and 70%)
-
RNase free water
RNA Extraction
-
(1)
Extraction buffer I was prewarmed at 65°C. Plant tissues (100 mg) were placed in pestle. PVP K-30 [mw = 40,000 (soluble); 7% of the extraction buffer] was added to it and ground to make fine powder.
-
(2)
Powdered tissue was transferred to a microcentrifuge tube containing 1 ml extraction buffer I, vortexed vigorously and incubated at 65°C for 10 min with intermittent vortexing every 2 min.
-
(3)
The mixture was further divided equally into two tubes. An equal volume of chloroform: isoamyl alcohol was added, vortexed briefly and centrifuged for 10 min at 13,000 rpm at room temperature to separate the organic and aqueous phase.
(Optional if dark color of aqueous phase persists): Upper aqueous phase was transferred to a fresh tube and 500 μl of extraction buffer I was added without adding PVP. Vortexed briefly and added equal amount of chloroform: isoamyl alcohol to separate the organic and aqueous phase.
-
(4)
The mixture was centrifuged for 10 min at 13,000 rpm at room temperature and the upper phase was transferred to a fresh tube.
-
(5)
The aqueous phase was transferred in a fresh tube and added 1 ml extraction buffer II, vortexed vigorously and left it undisturbed for 5 min at room temperature.
-
(6)
Equal volume of chloroform: isoamyl alcohol was added, vortexed briefly, and centrifuged for 10 min at 13,000 rpm at room temperature.
-
(7)
The upper phase was transferred to a new tube. For precipitation of RNA, 12 M of LiCl (to final concentration of 2 M), 3 volume of chilled absolute ethanol were added and incubated at −80°C for 2 h.
-
(8)
RNA was pelleted down by centrifuging the tube at 13,000 rpm at room temperature for 20 min.
-
(9)
Supernatant was discarded carefully without disturbing the pellet.
-
(10)
Absolute ethanol (1 ml) was added and vortexed briefly. Tubes were kept undisturbed at room temperature for 10 min to dissolve any salt contamination.
-
(11)
Samples were centrifuged at 13,000 rpm for 10 min and supernatant was carefully removed.
-
(12)
RNA pellet was washed with 70% ethanol and air dried.
-
(13)
RNA pellet was dissolved in RNase free water and store at −80°C till further use.
Assessment of RNA Quantity and Quality
Integrity of RNA was analyzed primarily by running 1 μg of each sample on 1.2% denaturating agarose gel containing formaldehyde and ethidium bromide. Concentration and purity of RNA were analyzed by UV–VIS spectrophotometer (Hitachi, Japan) determining the absorbance of RNA sample at wavelength of 280, 260, and 230 nm and ratio at A 260/280 and A 260/230 was calculated as described by Sambrook and Russell [17]. RNA quality and quantity were further analyzed by Agilent Bioanalyzer 2100 and RNA integrity number (RIN) was measured following the manufacturer’s instructions.
cDNA Synthesis and PCR
RNA was treated with DNase I (amplification grade, Invitrogen, USA) to remove any residual DNA contamination before proceeding to reverse transcription (RT). For cDNA synthesis, 2 μg of DNA free RNA was reverse transcribed with oligo-dT(18–20) primer and Superscript III reverse transcriptase (Invitrogen, USA) following the manufacturer’s instructions. Further, PCR was performed using the cDNA synthesized and 26S rRNA gene primers (Forward: 5′-CACAATGATAGGAAGAGCCGAC-3′ and Reverse 5′-CAAGGGAACGGGCTTGGCAGAATC-3′) [18]. PCR mixture contained 1× PCR buffer (10 mM Tris–HCl, 50 mM KCl, 1.5 mM MgCl2), 0.2 mM of dNTPs, 0.2 μM of each primer, 1 μl of cDNA template, 1U of Taq polymerase (New England Biolabs, USA), and RNase free ddH2O to make final volume as 25 μl. PCR was performed in a S1000™ Thermal cycler (Bio-Rad Laboratories, USA) with intial denaturation at 95°C for 3 min followed by 30 cycles consisting of 94°C for 30 s, 52°C for 30 s, and 72°C for 1 min with a terminal extension at 72°C for 7 min. Amplified PCR product (25 μl) was resolved on 1.2% agarose gel and stained with ethidium bromide.
