
Abstract
Substantial discoveries suggested that exosomes released from multiple sources of stem cells can affect the biological functions of target cells. In present period, the immunosuppressive properties of exosomes derived from bone marrow mesenchymal stem cells (BMMSCs-E) have been extensively recognized, but few studies have been reported about exosomes secreted from dental pulp stem cells (DPSCs-E) in the field of medical immunity. Hence, the aim of this study is to compare the immunomodulatory capacity of BMMSCs-E and DPSCs-E. Peripheral blood mononuclear cells (PBMCs) were co-cultured with them respectively and the proportion of regulatory T cells (Treg) was detected to increase. Subsequently, we stimulated CD4+T cells with BMMSCs-E and DPSCs-E to observe their effects on the polarizations, chemokines secretion, apoptosis, and proliferation of CD4+T cells. We found that DPSCs-E inhibited the differentiation of CD4+T cells into T helper 17 cells (Th17) and reduced the secretions of pro-inflammatory factors IL-17 and TNF-α, while promoted the polarization of CD4+T cells into Treg and increased the release of anti-inflammatory factors IL-10 and TGF-β. What’s more, these capabilities of DPSCs-E were stronger than those of BMMSCs-E. In addition, DPSCs-E were more effective in inducing apoptosis of CD4+T cells compared with BMMSCs-E, and DPSCs-E inhibited the proliferation of CD4+T cells, which is similar to BMMSCs-E. We draw a conclusion that DPSCs-E have stronger immune-modulating activities than BMMSCs-E, and may be a new therapeutic tool for the treatment of immunological diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mesenchymal stem cells (MSCs) are multipotent cells originally isolated from various tissues, such as bone marrow [1], cord blood [2], teeth [3], and adipose tissue [4]. They can differentiate into different cell lineages, namely osteoblasts, chondrocytes, adipocytes, and neurocytes under suitable conditions [5, 6]. MSCs not only possess regenerative potential, but also can interact with immunoregulatory cells, profoundly impact on their functions in vitro and in vivo [7, 8].
Bone marrow mesenchymal stem cells (BMMSCs) are the most widely studied stem cell subtype at the current stage, especially in the field of immunology. Numerous findings have shown that BMMSCs are candidates of cell-based therapy to treat autoimmune diseases such as rheumatoid arthritis (RA) [9], systemic lupus erythematosis (SLE) [10], graft-versus-host disease (GVHD) [11] and multiple sclerosis (MS) [12]. Dental pulp stem cells (DPSCs), one type of MSCs, originate from the dental pulp tissue with the capacity to differentiate into several cell lineages [13]. Compared with BMMSCs, DPSCs are easier accessible without serious technical and ethical problems, and are higher proliferative and more morphologically stable [14]. In existing medical literature, various results are reported in interactions between DPSCs and T helper cells. DPSCs induced the apoptosis of allogeneic T lymphocytes and the production of Treg via the Fas/Fas ligand interaction [15]. The secretion of soluble factors TGF-β and IL-10 from DPSCs were articulated to be involved in inhibiting hyperimmune reactions [16,17,18]. What is more interesting is that Makino Y indicated that the performance of DPSCs to ameliorate the imbalance of Th17/Treg is even stronger than BMMSCs and without any immune rejection [19]. Moreover, the therapeutic effect of DPSCs on SLE was superior to BMMSCs by inhibiting the Th17/Treg ratio [20].
Although MSCs have various therapeutic applications in human disease model mice, some researchers believe that stem cell therapy in clinical treatment of autoimmune diseases is still risky and controversial, such as obstruction of distal microvessels, tumorigenicity, virus contamination, and insufficient donor sources [21,22,23]. Therefore, much efforts have been devoted to develop alternative methods to cell-free therapy. Interestingly, many studies have demonstrated that transplanted stem cells contribute to therapeutic effects probably based on their secretion of exosomes [24, 25]. Exosomes are endosome-derived membranous nanovesicles with a size range of 30–150 nm secreted by all living cells. Exosomes usually contain different biological molecules such as proteins, lipids, cytokines, and RNAs [24, 25], which are responsible for intercellular signaling. In mice models of rheumatoid arthritis (RA) [26], graft-versus-host disease (GVHD) [27], systemic lupus erythematosus (SLE) [28] and uveitis [29], several laboratories have linked the protective effects observed with BMMSC-derived exosomes therapy directly to its immunosuppressive function. Besides vivo studies, Wancheng Chen also discovered BMMSC-derived exosomes could significantly suppress T cells activation and polarized activated CD4+T cells to Treg in vitro [30]. These findings would allow the development of biological therapeutics without the many risks and drawbacks associated with cell-based therapies. However, the easy-to-obtain mesenchymal stem cell subtype, namely dental pulp stem cells, whose release of exosomes have not been extensively studied in terms of immunosuppression heretofore.
