
Abstract
Recent studies show that morphine possesses protective preconditioning effects in different ischemia/reperfusion models. However, there is very little information about the antineuroinflammatory role of morphine and its protective effect against memory deficit. In the present study, we evaluated the role of morphine preconditioning in a model of mild neuroinflammation induced by intraperitoneal lipopolysaccharide (LPS) injection (1 mg/kg). Rats were trained on passive avoidance apparatus and challenged with LPS 20 h later. Four hours after LPS, rats were subjected to passive avoidance testing and then for the assessments of inflammatory and apoptotic cell death mediators in the hippocampus. LPS significantly increased the nuclear NF-κB and expression of COX-2, IL-1β, and TNF-α, augmented the activity of caspase-3 and PARP cleavage, and in parallel shortened the latencies to enter the dark compartment. Although morphine injection in a noninflammatory context was able to induce a neuroinflammatory response and memory loss, morphine preconditioning at the dose of 4 mg/kg significantly prevented the LPS-induced neuroinflammation and memory deficit. Morphine preconditioning was abolished by naloxone and, therefore, is dependent on opioid receptors. These results suggest that acute morphine injection, in spite of the induction of a neuroinflammatory response and amnesia per se, exerts an antineuroinflammatory role and protects from cell death and memory deficit in an inflammatory context.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
From the past three decades, the immune-privileged assumption of the central nervous system (CNS) has been changed (Turrin and Rivest 2006). Recently, cumulative evidence expresses the role of neuroinflammation in the progression of ischemia, brain trauma, stroke, and neurodegenerative diseases such as Alzheimer’s disease (Mucke and Eddleston 1993; Kreutzberg 1996; Brown and Bal-Price 2003).
Many physiological and behavioral functions are modulated by the opioidergic system, through three well-known classic opioid receptors, μ, δ, and κ (Kieffer 1995; Minami and Satoh 1995). Recently, it has been shown that toll-like receptor 4 (TLR-4) is also potentially involved in opioid functions and may act as another opioid receptor (Hutchinson et al. 2008). Morphine, an opioid agent with agonistic effects on all opioid receptors as well as TLR-4, has been well demonstrated to interfere in the immune system functions (Hutchinson et al. 2007, 2010a; Madera-Salcedo et al. 2011). Some studies show that morphine can possess the peripheral anti-inflammatory role at a therapeutic dose (Askari et al. 2008) or potentially enhance the peripheral inflammation at ultralow concentration (Pourpak et al. 2004). Several studies have also revealed the similar immunomodulatory effect of morphine in the CNS. For instance, morphine exerts a neuroprotective effect against human neuroblastoma oxidative stress and neuroinflammation (Rambhia et al. 2005), peroxynitrite-induced apoptosis in embryonic rat astrocyte (Kim et al. 2001), β-amyloid toxicity in HTP-11 neuroblastoma cell line (Pak et al. 2005), and lipopolysaccharide (LPS)- or 1-methyl-4-phenylpyridinium-induced dopaminergic neurotoxicity in rat primary mesencephalic neuron–glia cultures (Qian et al. 2007). Although glial activation and neuroinflammatory reaction is also reported to occur in response to opioids (Watkins et al. 2009), the potential neuroprotective effect of opioidergic compounds is associated with immune suppression (Kao et al. 2008). Therefore, the modulation of opioidergic system has gained attention as a new neuroprotective strategy to protect brain against some detrimental insults accompanied with neuroinflammatory reactions, the phenomenon named preconditioning. Preconditioning is defined as a protective event in which a prior stimulus or agent activates endogenous protective mechanisms against a further deleterious insult like stroke and ischemia (Barry and Zuo 2005). Divers stimuli, such as short episodes of ischemia and hypoxia (Gidday et al. 1994; Nandagopal et al. 2001) as well as low doses of endotoxin (Lin et al. 2009), have been shown to induce a preconditioning effect in the brain. Morphine preconditioning and its protective effects are also reported in some in vitro and in vivo studies (Lim et al. 2004; Zhao et al. 2006). However, regarding the ability of morphine to activate the TLR-4, it is not clear that morphine can synergistically enhance the neuroinflammatory response evoked by TLR-4 activation or can suppress it via a preconditioning effect and exert an antineuroinflammatory and protective role.
The endotoxin LPS is the best-known target of innate recognition which induces a robust inflammatory response by activating the phagocytic cells (Wright 1999) and is used to induce either peripheral or central inflammation in several experiments (Zujovic et al. 2001; Chakravarty and Herkenham 2005; Cunningham et al. 2005). LPS induces the expression of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 by macrophages/microglia, lymphocytes, and endothelial cells (Lynn and Golenbock 1992; Ulevitch and Tobias 1995) through its binding to membrane CD14 receptors, which transfer LPS to TLR-4 via myeloid differentiation protein 2 (Takeuchi and Akira 2001). TLR-4 has been known as a signal transducer receptor for LPS on immune cell surfaces (Hoshino et al. 1999; Qureshi et al. 1999). Interaction between LPS and TLR-4 also takes place in the circumventricular organs, which allow intracellular signaling and then rapid transcription of proinflammatory cytokines first within these organs and thereafter throughout the brain parenchyma during severe endotoxemia (Lacroix et al. 1998).
In the present study, using a combination of molecular and behavioral experiments, we evaluated the role of morphine pretreatment on LPS-induced neuroinflammation and apoptotic cell death in rat hippocampus. We also evaluated the effect of morphine preconditioning in LPS-induced memory impairment to show whether the potential anti-inflammatory and protective role of morphine is as much as to improve the memory retention in an inflammatory context.
Materials and Methods
Animals
Adult male Wistar rats weighing 220–250 g were purchased from the Neuroscience Research Center of Shahid Beheshti University of Medical Sciences, housed in groups of four under standard light and temperature regimes, and fed pelleted food and water ad libitum. After at least 1 week of habituation to handling, animals were admitted to the experimental procedures. All experiments were carried out in accordance with the National Institutes of Health guidelines for the use of experimental animals and approved by the local ethical committee. All efforts were made to minimize animal suffering and to reduce the number of animals used. Eight and four rats were used, respectively, for the behavioral and molecular experiments.