Rapid Amplification of cDNA Ends (RACE)
RACE ready cDNAs (5′ and 3′) were synthesized from 2 μg of total RNA using SMART™ RACE cDNA Amplification Kit (Clontech, USA). RACE PCR was performed for amplification of 5′ and 3′ ends of flavanone 3-hydroxylase (F3H) gene using the primers; 5′-GSP 5′-CCA AGT TTT GCC ATC ATC TCT GGT AGC C-3′ and 3′-GSP 5′-CAG TGA GGA TCT GAT GGG ACT AGC TTG C-3′ respectively along with the manufacturer’s provided primers in SMART™ RACE cDNA Amplification kit (Clontech, USA). PCR reaction mixture (25 μl) contained 1× Advantage 2 SA PCR Buffer (10 mM Tris–HCl pH 8.5, 50 mM KCl, 2 mM MgCl2), 0.2 mM dNTP mix, 0.2 mM GSP primer, 0.2 mM universal primer, 1 U Advantage 2 Polymerase Mix (Clontech, USA), 1 μl RACE ready cDNA as template (separate reactions for 3′ and 5′ RACE) and water. PCR was programmed with initial denaturation at 95°C for 3 min followed by 35 cycles of amplification at 95°C for 30 s, 65°C for 20 s, 68°C for 1 min, and final extension at 68°C for 10 min. Amplified products (25 μl each) were resolved on 1.5% agarose gels containing ethidium bromide, bands of expected sizes were excised and eluted from gel (GenElute gel extraction kit, Sigma), ligated into pGEM-T easy vector (Promega, USA) and transformed into E. coli DH5α competent cells. Recombinant plasmids with insert were isolated (GenElute plasmid isolation kit, Sigma) and sequenced using big dye terminator cycle sequencing kit v3.1 on an automated sequencer (ABI 310 Genetic analyzer, Applied Biosystems, USA).
mRNA Library Preparation and HiSeq 2000 Sequencing
RNA samples were sent to Microarray core facility, Huntsman Cancer Institute, University of Utah, Slat Lake City, UT, USA for RNA quality measurement on Agilent Bioanalyzer 2100, mRNA library preparation and sequencing. mRNA library was prepared using Illumina® TruSeq™ RNA Sample Preparation Kit (Illumina, USA). 50 cycled single end library sequencing was performed using HiSeq™ 2000 sequencing system (Illumina, USA).
Results and Discussion
P. emblica, being a rich source of secondary metabolites of medicinal importance, proves to be a good candidate to understand the biosynthesis of metabolites such as flavonoids and tannins at gene level. For such molecular studies, the foremost requirement is isolation of good quality of nucleic acids, so emphasis was to obtain RNA of high quality. Several previously published protocols were used on this plant but all of them yielded poor RNA in terms of quality as well as quantity. Although, there were methods for the isolation of RNA from plants belonging to family Euphorbiaceae, but either these were very time consuming such as for latex rich plant, Hevea sp. [19] which could not be effective for the P. emblica due to the metabolic differences in both the plants or combined with commercial kits to purify the RNA (from Jatropha sp.) [20, 21], which makes it expensive one. Here, we proposed a cost effective, less time consuming GTC and phenol based method to isolate high quality RNA without the use of silica column. GTC is a strong protein denaturant used to inactivate RNases [22, 23]. Phenol denatures the proteins and after centrifugation, proteins precipitate and make a layer at the interface between the organic and aqueous layers. It has been reported previously that in low pH environment RNA degradation is minimum and addition of a high salt concentration causes DNA to selectively precipitate at the interphase [24] along with proteins. CTAB, a strong detergent, break the plant cell wall and separate proteins from nucleic acid by making complexes with nucleic acids in high salt concentration. High salt concentration results into removal of polysaccharides, proteins and CTAB in a precipitation or co-precipitation during chloroform extraction [25]. PVPs role in the removal of secondary metabolites from nucleic acid preparations has been widely reported [26] and it also prevents browning effect of polyphenols [27]. Thus, we have developed a new protocol with two types of extraction buffers which stabilize the RNA as well as improve the purity. In this study, we maintained extraction buffers I and II at acidic pH by the addition of sodium citrate buffer and acid phenol, respectively, to stabilize RNA. Initially, use of high molar concentration of GTC in extraction buffer I resulted in highly viscous substance and hence RNA recovery was very poor. To overcome this problem, extraction buffer I was standardize with less molar concentration of GTC which successfully reduces viscosity as well as improved RNA recovery. Use of high amount of PVP, SDS, and salt in RNA extraction buffers renders a significant improvement in both yield and quality of RNA as compared to other methods such as Chang et al. [15], Ghawana et al. [16] and RNaeasy® Kit (Qiagen, Germany). The optional step removes all the colored components and resulted into colorless aqueous phase. Presence of phenol and SDS in extraction buffer II also improves the purity of RNA by removing residual proteins as well as secondary metabolites from aqueous phase. Chloroform: Isoamyl alcohol was used to remove PVP before addition of extraction buffer II since PVP is not compatible with phenol. In case of mature leaves and fruits, enriched with secondary metabolites, 2–3 repetitions were performed with extraction buffer II. To selectively precipitate RNA, LiCl in addition with ample amount of absolute ethanol was used, which reduced precipitation time.