In this paper, we isolated exosomes from BMMSCs and DPSCs, subsequently compared their differences in immunomodulatory by examining their effects on differentiation, secretion of cytokines, apoptosis, and proliferation of lymphocytes.
Materials and methods
Isolation of BMMSCs and DPSCs
All samples were acquired from healthy volunteers (age 25–35) after giving the informed consents which were approved by the Ethics Committee of the Affiliated Hospital of Nantong University. We isolate healthy dental pulp tissues (n = 8) from the caries-free teeth that need to be extracted due to orthodontics. The pulp was digested in 3 mg/mL collagenase type I for 1 h at 37 °C. Then, we obtained single-cell suspensions by passing the digested tissues through a 70-μm cell strainer (BD Falcon). Cell suspensions of dental pulp were placed at 25 cm2 culture dishes (Corning, USA) and cultured in high-glucose Dulbecco Modified Eagle Medium (HyClone, Logan, USA) with 10% fetal bovine serum (FBS; Gibco, Carlsbad, USA), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, USA) at 37 °C in a 5% CO2 incubator. The medium was changed every 3 days. Cells were passaged at the ratio of 1:3 when they reached 85–90% confluence.
BMMSCs were isolated from BM aspirates of healthy people (n = 8). Five milliliters of heparinized BM were mixed with equivalent phosphate-buffered saline (PBS). Next, the resuspended cells were placed in the upper layer of Ficoll solution (1.077 g/mL) and centrifuged at 1000×g for 30 min under ordinary temperature. The mononuclear cells at the interface were collected and cultured in low-glucose Dulbecco Modified Eagle Medium (HyClone, Logan, USA) with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Then, the cells were seeded into 25 cm2 dish at a density of 2 × 107 cells and cultured at 37 °C and 5% CO2. After 5 days, nonadherent cells were removed by changing the culture medium and the medium was changed every 3 days. Finally, cells were passaged at the ratio of 1:3 when they reached 85–90% confluence. The third or fourth passage cells were used in the all experiments.
Cell surface markers analysis of BMMSCs and DPSCs
Two cell populations were immunophenotypically characterized by flow cytometry using the Human MSC Analysis Kit: the MSC positive cocktail (CD90-FITC, CD73-APC, CD105-Percp-Cy5.5), the negative MSC cocktail (CD45-PE, CD34-PE, PE, CD11b-PE, CD19-PE and HLA-DR-PE) (BD Biosciences, Oxford, UK). Data were analyzed with FlowJo Analysis Software (FlowJo).
Osteogenic differentiation
BMMSCs and DPSCs were plated into 12-well culture plate (Corning, USA) at a density of 2 × 105 cells/well. After cell adhesion, the culture medium was replaced with osteogenic medium supplemented with 10% FBS, 50 μg/mL ascorbic acid, 100 nM dexamethasone, and 5 mM β-glycerophosphate. The media were changed every 2 days. One week later, the total protein was used for Western blot. Two weeks later, the collected cells were then used for prepared for Alizarin red staining and alkaline phosphatase (ALP) assay.
Adipogenic differentiation
BMMSCs and DPSCs were plated into 12-well culture plate (Corning, USA) at a density of 2 × 105 cells/well. After cell adhesion, the culture medium was replaced with adipogenic medium supplemented with 10% FBS, 60 μmol/L indomethacin, 1 μmol/L dexamethasone, 5 μg/mL insulin, and 0.5 mmol/L IBMX. The media were changed every 2 days. After 2 weeks, the expression level of PPARγ in cells were detected by Western blot.
Alizarin red staining and ALP assay
The mineralization potential of the cells was assessed via Alizarin red staining when cells were cultured with osteogenic medium for 2 weeks. The cells were fixed with 4% paraformaldehyde for 1 h and then incubated with 40 mM Alizarin red S (Sigma) for 15 min in the dark. ALP staining were carried out after osteogenic induction for 2 weeks using the ALP Assay Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions.