Intracerebroventricular Cannulation
Rats were anesthetized with chloral hydrate (400 mg/kg, intraperitoneal [i.p.]) (Liao et al. 2003) and positioned in a stereotaxic apparatus (Stoelting Co., Wood Dale, IL, USA). A 23-gauge, stainless steel tubing guide cannula was implanted in the right lateral ventricle of the rat according to the coordinates of the atlas of Paxinos and Watson (2007), relative to the bregma: posterior −0.6 mm, lateral +1.6 mm, and ventral 3.7 mm from the surface of the skull. The guide cannula was anchored to the skull using mounting screws and dental caulk. The animals were allowed 3 days postoperative recovery before being used for the experiments.
LPS Injection and Drug Administration
In this study, we had 14 experimental groups. In groups 1 and 2, the rats received i.p. injection of vehicle (sterile phosphate-buffered saline [PBS], 1 mL/kg) or LPS (Sigma, St. Louis, MO, USA; Escherichia coli serotype 055:B5, 1 mg in 1 mL PBS, at the dose of 1 mg/kg) and 4 h later were subjected to molecular assessment of neuroinflammation. In group 3, lpsRS (Invivogen, San Diego, CA, USA; TLR-4 antagonist, dissolved in PBS) was injected at the dose of 40 μg/8 μL/rat, i.c.v, 35 min before LPS injection. In group 4, naloxone (Temad, Tehran, Iran; opioid receptor antagonist, dissolved in PBS) was injected at the dose of 4 mg/kg, i.p., 35 min before and 90 min after LPS injection. Groups 3 and 4 were also only subjected to molecular assessment of neuroinflammation.
In groups 5 and 6, the rats received i.p. injection of LPS or PBS, 20 h after passive avoidance training, and 4 h later were subjected to memory retrieval testing and then molecular assessments.
In groups 7, 8, and 9, morphine sulfate (Temad, Tehran, Iran; dissolved in PBS) at the doses of 4, 7, and 10 mg/kg was injected i.p. 30 min before PBS injection. In groups 10, 11, and 12, morphine sulfate at the doses of 4, 7, and 10 mg/kg was injected i.p. 30 min before LPS injection.
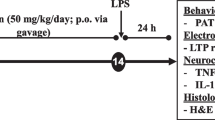
According to the significant anti-inflammatory and protective role of morphine at the dose of 4 mg/kg, we designed the last two groups: group 13 in which morphine pretreatment was combined with naloxone, 4 mg/kg, i.p., 35 min before and 85 min after LPS injection and group 14 in which morphine pretreatment was combined with lpsRS, 40 μg/8 μL/rat, i.c.v, 35 min before LPS injection. The experimental timescale is shown schematically in Fig. 1.

Schematic representation of the experimental timescale
Passive Avoidance Training and Testing
Passive (inhibitory) avoidance task was selected as the tool for the assessment of memory retrieval in rats as described previously (Pakpour et al. 2010). All behavioral training and testing were performed between 10 am to 15 pm, during the light period, and a habituation period of 1 h to the conditions of the experimental room preceded all the behavioral procedures.
The test was performed in two consecutive days, an acquisition trial on the first day and a retention test 24 h later. On the training day, animals were placed into the light chamber of a passive avoidance apparatus (shuttle box), facing away from the door, which was in the closed position. After 10 s of habituation, the sliding door was opened, allowing the rat to enter the dark chamber. After entering the dark compartment, the rat was given a scrambled foot shock (1 mA for 1.5 s) through the stainless steel grid floor. The rats that reentered the dark chamber within a period of 120 s were omitted from this study.
Twenty hours after the training session, rats were randomly assigned to one of the ten abovementioned groups (groups 5–14) and were given injections of either BPS or LPS with or without morphine and/or morphine plus naloxone or lpsRS pretreatments. In all these groups, 4 h after LPS or PBS injection, rats were placed in the light chamber for 10 s, after which the door was opened and they were given access to the dark compartment of the apparatus within a period of 300 s. Memory retrieval was assessed by measuring the latency to enter the dark compartment.
Immunoblotting of Inflammatory and Apoptosis Markers
Four hours after LPS injection or immediately after behavioral tests, the rats were decapitated and the brains were immersed in cold PBS. The hippocampi were then isolated from the brain, quickly immersed in liquid nitrogen, and stored at −70°C for later use. We randomly used four rats per group when molecular experiments were performed.
Nuclear and Cytosolic Protein Extraction
Nuclear and cytosolic extracts were prepared as described previously (Pradillo et al. 2005; Wang et al. 2006). In brief, hippocampus tissues were homogenized with 300 μL of cold buffer A (10 mM HEPES [pH 7.9], 1 mM EDTA, 1 mM EGTA, 10 mM KCl, 1 mM DTT, supplemented with complete protease inhibitor tablet; Roche, Nutley, NJ, USA) using a micro-homogenizing system (Micro Smash MS-100) at 4°C for 45 s at 3,500 rpm. The cell suspension was incubated on ice for 15 min and then Nonidet P-40 was added to reach a 0.6% concentration. The tubes were gently vortexed for 15 s and then centrifuged at 4°C for 5 min at 8,000×g. The supernatant was collected and stored at −70°C for Western blot analysis of cytosolic proteins. The pellets were resuspended in 100 μL of cold buffer B (buffer A, supplemented with 20% glycerol and 0.4 M KCI) and gently shaken for 30 min at 4°C. Nuclear protein extracts were obtained by centrifugation at 13,000×g for 5 min, and the supernatant was stored at −70°C for Western blot analysis of nuclear proteins.
Western Blot Analysis for COX-2, TNF-α, IL-1β, and Cleaved Caspase-3
The protein concentration of cytosolic extracts was determined by the Bradford assay and equivalent amounts (40 μg protein) from each sample were subjected to 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride membrane. After incubation in blocking solution (2% nonfat dry milk in TBST buffer: 20 mM Tris–HCl, 150 mM NaCl, and 0.05% Tween 20) for 75 min at room temperature, membranes were incubated with the primary antibodies against TNF-α (Cell Signaling, Danvers, MA, USA; 1:1,000 dilution), COX-2 (ABR-Affinity BioReagents, Golden, CO, USA; 1:10,000 dilution), and cleaved caspase-3 (Cell Signaling, Danvers, MA, USA; 1:1,000 dilution) at 4°C overnight. The next day, membranes were washed three times in TBST and further incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Cell Signaling, Danvers, MA, USA; 1:10,000 dilutions) for 75 min at room temperature. After several washes, the blots were developed using the ECL Advanced Western Blotting Detection Kit (GE Healthcare, Amersham, Buckinghamshire, UK) following the manufacturer’s instructions and exposed onto Kodak X-ray films. TNF-α incubated membranes were then incubated in stripping buffer (100 mM 2-mercaptoethanol, 2% (w/v) SDS, 62.5 mM Tris–HCl pH 6.7) for 5 min at 50°C and reprobed with primary antibody against IL-1β (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1:250 dilution). To obtain loading controls, all the blots were incubated in stripping buffer and reprobed with the primary antibody recognizing β-actin protein (Cell Signaling, Danvers, MA, USA; 1:1,000 dilution).The relative expression of protein bands were quantified by scanning of the X-ray films and densitometric analysis with the ImageJ software.