Quantity and Quality of RNA
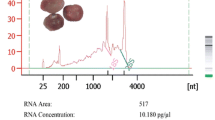
To illustrate the utility of the method, RNA integrity was assessed by electrophoresis and spectrophotometer. Denaturing formaldehyde–agarose gel showed very poor quality of RNA isolated by other protocols. The RNA was either degraded or yield was very low (Fig. 1). Whereas, RNA isolated by our protocol clearly shows distinct bands of 28S and 18S rRNA without any degradation and DNA contamination (Fig. 2a). Similarly, small RNA bands were also intact as compared to other protocols. High quality RNA without any genomic DNA contamination was obtained at pH 4.5 while at pH 8.0, high DNA contamination was observed (Fig. 2b). Quantity and purity of RNA were found to be superior using this method as compared to previously reported methods (Table 1). A 260/280 ratio for RNA isolated from different plant parts varied from 1.81 to 2.32, far better than the other methods used. Ratio of A 260/230 between 2.18 to 2.33 also indicated high purity of RNA with very small polysaccharides and phenolic contamination. A 260/280 ratio for RNA isolated by this protocol from young leaves (2.17), flowers (2.11), and mature leaves (2.07) are very high as compared to Ghawana et al. [16] (1.71, 0.80, 0.48), Chang et.al. [15] (1.03, 1.09, 0.32), and RNeasy® protocol (0.87, 0.32, 0.24). Also, A 260/230 ratio for young leaves (2.31), flowers (2.23), and mature leaves (2.29) were also very good than other methods (Table 1). Analysis using Agilent Bioanalyzer 2100 also confirmed high quality RNA (RIN 8.0) isolated by this protocol (Fig. 3). RNA samples with RIN greater than 5.0 can be used for quantitative RT-PCR [28], and greater than 7.0 is the standard RIN for construction of cDNA libraries [29].


a Isolation of RNA from different tissues of P. emblica using the method described in this study. Ethidium bromide-stained denaturating agarose gel shows distinct bands of 28S and 18S rRNA bands with no visible DNA contamination. YL young leaves, ML mature leaves, Fl flowers, Y Ft young fruit, M Ft mature fruit. b Effect of extraction buffer pH on RNA isolation from leaf tissues. DNA band is clearly visible when pH of extraction buffers was maintained around 8.0

Electropherogram of RNA generated by Agilent Bioanalyzer 2100 for a leaf and b flower. RIN was found to be 8
PCR Amplification
cDNA was successfully synthesized using RNA and reverse transcriptase enzyme. PCR amplification yielded approximately 530 bp fragments of 26S rRNA (Fig. 4a) using the cDNA synthesized from various plant tissues. RACE ready cDNA was prepared and RACE PCR was performed to amplify 5′ and 3′ ends of F3H gene using gene specific primers. Amplicons yielded by 5′and 3′ RACE PCR (Fig. 4b) were cloned in pGEM-T® easy vector and sequenced. Sequence data indeed showed that cloned fragments belonged to F3H gene (data not shown).