Western blot analysis
Proteins were extracted from exosomes using RIPA buffer supplemented with 1 mM PMSF and loading buffer (Invitrogen, Thermo Fisher Scientific, MA, USA). Protein concentrations were determined using a BCA protein assay kit (Beyotime, Shanghai, China), according to the manufacturer’s specifications. Equal amounts of protein (100 μg) was subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The separated proteins were transferred onto polyvinylidene fluoride (PVDF, Invitrogen, Thermo Fisher Scientific, MA, USA) membranes in a blotting apparatus (Bio-Rad, CA) at 300 mA for 50 min. Membranes were blocked with 5% not-fat milk for 2 h at 37 °C and incubated overnight at 4 °C with primary antibodies: GADPH (1:1000, anti-mouse; Santa Cruz, USA), RUNX2 (1:800, anti-mouse; Sigma, USA), PPARγ (1:800, anti-mouse; Sigma, USA), CD9 (1:800, anti-mouse; Sigma, USA), and CD63 (1:200, anti-rabbit; Santa Cruz, USA). After washing with TBST for three times, the membranes were probed with HRP-conjugated anti-rabbit or anti-mouse IgG antibody (Beyotime, Shanghai, China) for 2 h at room temperature. GAPDH was used as the normalizing control. Finally, the protein bands were visualized using Pierce™ ECL Plus Western Blotting Substrate (Thermo Fisher Scientific, USA).
PBMC and CD4+T cells preparation
PBMCs were obtained using fresh whole blood from 10 young healthy volunteers (age 25–35) with informed consents by density gradient centrifugation using the Ficoll-Hypaque. PBMCs were stimulated with 5 μg/mL of ConA (Sigma-Aldrich, USA). CD4+T cells were isolated using EasySepTM CD4+T cells negative selection kit (Stem Cell Technologies, Canada), according to the manufacturer’s instructions. Culture plates were coated with 2 μg/mL of anti-CD3 mAb and 4 μg/mL of anti-CD28 mAb (BioLegend, USA). Isolated cells were cultured in RPMI-1640 medium (Gibco, USA) with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco, USA) in 5% CO2 atmosphere at 37 °C. The PBMCs or CD4+T cells used in each of our experiments were derived from the peripheral blood of the same volunteer, but not from the mixed peripheral blood of several volunteers.
Isolation and identification of exosomes
BMMSCs or DPSCs were seeded to confluence in 100 mm cell culture dishes (Corning, USA). At 80% confluence, the growth medium was replaced with serum-free medium (DMEM). After 48 h, the supernatant was collected and submitted to differential centrifugation to acquire exosomes. Briefly, the supernatant was centrifuged at 300×g for 10 min firstly and then centrifuged at 16500×g for 30 min at 4 °C. The supernatant was next passed through a 0.2 μm filter (Millicell, Germany). Next, the filter liquor was ultracentrifuged at 4 °C at 100,000×g for 70 min, then the sediment was rinsed with PBS, and ultracentrifuged again at 4 °C at 100,000×g for 70 min finally. Exosomes was evaluated by transmission electron microscopy (TEM) and nanoparticle tracking analysis (NTA). Total protein of exosomes was detected by a BCA kit (Beyotime, Shanghai, China), and CD9 (anti-rabbit; Sigma, USA), CD63 (anti-mouse; Santa Cruz, USA) marker was identified by Western blot. Exosomes were isolated from stem cells extracted from each volunteer, rather than from pooled volunteer-derived stem cells.
Co-culture of exosomes and T cells
PBMCs or CD4+T cells were plated on 24-well culture plate (Corning, USA) at the density of 1 × 106 cells/well, then stimulated with 80 μg/mL DPSCs-E or BMMSCs-E and the corresponding stimulants mentioned above. After 3 days, the suspended cells were collected for the following experiments. The supernatant of cell cultures was collected and kept at − 80 °C for later examinations of ELISA.
Incorporation of exosomes
Exosomes were labeled with PKH67 green fluorescence lipid dye (Sigma, USA) according to the manufacturer’s instructions. PKH67-labeled exosomes were co-cultured with 2 × 105 CD4+T cells in a 24-well plate for 6 h at 37 °C and 5% CO2. Subsequently, CD4+T cells were fixed in 4% paraformaldehyde (PFA) solution, and the nuclei were stained with DAPI (Beyotime Institute of Biotechnology, China). The labeled exosomes in the CD4+T cells were imaged under a fluorescence microscope.