Western Blot Analysis for NF-κB and PARP
The protein concentration of nuclear extracts was determined by the Bradford assay and equivalent amounts (60 μg protein) from each sample were subjected to SDS-PAGE and then immunoblotting using primary antibodies against NF-κB (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; 1/1,000 dilution) and PARP (Cell Signaling, Danvers, MA, USA; 1:1,000 dilution), as described above.
Statistical Analysis
Data from all the experiments are expressed as the mean ± SEM. Statistical significance was assessed with one-way analysis of variance followed by Tukey’s post hoc multiple comparison tests using the SPSS software. Significance was taken at P < 0.05.
Results
LPS-Induced Neuroinflammatory Response is Prevented by Naloxone and lpsRS
There are pieces of evidence showing that systemic LPS challenge induces an acute neuroinflammatory reaction. To address this issue, we first determined the hippocampal expression of TNF-α and IL-1β, as proinflammatory cytokines, 4 h after the i.p. injection of LPS. As it is shown in Fig. 2, LPS significantly increased the hippocampal expression levels of TNF-α and IL-1β, compared with the vehicle-injected group (control). The main objectives of the present study were to determine the potential anti-inflammatory and protective role of morphine pretreatment and to evaluate whether this role is mediated through opioid receptors. To answer the latter issue, we intended to use naloxone for the pharmacologic inhibition of opioid receptors. Considering the recent findings showing that naloxone possesses TLR-4 antagonistic effects in addition to its pronounced antagonistic effects on opioid receptors, it was necessary to evaluate firstly the effect of naloxone on LPS-induced neuroinflammation. Therefore, we designed a preliminary group receiving LPS with naloxone in the same schedule which was assumed for the group receiving morphine plus naloxone treatment. As it is shown in Fig. 2, naloxone was able to significantly decrease the LPS-induced inflammatory response. To subtract the antagonistic effect of naloxone on TLR-4 from opioid receptors, we designed another group receiving LPS with lpsRS in the same schedule which was assumed for the group receiving morphine plus lpsRS. This TLR-4 antagonist did also prevent the LPS-induced neuroinflammatory response, and unexpectedly, there was no significant difference between the anti-inflammatory role of lpsRS and naloxone.

Effects of naloxone and TLR-4 blockade on LPS-induced neuroinflammatory response. Hippocampal expression of proinflammatory cytokines was assessed 4 h after the i.p. injection of LPS (1 mg/kg) by Western blotting. LPS significantly increased the expression levels of TNF-α and IL-1β, compared with the PBS-injected group (control). Pretreatment with naloxone (4 mg/kg, i.p., 35 min before and 85 min after LPS injection), as well as lpsRS (a TLR-4 antagonist; 40 μg/rat, intracerebroventricular (i.c.v.), 35 min before LPS injection) significantly protected against LPS-induced neuroinflammatory response. The quantitative data, presented as the mean ± SEM, were obtained from densitometric analysis of scanned X-ray films and normalized to corresponding immunoblots of β-actin as the loading control. ***P < 0.001 versus the control group; ## P < 0.01 and ### P < 0.001 versus the LPS group
These results were disappointing for the experiments designed on the basis of the opioid receptor antagonist naloxone. However, it was still interesting to know what will happen if we administer morphine and naloxone in LPS-injected animals.
Morphine Pretreatment Prevents the LPS-Induced Neuroinflammatory Response
To determine whether morphine pretreatment can prevent the LPS-induced neuroinflammation, we first examined the effect of three doses of morphine on the expression level of inflammatory proteins in a noninflammatory context. Four hours after PBS injection in morphine-pretreated groups, Western blot analysis revealed that morphine at the dose of 4 mg/kg has no significant effect on TNF-α and IL-1β expression compared with the control group (Fig. 3a). However, morphine at the doses of 7 and 10 mg/kg significantly increased TNF-α expression and at the dose of 7 mg/kg significantly increased IL-1β expression (Fig. 3a). Although the statistical data from the comparison between morphine-injected groups and LPS group are not shown in the figures, in most cases, the inflammatory response triggered by a single morphine injection was not as much as that triggered by LPS.


Effects of morphine pretreatment on LPS-induced neuroinflammatory response. The hippocampal expression of TNF-α and IL-1β (a), COX-2 (b), and nuclear level of NF-κB (c) were determined by Western blotting. All the panels show the representative immunoblots of the control group, LPS (1 mg/kg, i.p.) group, groups receiving morphine i.p. injections at the doses of 4, 7, and 10 mg/kg (M4, M7, and M10, respectively) 30 min before i.p. PBS injection, groups receiving morphine pretreatments at the doses of 4, 7, and 10 mg/kg (M4-LPS, M7-LPS, and M10-LPS, respectively) 30 min before i.p. LPS injection, group receiving morphine at the dose of 4 mg/kg plus lpsRS (40 μg/rat, i.c.v.) 30 and 35 min, respectively, before LPS injection (lpsRS-M4-LPS), and group receiving morphine at the dose of 4 mg/kg 30 min before LPS plus naloxone (4 mg/kg, i.p.) 35 min before and 85 min after LPS injection (N-M4-LPS). The quantitative data, presented as the mean ± SEM, were obtained from densitometric analysis of scanned X-ray films and normalized to corresponding immunoblots of β-actin as the loading control. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the control group; # P < 0.05, ## P < 0.01, and ### P < 0.001 versus the LPS group; & P < 0.05, && P < 0.01, and &&& P < 0.001 versus the M4-LPS group
Morphine pretreatment at all three doses significantly decreased the expression level of TNF-α compared with the vehicle-treated LPS-injected group. This effect was only significant at the doses of 4 and 7 mg/kg morphine on the expression of IL-1β (Fig. 3a). There was also a significant difference between the anti-inflammatory effects of morphine at the doses of 4 and 10 mg/kg on both TNF-α and IL-1β expression, and therefore, the 4-mg/kg dose of morphine was selected for further mechanistic analysis.