a Amplification of 26S rRNA fragments using the cDNA synthesized from young leaves (YL), mature leaves (ML), flowers (Fl), young fruit (YFt), and mature fruit (MFt). b Amplification of 5′ and 3′ ends of F3H gene using RACE ready cDNA and gene specific primers
mRNA Library Preparation and Sequencing
Large scale transcriptome analysis using high-throughput sequencing technologies is a widely used approach to study and compare gene expression between different tissues or developmental stages of plants. mRNA library was prepared using the RNA isolated by this method and approximately 20 million 50 cycled single end sequences were generated using Illumina’s next generation sequencing platform, HiSeq 2000. The sequences are in process of bioinformatics analysis for their annotation and further characterization (data will be published elsewhere).
In summary, the present protocol was used to isolate high quality RNA from the different parts of P. emblica. Isolated RNA has shown to be biologically active and suitable for many downstream processes like RT, PCR, RACE, and library preparation for large scale transcriptome analysis. The developed method is simple, reliable, and reproducible without any difficulty.
Abbreviations
- CTAB:
-
Cetyl trimethyl ammonium bromide
- DEPC:
-
Diethyl pyrocarbonate
- EDTA:
-
Ethylenediamine tetraacetic acid
- GTC:
-
Guanidine thiocyanate
- LiCl:
-
Lithium chloride
- PCR:
-
Polymerase chain reaction
- PVP:
-
Polyvinylpyrrolidone
- RT:
-
Reverse transcriptase
- SDS:
-
Sodium dodecyl sulfate
References
Rajak, S., Banerjee, S. K., Sood, S., Dinda, A. K., Gupta, Y. K., Gupta, S. K., et al. (2004). Emblica officinalis causes myocardial adaptation and protects against oxidative stress in ischemic-reperfusion injury in rats. Phytotherapy Research, 18, 54–60.
El-Mekkawy, S., Meselhy, M. R., Kusumoto, I. T., Kadota, S., Haltori, M., & Namba, T. (1995). Inhibitor effects of Egyptian folk medicine on human immunodeficiency viruses (HIV) reverse transcriptase. Chemical Pharmaceutical Bulletin, 43, 641–648.
Khanna, P., & Bansal, R. (1975). Phyllantidine and phyllantine from Emblica officinalis Gaertn. leaves, fruits, and in vitro tissue cultures. Indian Journal of Experimental Biology, 13, 82–83.
Lambertini, E., Piva, R., Khan, M. T., Lampronti, I., Bianchi, N., Borgatti, M., et al. (2004). Effects of extracts from Bangladesh medicinal plants on in vitro proliferation of human breast cancer cell lines and expression of estrogen receptor alpha gene. International Journal of Oncology, 24, 419–423.
Al-Rehaily, A. J., Al-Howiriny, T. A., Al-Sohaibani, M. O., & Rafatullah, S. (2002). Gastroprotective effects of Emblica officinalis on in vitro test models in rats. Phytomedicine, 9, 515–522.
Roy, A. K., Dhir, H., & Sharma, A. (1991). Comparative efficacy of Phyllanthus emblica fruit extract and ascorbic acid in modifying hepatotoxic and renotoxic effects induced by metals in vivo. International Journal of Crude Drug Research, International Journal of Pharmacology (Netherlands)., 29, 117–126.
Sai Ram, M., Neetu, D., Yogesh, B., Anju, B., Dipti, P., Pauline, T., et al. (2002). Cyto-protective and immunomodulating properties of Amla (Emblica officinalis) on lymphocytes: An in vitro study. Journal of Ethnopharmacology, 81, 5–10.
Barthakur, N. N., & Arnold, N. P. (1991). Chemical analysis of the emblic (Phyllanthus emblica L.) and its potential as a food source. Scientia Horticulturae, 47, 99–105.
Loomis, M. D. (1974). Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Methods in Enzymology, 31, 528–544.
Thakur, R. S., Puri, H. S., & Akhtar, H. (1989). Major medicinal plants of India. Lucknow: Central Institute of Medicinal and Aromatic Plants.
Chaudhuri, R. K. (2004). Standardised extract of Phyllanthus emblica: A skin lightener with antiaging benefits. Proceedings PCIA conference, Guangzhou, China, 9–11 March.
Birtic, S., & Kranner, I. (2006). Isolation of high-quality RNA from polyphenols, polysaccharide and lipid-rich seeds. Phytochemical Analysis, 17, 144–148.