Flow-cytometry analysis of PBMC and CD4+T cells
PBMCs or CD4+T cells were quantified by flow cytometry (eBioscience, USA). Data were analyzed using FlowJo 6.0 software (Tree StarInc, Ashland, OR, USA). To detect Treg, PBMCs or CD4+T cells (1 × 106 cells /sample) were stained with FITC-conjugated anti-CD4 antibody and APC-conjugated anti-CD25 antibody (BD Biosciences, Oxford, UK) for 30 min at 4 °C in the dark. Next, CD4+T cells were fixed, permeabilized and stained with PE-conjugated anti-FOXP3 antibody (BD Biosciences, Oxford, UK). To detect Th17, CD4+T cells (1 × 106 cells/sample) were stimulated with phorbol myristate acetate (PMA;50 ng/mL; Sigma, USA) and ionomycin (500 ng/mL; Sigma, USA) for 6 h, with brefeldin A (10 μg/mL; Calbiochem, Germany) for 4 h. Then CD4+T cells were incubated with APC-conjugated anti-CD4 antibody (BD Biosciences, Oxford, UK) for 30 min at 4 °C under a shield. After fixation and permeabilization, cells were stained with PE-conjugated anti-IL-17 antibodies (BD Biosciences, Oxford, UK).
Cytokine quantification
Cell supernatants preserved at − 80 °C were took out to analyze the release of proinflammatory and anti-inflammatory factors. The level of IL-10, IL-17, TGF-β, and TNF-α in supernatants were performed by commercially available human IL-10 quantikine ELISA Kit, human IL-17 quantikine ELISA kit, human TGF-β Quantikine ELISA Kit, and human TNF-α quantikine ELISA kit (R&D Systems, USA) separately as per manufacturer’s instructions. Before measuring the concentration of IL-17, CD4+T cells need to be stimulated in vitro with phorbol myristate acetate (PMA;50 ng/mL; Sigma, USA) and ionomycin (500 ng/mL; Sigma, USA) for 6 h, with brefeldin A (10 μg/mL; Calbiochem, Germany) for 4 h.
Real-time polymerase chain reaction analysis
Total RNA of CD4+T cells were collected using TRIzol (Invitrogen, Thermo Fisher Scientific, MA, USA), following manufacturer’s instructions. For IL-10, IL-17, TGF-β, TNF-α, FOXP3, and RORC mRNA detection, complementary DNA (cDNA) was synthesized using RevertAid RT Reverse Transcription Kit (Invitrogen, Thermo Fisher Scientific, MA, USA). AceQ qPCR SYBR Green Master Mix (without ROX) (Vazyme, Shanghai, China) was then used to quantitative PCR of genes. GAPDH was used for normalization. The reaction program was set initially at 95 °C for 30 s, followed by other 40 cycles composed of 95 °C for 10 s and 60 °C for 30 s, with a final dissociation stage of 95 °C for 15 s, 60 °C for 1 min and 95 °C for 15 s. We use a light cycler 480 Real-Time PCR System (Roche Diagnostic, Mannheim, Germany) to test these levels.
The primer sequences used in the experiment were as follows: GAPDH: 5′-GAAGGTGAAGGTCGGAGTC-3′, 5′-GAAGATGGTGATGGGATTTC-3′; RORC: 5′-AAGGCCAAGAGCATCTCAGAC-3′, 5′-GCTGTGGCCTCAAGGATAAG-3′; FOXP3: 5′-GCTTCATCTGTGGCATCATC-3′, 5′-TGGAGGAACTCTGGGAATGT-3′; IL-10: 5′-GTGATGCCCCAAGCTGAGA-3′, 5′-CACGGCCTTGCTCTTGTTTT-3′; IL-17: 5′-CGGACTGTGATGGTCAACCT-3′, 5′-GACCAGGATCTCTTGCTGGA-3′; TGF-β: 5′-CAATTCCTGGCGATACCTCGA-3′, 5′-GCACAACTCCGGTGACATCAA-3′; TNF-α: 5′-ACGTGGAACTGGCAGAAGAG-3′, 5′-GGTTGTCTTTGAGATCCATGC-3′. The 2−∆∆Ct method was used for relative quantification of the gene expression.