To have more evidence showing the inflammatory responses triggered by a single morphine injection and the anti-inflammatory role of morphine pretreatment in an inflammatory context, we further analyzed the nuclear level of NF-κB and also the cytosolic level of COX-2. As it is shown in Fig. 3b, c, LPS injection significantly increased the hippocampal nuclear NF-κB as well as COX-2 expression. Morphine pretreatment in PBS-injected rats at all three doses had no significant effects on the nuclear level of NF-κB, but at the doses of 7 and 10 mg/kg significantly increased the hippocampal levels of COX-2 (Fig. 3b, c). Morphine pretreatment at all three doses significantly reduced the nuclear level of NF-κB in LPS-injected rats and also at the doses of 4 and 7 mg/kg significantly decreased the hippocampal COX-2 levels compared with the LPS group. In these two cases, the anti-inflammatory role of morphine at the dose of 4 mg/kg was also more pronounced than at the dose of 10 mg/kg (Fig. 3b, c).
Morphine Pretreatment Prevents the LPS-Induced Caspase-3 Activation and PARP Cleavage
We assessed the occurrence of apoptotic cell death by measuring the activation of caspase-3 and also nuclear PARP cleavage in the rat hippocampus 4 h after LPS injection. LPS-injected rats showed significantly higher levels of cleaved caspase-3 (Fig. 4a) and also nuclear PARP cleavage in comparison with the PBS-injected rats (Fig. 4b). Morphine-treated rats at the doses of 7 and 10 mg/kg also showed significantly higher levels of cleaved caspase-3 in their hippocampus cell lysate in comparison with the control group. However, morphine pretreatment at all three doses prevented the activation of caspase-3 in LPS-injected rats, the effect which was also more pronounced at the dose of 4 mg/kg morphine (Fig. 4a).

Effects of morphine pretreatment on LPS-induced apoptotic cell death. The level of cleaved caspase-3 in the hippocampus cell lysate and the level of PARP cleavage in the nuclear fraction were determined by Western blotting. a All the panels show the representative immunoblots of the control group, LPS (1 mg/kg, i.p.) group, groups receiving morphine i.p. injections at the doses of 4, 7, and 10 mg/kg (M4, M7, and M10, respectively) 30 min before i.p. PBS injection, groups receiving morphine pretreatment at the doses of 4, 7, and 10 mg/kg (M4-LPS, M7-LPS, and M10-LPS, respectively) 30 min before i.p. LPS injection, group receiving morphine at the dose of 4 mg/kg plus lpsRS (40 μg/rat, i.c.v.) 30 and 35 min, respectively, before LPS injection (lpsRS-M4-LPS), and group receiving morphine at the dose of 4 mg/kg 30 min before LPS plus naloxone (4 mg/kg, i.p.) 35 min before and 85 min after LPS injection (N-M4-LPS). b The panels show the representative immunoblots of the control group, LPS (1 mg/kg, i.p.) group, group receiving lpsRS (40 μg/rat; i.c.v.) 5 min before LPS (lpsRS-LPS), group receiving naloxone (4 mg/kg; i.p.) 35 min before and 120 min after LPS (N-LPS), group receiving morphine at the dose of 4 mg/kg 30 min before LPS (M4-LPS), and group receiving morphine at the dose of 4 mg/kg 30 min before LPS plus naloxone (4 mg/kg, i.p.) 35 min before and 120 min after LPS injection (N-M4-LPS). The quantitative data, presented as the mean ± SEM, were obtained from densitometric analysis of scanned X-ray films and normalized to corresponding immunoblots of β-actin as the loading control. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the control group; # P < 0.05, ## P < 0.01, and ### P < 0.001 versus the LPS group; & P < 0.05, && P < 0.01, and &&& P < 0.001 versus the M4-LPS group
Morphine pretreatment at the dose of 4 mg/kg did also significantly reduce the hippocampal nuclear PARP cleavage in LPS-injected rats, and this protective effect of morphine was comparable to that observed in naloxone- or lpsRS-treated, LPS-injected groups. Naloxone and lpsRS, in parallel to their anti-inflammatory effects, significantly diminished the PARP cleavage in LPS-injected rats (Fig. 4b).
Anti-inflammatory and Protective Role of Morphine is Mediated through Opioid Receptors
The results mentioned above indicate that morphine can significantly reduce the LPS-induced neuroinflammatory response and apoptotic cell death in the rat hippocampus, the effect which is also recapitulated by a TLR-4 antagonist and also naloxone, a mixed opioid and TLR-4 antagonist. Although we first intended to use naloxone as an opioid receptor antagonist to investigate the role of opioid receptors in the inflammatory/neurotoxic or antineuroinflammatory/protective effects of morphine in LPS-injected rats, after reaching the above-described results, we got also interested to know whether there is any additive or synergistic effect between the anti-inflammatory/protective role of naloxone and morphine.
To answer this issue, we had two more groups of LPS-injected rats which received morphine at the dose of 4 mg/kg plus naloxone or lpsRS. Surprisingly, naloxone, but not lpsRS, reversed the anti-inflammatory and protective role of morphine. As it is shown in Fig. 3a, b, naloxone antagonized the inhibitory effect of morphine 4 mg/kg and reversed the expression level of TNF-α and COX-2 to some extent near to LPS group. This effect on NF-κB and IL-1β was slightly different, and in this group, although the levels of NF-κB and IL-1β were significantly higher than the morphine 4 mg/kg plus LPS group, were also significantly less than the LPS group (Fig. 3c, a, respectively). This observation may be due to the antagonistic effects of naloxone on TLR-4 and the anti-inflammatory effects exerted through this mechanism, as proved and shown in this study. The same results were also observed on the activation of caspase-3 and PARP cleavage. Naloxone antagonized the protective effect of morphine 4 mg/kg and increased the level of cleaved caspase-3 as well as PARP cleavage. However, lpsRS neither antagonized nor synergistically increased the anti-inflammatory and protective effects of morphine (Fig. 4a, b).