Lal, L., Sahoo, R., Gupta, R. K., Sharma, P., & Kumar, S. (2001). RNA isolation from tea leaves, a high polyphenolic containing tissue, and extension of the procedure to diverse plant species. Plant Molecular Biology Reporter, 19, 181a–187f.
Jaakola, L., Pirttila, A. M., Halonnen, M., & Hohtola, A. (2001). Isolation of high quality RNA from Bilberry (Vaccinum myrtillus L.) fruit. Molecular Biotechnology, 19, 201–203.
Chang, S., Puryear, J., & Cairney, J. (1993). A simple and efficient method for isolating RNA from pine trees. Plant Molecular Biology Reporter, 11, 113–116.
Ghawana, S., Paul, A., Kumar, H., Kumar, A., Singh, H., Bhardwaj, P. K., et al. (2011). An RNA isolation system for plant tissues rich in secondary metabolites. BMC Research Notes, 4, 85–89.
Sambrook, J., & Russell, D. W. (2001). Molecular cloning: A laboratory manual. New York, NY: Cold Spring Harbor Laboratory Press.
Singh, K., Raizada, J., Bhardwaj, P., Ghawana, S., Rani, A., Singh, H., et al. (2004). 26 S rRNA-based internal control gene primer pair for reverse transcription-polymerase chain reaction-based quantitative expression studies in diverse plant species. Analytical Biochemistry, 335, 330–333.
Wang, X. C., Tain, W. M., & Li, Y. X. (2008). Development of an efficient protocol of RNA isolation from recalcitrant tree tissues. Molecular Biotechnology, 38, 57–64.
Sangha, J. S., Gu, K., Kaur, J., & Yin, Z. (2010). An improved method for RNA isolation and cDNA library construction from immature seeds of Jatropha curcas L. BMC Research Notes, 3, 126–131.
Kumar, G. R. K., Eswaran, N., & Johnson, T. S. (2011). Isolation of high-quality RNA from various tissues of Jatropha curcas for downstream applications. Analytical Biochemistry, 413, 63–65.
Bilgin, D. D., DeLucia, E. H., & Clough, S. J. (2009). A robust plant RNA isolation method suitable for Affymetrix GeneChip analysis and quantitative real-time RT-PCR. Nature Protocol, 4, 333–340.
Chomczynski, P., & Sacchi, N. (2006). The single step method of RNA isolation by acid guanidinium thiocyanate phenol chloroform extraction: Twenty something years on. Nature Protocol, 1, 581–585.
Noonberg, S. B., Scott, G. K., & Benz, C. C. (1995). Effect of pH on RNA degradation during guanidinium extraction. Biotechniques, 19, 31–33.
Fang, G., Hammer, S., & Gurmet, R. (1992). A quick and inexpensive method for removing polysaccharides from plant genomic DNA. Biofeedback, 13, 52–54.
Chen, G. Y. J., Jin, S., & Goodwin, P. H. (2000). An improved method for the isolation of total RNA from Malva pusilla tissues infected with Colletotrichum gloeosporioides. Journal of Phytopathology, 148, 57–60.
Emine, Z. Y., & Sule, P. (2004). Purification and characterization of pear (Pyrus communis) polyphenol oxidase. Turkish Journal of Chemistry, 28, 547–557.
Abbott, E., Hall, D., Hamberger, B., & Bohlmann, J. (2010). Laser microdissection of conifer stem tissues: Isolation and analysis of high quality RNA, terpene synthase enzyme activity, and terpenoid metabolites from resin ducts and cambial zone tissue of white spruce (Picea glauca). BMC Plant Biology, 10, 106–121.
Fleige, S., & Pfaffi, M. W. (2006). RNA integrity and the effect on the real-time qRT-PCR performance. Molecular Aspects of Medicine, 27, 126–139.
Acknowledgments
We sincerely acknowledge the Department of Science and Technology (DST), India, for financial support to carry out this research work. Authors are also thankful to Dr. Brian Dalley, Director, Microarray core facility, University of Utah, Salt Lake City, USA for their valuable services for mRNA library preparation and high-throughput sequencing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kumar, A., Singh, K. Isolation of High Quality RNA from Phyllanthus emblica and Its Evaluation by Downstream Applications. Mol Biotechnol 52, 269–275 (2012). https://doi.org/10.1007/s12033-011-9492-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12033-011-9492-5