Apoptosis assays
CD4+T cells stimulated with 2 μg/mL of anti-CD3 mAb and 4 μg/mL of anti-CD28 mAb co-cultured with or without exosomes for 3 days. Ann 647/propidium iodide (PI) staining (FACMAS, China) was performed to determine cell apoptosis and death according to manufacturer’s directions. Cells were labeled for 15 min under dark and washed twice in PBS, then data were analyzed with FlowJo Analysis Software (FlowJo).
Cell proliferation
CD4+T cells were cultured in a 96-well culture plate (Corning, USA) at the density of 4 × 105 cells/well. Cells stimulated with 2 μg/mL of anti-CD3 mAb and 4 μg/mL of anti-CD28 mAb co-cultured with or without exosomes for 3 days. CD4+T cells proliferation was estimated using a cell counting kit-8 (CCK8) assay kit (Beyotime, Shanghai, China), according to manufacturer’s product information. The absorbance at 450 nm was measured with a microplate reader.
Statistical evaluation
All the experiments were repeated at least three times independently and the data were presented as means ± SD. Paired t test was applied for comparison between the two groups. All statistical evaluation performed with GraphPad Prism 6.01. Value of P < 0.05 was considered statistically significant.
Results
Identification of BMMSCs and DPSCs
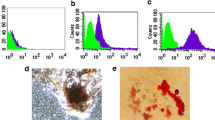
Two stem cell subtypes we isolated from bone marrow and dental pulp tissue showed a characteristic fibroblastic spindle-like morphology under a microscope (Fig. 1a). Subsequently, we used flow cytometry analysis to demonstrate that both BMMSCs and DPSCs were positive for mesenchymal markers CD90, CD73, and CD105, while did not express hematopoietic lineage markers CD45, CD34, CD11b, CD19, and HLA-DR (Fig. 1b). In addition, alkaline phosphatase (ALP) and alizarin red staining showed BMMSCs and DPSCs formed mineralized matrix under osteogenic conditions. The two stem cell subtypes expressed highly osteogenic marker gene RUNX2 (Fig. 1c, d). Moreover, after 14 days of adipogenic induction, the expression of adipogenic marker gene PPARγ was upregulated (Fig. 1e). These results indicated that BMMSCs and DPSCs were successfully isolated.

Identification of BMMSCs and DPSCs. a The morphology of BMMSCs and DPSCs was observed under an optical microscope (magnification × 200). b The phenotypes of BMMSCs and DPSCs were examined by flow cytometry. c The calcification ability and ALP activity after two weeks osteogenic differentiation were detected by Alizarin red staining and ALP staining. d Osteogenesis-related transcription factor RUNX2 was measured by western blot at one week of osteogenic induction. e Adipogenic-related transcription factor PPARγ was measured by Western blot at 2 weeks of osteogenic induction
The characterization of exosomes derived from BMMSCs and DPSCs
To compare the inhibitory effects of BMMSC-derived exosomes (BMMSCs-E) and DPSC-derived exosomes (DPSCs-E) on the activity of immunocytes, we first need to effectively isolate exosomes from BMMSCs and DPSCs. Transmission electron microscopy (TEM) indicated that the vesicles we obtained were round cup-shaped structure (Fig. 2a). By nanoparticle tracking analysis (NTA) technique, we observed the diameter distribution of these particles varied from 30 to 250 nm with a mean size of 135 nm, which was consistent with published reports (Fig. 1b). Besides, the key exosomal membrane proteins including CD9 and CD63 were positive in BMMSCs-E and DPSCs-E via Western blot results (Fig. 2c). All analyses revealed that the main components of our purified microvesicles were exosomes.

The characterization of BMMSCs-E and DPSCs-E. a Transmission electron microscopy (TEM) was used to image the structure of exosomes (Scale bar = 200 nm). b Nanoparticle tracking analysis (NTA) displayed particle size distribution of exosomes. c The same amounts of proteins from BMMSCs-E and DPSCs-E were detected the expression of exosomal marker proteins CD63 and CD9 by Western blot
Comparison Between the effects of BMMSCs-E and DPSCs-E on the ratio of Treg in PBMC
To test the impact of BMMSCs-E and DPSCs-E on immunity, PBMCs were stimulated with them respectively for three days, then we demonstrated that both BMMSCs-E and DPSCs-E could increase the proportion of Treg in PBMCs, and DPSCs-E were a bit more effective (Fig. 3a, b). Furthermore, qRT-PCR showed that Treg master transcription factor FOXP3 mRNA level in the PBMC exposed to DPSCs-E were higher than those exposed to BMMSCs-E (Fig. 3c).