Morphine Pretreatment Protects from LPS-Induced Memory Deficit
In the present study, we also tried to investigate the effect of morphine on LPS-induced memory deficit. As it is shown in Fig. 5, LPS significantly impaired the inhibitory avoidance memory retrieval and decreased the latency to enter the dark compartment in comparison with the control group. Morphine injection in a noninflammatory context also impaired memory retrieval (Fig. 5). Although all three doses of morphine significantly deceased the entry latency, this effect was more pronounced at the doses of 4 and 7 mg/kg than the dose of 10 mg/kg. In spite of the potential amnesic effect, morphine pretreatment at the doses of 4 and 10 mg/kg significantly prevented the LPS-induced memory deficit. In agreement with the above-described results, the protective role of morphine on LPS-induced memory impairment was also reversed by naloxone (Fig. 5).

Effects of morphine on passive avoidance memory retrieval in noninflammatory and inflammatory contexts. Twenty hours after passive avoidance training, rats were assigned to ten groups and given injections of either BPS or LPS (1 mg/kg) with or without morphine and/or morphine plus naloxone or lpsRS pretreatments. The entry latency (mean ± SEM) into the dark compartment was assessed 4 h later. Shorter entrance latency indicates impaired memory retention. Morphine injection at the doses of 4, 7, and 10 mg/kg as well as LPS impaired the passive avoidance memory retrieval. Morphine pretreatment at the doses of 4 and 10 mg/kg protected the rats from LPS-induced memory deficit. Naloxone, but not lpsRS, halted the protective role of morphine 4 mg/kg in LPS-injected rats. *P < 0.05 and ***P < 0.001 versus the control group; ## P < 0.01 and ### P < 0.001 versus the LPS group; && P < 0.01 versus the M4-LPS group
Discussion
In the current study, we showed the antineuroinflammatory and memory recall-improving role of morphine preconditioning in LPS-induced neuroinflammation and also showed that these effects are mediated through opioid receptors.
Acute peripheral infection has been demonstrated to induce neuroinflammatory responses in several studies (Chakravarty and Herkenham 2005; Cunningham et al. 2005). It is now clear that, immediately after i.p. injection of LPS, it can be recognized by immune cells plasma membrane-bounded TLR-4 and TNF-α will be instantly secreted (Supajatura et al. 2001; Piliponsky et al. 2010). LPS/TLR-4 interaction has been also proved in the CNS immediately following the expression of proinflammatory cytokines during severe endotoxemia (Laflamme and Rivest 2001). Our data showed a significant increase in TNF-α and IL-1β levels in the rat hippocampus 4 h after LPS administration and conceded the previous studies and also our experiments to simulate neuroinflammation. TNF-α and IL-1β are proinflammatory cytokines that are produced by microglia as well as in peripheral tissues (Merrill and Benveniste 1996; Bao et al. 2001).
Recently, it has been shown that naloxone, in addition to its antagonistic effects on opioid receptors, possesses TLR-4 antagonistic effects (Hutchinson et al. 2008) and has successively calmed neuroinflammation in several studies (Liao et al. 2003; Lin et al. 2010). We assessed the role of naloxone pretreatment in LPS-induced neuroinflammation and, as was expected, it was able to suppress the neuroinflammatory response as well as a TLR-4 antagonist. These data conceded previous documents about TLR-4 antagonistic and anti-inflammatory effects of naloxone and also raised the question whether the combination of naloxone with morphine will potentiate or abolish the preconditioning effect of morphine.
Regarding the inflammatory versus anti-inflammatory and apoptotic versus protective effects, morphine has received intensive long-lasting studies. However, there is no data comparing the in vivo effects of acute morphine injection in noninflammatory and inflammatory background. In this study, we showed that a single morphine injection at three different therapeutic doses exerts a proinflammatory response accompanied with apoptotic cell death and passive avoidance memory impairment. These results were expected because of the potential agonistic effects of morphine on TLR-4 and activation of microglial cells (Hutchinson et al. 2007). There are also pieces of evidence showing the induction of apoptotic cell death in the hippocampus and memory impairment in response to morphine (Spain and Newsom 1991; Zarrindast et al. 2006). It has been also demonstrated that chronic application of morphine triggers apoptosis in the rat spinal cord and different brain areas (Boronat et al. 2001; Mao et al. 2002; Atici et al. 2004; Chen et al. 2008), in human and rodent peripheral immune system-related cells (Singhal et al. 1998; Yin et al. 1999), and in embryonic neuronal and glial cells (Hu et al. 2002; Nasiraei-Moghadam et al. 2010). Our results proved that morphine induces a neuroinflammatory response and apoptosis even in acute administration; however, this effect seems to be partly dose-dependent and to be enhanced as the morphine dose is increased. Furthermore, the results of this study showed that morphine administration before the induction of a neuroinflammatory reaction exerts an opposite protective preconditioning effect. In general, preconditioning-induced protection describes a phenomenon in which a prior exposure to a stimulus or agent renders protection against a subsequent detrimental insult. For example, it has been shown that mild activation of TLR-4 by LPS or TLR-9 by its ligand (CpG oligodeoxynucleotide) in different cerebral ischemia models reprograms the response to injury and improves the outcome (Stevens et al. 2008; Vartanian et al. 2011). In these experiments, the preconditioning stimuli induce an inflammatory reaction, which occurs with morphine as well.
An essential feature of the early innate immune response and activation of TLR-4 signaling by LPS is the secretion of proinflammatory cytokines via the activation of signal transduction pathways, including NF-κB, which ultimately leads to the transcription of chemokines, proinflammatory cytokines, and enzymes like IL-1β, TNF-α, and COX-2 (D’Acquisto et al. 1997; Liu and Malik 2006). Our results showed that NF-κB signaling raises 4 h after LPS injection and is accompanied with the elevation of other inflammatory factors. The morphine preconditioning effect prevents the activation of this proinflammatory transcription factor in response to LPS challenge and also reduces the expression of IL-1β, TNF-α, and COX-2. Although there are some pieces of evidence showing the induction of NF-κB signaling in response to chronic morphine administration (Frässdorf et al. 2005; Roy et al. 2006; El-Hage et al. 2008), we showed that a single morphine injection causes no significant change in the basal level of nuclear NF-κB in the noninflammatory context. In agreement with other documents showing the induction of COX-2 in neuroinflammatory conditions (Giovannini et al. 2003; Sugimoto and Iadecola 2003; Candelario-Jalil et al. 2005), our results showed that higher doses of morphine as well as LPS challenge increases COX-2 expression in the rat hippocampus and that the morphine preconditioning effect prevents the induction of COX-2 in response to LPS.
Caspase-3 is considered to be the main executioner caspase involved in apoptotic cell death (Jänicke et al. 1998; Slee et al. 2001) and the detection of an 89- or 24-kDa caspase cleavage fragment of PARP-1 is described as a hallmark of apoptosis (Lazebnik et al. 1994). Here, we showed that, 4 h after LPS challenge, the level of cleaved caspase-3 as well as nuclear PARP cleavage is increased in the hippocampus and this is in agreement with other studies showing that LPS induces apoptosis (Comstock et al. 1998; Joshi et al. 2003). Although the apoptotic effects of morphine have been reported in a variety of in vitro and in vivo models, there are some studies showing an antiapoptotic and protective effect of morphine preconditioning or postconditioning on ischemia (Okubo et al. 2004; Wang et al. 2009). Here, we showed that, although an acute dose of morphine is able to induce apoptotic cell death in the rat hippocampus, its preconditioning effect can protect the hippocampus neuronal cells from the activation of caspase-3 and PARP cleavage. This is in contrast with a recent study showing that morphine alone does not directly induce caspase activity, but enhances the LPS-induced caspase activity in spleen-, thymus-, and bone marrow-derived immune cells (Olin et al. 2010).
There are pieces of evidence showing that learning and memory processes can be affected by inflammatory reactions within the brain (Yirmiya and Goshen 2011). Some documents also mention a crucial role for NF-κB signaling in long-term memory development (Albensi and Mattson 2000; Mattson et al. 2000; Kassed et al. 2002; Dash et al. 2005). There are also some studies revealing that pretraining and pretest administration of morphine at the dose of 5 mg/kg, s.c., impairs the passive avoidance memory retrieval (Zarrindast et al. 2006). Here, we showed that all three doses of morphine as well as LPS challenge impair the passive avoidance memory retention and, interestingly, that morphine preconditioning can protect the rats from LPS-induced memory deficit. Other investigators have also showed that morphine preconditioning or postconditioning in ischemia/reperfusion models can improve both short-term and long-term memory performance (Pateliya et al. 2008; Rehni et al. 2008).
After confirming the anti-inflammatory and protective role of morphine in LPS-induced neuroinflammation at both behavioral and molecular levels, we used naloxone to evaluate if these effects are mediated through opioid receptors. Our results showed that all of the protective preconditioning effects of morphine can be blocked with the opioid receptor antagonist. There are also some documents showing that the preconditioning effect of morphine in ischemia/reperfusion models can be attenuated by naloxone and is dependent on the activation of opioid receptors (McPherson and Yao 2001; Okubo et al. 2004; Gwak et al. 2010). Regarding the fact that naloxone, in addition to its antagonistic effects on opioid receptors, has also the same effects on TLR-4 (Hutchinson et al. 2007, 2010a, b), the conversion of the protective role of morphine may be due to the blockage of TLR-4 by naloxone and exertion of the innate proinflammatory and neurotoxic role of morphine. To exclude this issue and be sure that the conversion of the protective role of morphine by naloxone is only because of opioid receptor blockage, we used a pure TLR-4 antagonist. Interestingly, the concurrent use of lpsRS with morphine neither blocked the protective effect of morphine nor additively or synergistically enhanced it. It, therefore, confirmed that the preconditioning effect of morphine in LPS-induced neuroinflammation is mediated through opioid receptors. In addition, the observation that the TLR-4 antagonist did not enhance the protective effect of morphine can further emphasize that the preconditioning effect of morphine is highly due to the activation of opioid receptor signaling pathways, not because of a simple competition between morphine and LPS on TLR-4.
Conclusion
It can be concluded that morphine has a preconditioning effect on LPS-induced neuroinflammation. Although morphine has proinflammatory, neurotoxic, and amnesic effects in a noninflammatory context, it can prevent the induction of neuroinflammatory response by LPS and also protect the hippocampal cells from apoptosis and also passive avoidance memory deficit in rats. The preconditioning protective role of morphine is dependent on opioid receptors. However, regarding the potential effect of morphine on TLR-4, further studies are required to delineate why morphine has no additive effects on LPS-induced neuroinflammation and reversely can prevent it.
References
Albensi BC, Mattson MP (2000) Evidence for the involvement of TNF and NF B in hippocampal synaptic plasticity. Synapse 35:151–159
Askari N, Mahboudi F, Haeri Rohani A et al (2008) Effects of single administration of morphine on G protein mRNA level in the presence and absence of inflammation in the rat spinal cord. Scand J Immunol 67:47–52
Atici S, Cinel L, Cinel I et al (2004) Opioid neurotoxicity: comparison of morphine and tramadol in an experimental rat model. Int J Neurosci 114:1001–1011
Bao L, Zhu Y, Elhassan AM et al (2001) Adjuvant-induced arthritis: IL-1 [beta], IL-6 and TNF-[alpha] are up-regulated in the spinal cord. Neuroreport 12:3905
Barry U, Zuo Z (2005) Opioids: old drugs for potential new applications. Curr Pharm Des 11:1343–1350
Boronat MA, García-Fuster MJ, García-Sevilla JA (2001) Chronic morphine induces up–regulation of the pro–apoptotic Fas receptor and down–regulation of the anti-apoptotic Bcl-2 oncoprotein in rat brain. Br J Pharmacol 134:1263–1270
Brown GC, Bal-Price A (2003) Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol 27:325–355
Candelario-Jalil E, Mhadu N, González-Falcón A et al (2005) Effects of the cyclooxygenase-2 inhibitor nimesulide on cerebral infarction and neurological deficits induced by permanent middle cerebral artery occlusion in the rat. J Neuroinflammation 2:3
Chakravarty S, Herkenham M (2005) Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J Neurosci 25:1788
Chen Q, Cui J, Zhang Y, Yu LC (2008) Prolonged morphine application modulates Bax and Hsp70 levels in primary rat neurons. Neurosci Lett 441:311–314
Comstock KL, Krown KA, Page MT et al (1998) LPS-induced TNF-[alpha] release from and apoptosis in rat cardiomyocytes: obligatory role for CD14 in mediating the LPS response. J Mol Cell Cardiol 30:2761–2775
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH (2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25:9275
D’Acquisto F, Iuvone T, Rombolą L, Sautebin L, Di Rosa M, Carnuccio R (1997) Involvement of NF-[kappa] B in the regulation of cyclooxygenase-2 protein expression in LPS-stimulated J774 macrophages. FEBS Lett 418:175–178
Dash PK, Orsi SA, Moore AN (2005) Sequestration of serum response factor in the hippocampus impairs long term spatial memory. J Neurochem 93:269–278
El-Hage N, Bruce-Keller AJ, Yakovleva T et al (2008) Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-κB trafficking and transcription. PLoS One 3:e4093
Frässdorf J, Weber NC, Obal D et al (2005) Morphine induces late cardioprotection in rat hearts in vivo: the involvement of opioid receptors and nuclear transcription factor B. Anesth Analg 101:934
Gidday JM, Fitzgibbons JC, Shah AR, Park T (1994) Neuroprotection from ischemic brain injury by hypoxic preconditioning in the neonatal rat. Neurosci Lett 168:221–224
Giovannini M, Scali C, Prosperi C, Bellucci A, Pepeu G, Casamenti F (2003) Experimental brain inflammation and neurodegeneration as model of Alzheimer’s disease: protective effects of selective COX-2 inhibitors. Int J Immunopathol Pharmacol 16:31
Gwak MS, Li L, Zuo Z (2010) Morphine preconditioning reduces lipopolysaccharide and interferon-[gamma]-induced mouse microglial cell injury via [delta] 1 opioid receptor activation. Neuroscience 167:256–260
Hoshino K, Tsutsui H, Kawai T et al (1999) Cutting edge: generation of IL-18 receptor-deficient mice: evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J Immunol 162:5041
Hu S, Sheng WS, Lokensgard JR, Peterson PK (2002) Morphine induces apoptosis of human microglia and neurons. Neuropharmacology 42:829–836
Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR (2007) Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. Sci World 98:111
Hutchinson M, Coats B, Lewis S et al (2008) Proinflammatory cytokines oppose opioid-induced acute and chronic analgesia. Brain, Behav, Immun 22:1178–1189
Hutchinson MR, Loram LC, Zhang Y et al (2010a) Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity. Neuroscience 168:551–563
Hutchinson MR, Zhang Y, Shridhar M et al (2010b) Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain, Behav, Immun 24:83–95
Jänicke RU, Sprengart ML, Wati MR, Porter AG (1998) Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 273:9357
Joshi VD, Kalvakolanu DV, Cross AS (2003) Simultaneous activation of apoptosis and inflammation in pathogenesis of septic shock: a hypothesis1. FEBS Lett 555:180–184
Kao TK, Ou YC, Liao SL et al (2008) Opioids modulate post-ischemic progression in a rat model of stroke. Neurochem Int 52:1256–1265
Kassed C, Willing A, Garbuzova-Davis S, Sanberg P, Pennypacker K (2002) Lack of NF-[kappa] B p50 exacerbates degeneration of hippocampal neurons after chemical exposure and impairs learning. Exp Neurol 176:277–288
Kieffer BL (1995) Recent advances in molecular recognition and signal transduction of active peptides: receptors for opioid peptides. Cell Mol Neurobiol 15:615–635
Kim MS, Cheong YP, So HS et al (2001) Protective effects of morphine in peroxynitrite-induced apoptosis of primary rat neonatal astrocytes: potential involvement of G protein and phosphatidylinositol 3-kinase (PI3 kinase). Biochem Pharmacol 61:779–786
Kreutzberg GW (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci 19:312–318
Lacroix S, Feinstein D, Rivest S (1998) The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol 8:625–640
Laflamme N, Rivest S (2001) Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J 15:155
Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347
Liao S, Chen W, Raung S, Chen C (2003) Neuroprotection of naloxone against ischemic injury in rats: role of mu receptor antagonism. Neurosci Lett 345:169–172
Lim YJ, Zheng S, Zuo Z (2004) Morphine preconditions Purkinje cells against cell death under in vitro simulated ischemia–reperfusion conditions. Anesthesiology 100:562
Lin HY, Huang CC, Chang KF (2009) Lipopolysaccharide preconditioning reduces neuroinflammation against hypoxic ischemia and provides long-term outcome of neuroprotection in neonatal rat. Pediatr Res 66:254
Lin S, Tsai R, Tai Y et al (2010) Ultra-low dose naloxone upregulates interleukin-10 expression and suppresses neuroinflammation in morphine-tolerant rat spinal cords. Behav Brain Res 207:30–36
Liu SF, Malik AB (2006) NF-κB activation as a pathological mechanism of septic shock and inflammation. Am J Physiol-Lung Cell Mol Physiol 290:L622–L645
Lynn W, Golenbock D (1992) Lipopolysaccharide antagonists. Immunol Today 13:271–276
Madera-Salcedo IK, Cruz SL, Gonzalez-Espinosa C (2011) Morphine decreases early peritoneal innate immunity responses in Swiss-Webster and C57BL6/J mice through the inhibition of mast cell TNF-[alpha] release. J Neuroimmunol 232:101–107
Mao J, Sung B, Ji RR, Lim G (2002) Neuronal apoptosis associated with morphine tolerance: evidence for an opioid-induced neurotoxic mechanism. J Neurosci 22:7650
Mattson MP, Culmsee C, Yu ZF, Camandola S (2000) Roles of nuclear factor B in neuronal survival and plasticity. J Neurochem 74:443–456
McPherson BC, Yao Z (2001) Morphine mimics preconditioning via free radical signals and mitochondrial KATP channels in myocytes. Circulation 103:290–295
Merrill JE, Benveniste EN (1996) Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci 19:331–338
Minami M, Satoh M (1995) Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci Res 23:121–145
Mucke L, Eddleston M (1993) Astrocytes in infectious and immune-mediated diseases of the central nervous system. FASEB J 7:1226
Nandagopal K, Dawson TM, Dawson VL (2001) Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther 297:474
Nasiraei-Moghadam S, Kazeminezhad B, Dargahi L, Ahmadiani A (2010) Maternal oral consumption of morphine increases Bax/Bcl-2 ratio and caspase 3 activity during early neural system development in rat embryos. J Mol Neurosci 41:156–164
Okubo S, Tanabe Y, Takeda K et al (2004) Ischemic preconditioning and morphine attenuate myocardial apoptosis and infarction after ischemia–reperfusion in rabbits: role of delta-opioid receptor. Am J Physiol Heart Circ Physiol 287:H1786
Olin MR, Roy S, Molitor T (2010) In vivo morphine treatment synergistically increases LPS-induced caspase activity in immune organs. J Neuroimmune Pharm 5:546–552
Pak T, Cadet P, Mantione KJ, Stefano GB (2005) Morphine via nitric oxide modulates beta-amyloid metabolism: a novel protective mechanism for Alzheimer’s disease. Med Sci Monit 11:BR357–BR366
Pakpour B, Ahmadi S, Nayer-Nouri T, Oryan S, Zarrindast MR (2010) Inhibitory avoidance memory deficit induced by scopolamine: interaction with glutamatergic system in the nucleus accumbens. Behav Pharmacol 21:719
Pateliya BB, Singh N, Jaggi AS (2008) Possible role of opioids and K ATP channels in neuroprotective effect of postconditioning in mice. Biol Pharm Bull 31:1755–1760
Paxinos G, Watson C (2007) The rat brain in stereotaxic coordinate, sixth ed. Academic press, New York
Piliponsky AM, Chen CC, Grimbaldeston MA et al (2010) Mast cell-derived TNF can exacerbate mortality during severe bacterial infections in C57BL/6-KitW-sh/W-sh mice. Am J Pathol 176:926
Pourpak Z, Ahmadiani A, Alebouyeh M (2004) Involvement of interleukin 1 in systemic morphine effects on paw oedema in a mouse model of acute inflammation. Scand J Immunol 59:273–277
Pradillo JM, Romera C, Hurtado O et al (2005) TNFR1 upregulation mediates tolerance after brain ischemic preconditioning. J Cereb Blood Flow Metab 25:193–203
Qian L, Tan K, Wei S et al (2007) Microglia-mediated neurotoxicity is inhibited by morphine through an opioid receptor-independent reduction of NADPH oxidase activity. J Immunol 179:1198
Qureshi S, Larivière L, Leveque G et al (1999) Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). The J Exp Med 189:615
Rambhia S, Mantione KJ, Stefano GB, Cadet P (2005) Morphine modulation of the ubiquitin–proteasome complex is neuroprotective. Med Sci Monit: Int Med J Exp Clin Res 11:BR386
Rehni AK, Singh TG, Jaggi AS, Singh N (2008) Pharmacological preconditioning of the brain: a possible interplay between opioid and calcitonin gene related peptide transduction systems. Pharmacol Rep 60:904–913
Roy S, Wang J, Kelschenbach J, Koodie L, Martin J (2006) Modulation of immune function by morphine: implications for susceptibility to infection. J Neuroimmune Pharmacol 1:77–89
Singhal PC, Sharma P, Kapasi AA, Reddy K, Franki N, Gibbons N (1998) Morphine enhances macrophage apoptosis. J Immunol 160:1886
Slee EA, Adrain C, Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem 276:7320
Spain JW, Newsom GC (1991) Chronic opioids impair acquisition of both radial maze and Y-maze choice escape. Psychopharmacology 105:101–106
Stevens SL, Ciesielski TM, Marsh BJ et al (2008) Toll-like receptor 9: a new target of ischemic preconditioning in the brain. J Cereb Blood Flow Metab 28:1040–1047
Sugimoto K, Iadecola C (2003) Delayed effect of administration of COX-2 inhibitor in mice with acute cerebral ischemia. Brain Res 960:273–276
Supajatura V, Ushio H, Nakao A, Okumura K, Ra C, Ogawa H (2001) Protective roles of mast cells against enterobacterial infection are mediated by Toll-like receptor 4. J Immunol 167:2250
Takeuchi O, Akira S (2001) Toll-like receptors; their physiological role and signal transduction system. Int Immunopharmacol 1:625–635
Turrin NP, Rivest S (2006) Molecular and cellular immune mediators of neuroprotection. Mol Neurobiol 34:221–242
Ulevitch R, Tobias P (1995) Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol 13:437–457
Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP (2011) LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation 8:140
Wang Z, Kang J, Li Y, Yuan Z, Liu S, Sun L (2006) The effects of dexamethasone on rat brain cortical nuclear factor kappa B (NF-[kappa] B) in endotoxic shock. Toxicol Appl Pharmacol 214:263–269
Wang Z, Zhao H, Peng J et al (2009) Anti-apoptotic effect of morphine postconditioning on myocardial ischemia–reperfusion injury and its anti-oxidative stress mechanism. J Sun Yat-Sen Univ (Med Sci) 5
Watkins LR, Hutchinson MR, Rice KC, Maier SF (2009) The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci 30:581–591
Wright SD (1999) Toll, a new piece in the puzzle of innate immunity. The J Exp Med 189:605
Yin D, Mufson RA, Wang R, Shi Y (1999) Fas-mediated cell death promoted by opioids. Nature 397:218
Yirmiya R, Goshen I (2011) Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, Behav, Immun 25:181–213
Zarrindast MR, Fazli-Tabaei S, Ahmadi S, Yahyavi SH (2006) Effect of lithium on morphine state-dependent memory of passive avoidance in mice. Physiol Behav 87:409–415
Zhao P, Huang Y, Zuo Z (2006) Opioid preconditioning induces opioid receptor-dependent delayed neuroprotection against ischemia in rats. J Neuropathol Exp Neurol 65:945
Zujovic V, Schussler N, Jourdain D, Duverger D, Taupin V (2001) In vivo neutralization of endogenous brain fractalkine increases hippocampal TNF [alpha] and 8-isoprostane production induced by intracerebroventricular injection of LPS. J Neuroimmunol 115:135–143
Acknowledgments
The authors are grateful to the Neuroscience Research Center of Shahid Beheshti University of Medical Sciences for the financial support of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rostami, F., Oryan, S., Ahmadiani, A. et al. Morphine Preconditioning Protects Against LPS-Induced Neuroinflammation and Memory Deficit. J Mol Neurosci 48, 22–34 (2012). https://doi.org/10.1007/s12031-012-9726-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-012-9726-4