BMMSCs-E and DPSCs-E increased the ratio of Treg in PBMC. a PBMC stimulated by 5 μg/mL ConA were treated with BMMSCs-E and DPSCs-E for 72 h, then the proportion of CD4+CD25+FOXP3+Treg was measured by flow cytometry. b The statistical view of the percentage of Treg in PBMCs. c Collect the total RNA of PBMC treated differently, qRT-PCR determined the mRNA expression level of FOXP3. Comparisons between two groups were performed with paired t test. Values are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. All results were obtained at least three independent experiments
Comparison of BMMSCs-E and DPSCs-E on the regulation of Th17/Treg ratio in CD4+T cells
In order to further verify the immunomodulatory capacity of exosomes derived from the two stem cell subtypes, we added the same amount of BMMSCs-E and DPSCs-E into CD4+T cells to observe cell polarizations. Flow cytometry analysis suggested that both treatment groups could increase the frequency of Treg compared with the control group, while the percentage of Th17 was decreased. It was noteworthy that DPSCs-E is more effective in improving the imbalance of Th17/Treg than BMMSCs-E (Fig. 4a, b). In addition, we collected CD4+T cells to detect the mRNA expression of Treg-related transcription factor FOXP3 and Th17-related transcription factor RORC via qRT-PCR, which results were correspond to the flow cytometry (Fig. 4c, d). Based on these findings we concluded that DPSCs-E show stronger immunosuppression function in restoring the imbalance of Th17/Treg.

BMMSCs-E and DPSCs-E inhibited the ratio of Th17/Treg in CD4+T cells. a, c CD4+T cells were stimulated with BMMSCs-E and DPSCs-E coated with anti-CD3 mAb and anti-CD28 mAb for 72 h, the proportion of CD4+CD25+FOXP3+Treg and CD4+IL-17+Th17 was measured by flow cytometry. b, d The statistical views of the percentage of Treg and Th17 in CD4+T cells. e, f Collect the total RNA of CD4+T cells treated differently, qRT-PCR determined the mRNA expression level of FOXP3 and RORC. Comparisons between two groups were performed with paired t-test. Values are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. All results were obtained at least three independent experiments
The effects of BMMSCs-E and DPSCs-E on cytokines secretion
At next time step, we investigated the effect of BMMSCs-E and DPSCs-E on the secretion of proinflammatory cytokines and anti-inflammatory cytokines in CD4+T cells. The results pointed out that the concentration of IL-10 and TGF-β in DPSCs-E group were higher than those in BMMSCs-E group, and the two groups were both higher compared with control group (Fig. 5a, b). Contrary to anti-inflammatory cytokines, CD4+T cells secreted minimal amount of proinflammatory cytokines IL-17 and TNF-α under DPSCs-E stimulation (Fig. 5c, d). Furthermore, IL-10 and TGF-β mRNA expression level was highest in CD4+T cells stimulated with DPSCs-E, while IL-17 and TNF-α transcription level was lowest (Fig. 5e–h). These results also clarified that DPSCs-E possess better anti-inflammatory properties compared with BMMSCs-E.

BMMSCs-E and DPSCs-E treatments promoted the secretion of IL-10 and TGF-β, but suppress the levels of IL-17 and TNF-α. a, b The concentration of the anti-inflammatory cytokines IL-10 and TGF-β were evaluated by ELISA in supernatant of CD4+T cells treated with BMMSCs-E and DPSCs-E separately for 72 h. c, d The levels of the proinflammatory cytokine IL-17 and TNF-α were measured by ELISA in culture medium of CD4+T cells treated with BMMSCs-E and DPSCs-E separately for 72 h. e–h Collected the total RNA of CD4+T cells to determine the mRNA levels of IL-10, TGF-β, IL-17 and TNF-α by qRT-PCR. Comparisons between two groups were performed with paired t test. Values are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. All results were obtained at least three independent experiments
Comparison of the effects of BMMSCs-E and DPSCs-E on the apoptosis and proliferation of CD4+T cells
We investigated the capability of the two exosomes on proliferation and apoptosis of CD4+T cells furtherly. Compared with BMMSCs-E, DPSCs-E has a stronger property of inducing the apoptosis of CD4+T cells (Fig. 6a, b). However, the inhibitory effect of BMMSCs-E and DPSCs-E on proliferation of CD4+T cells was similar (Fig. 6c). Finally, in order to ensure the extracted exosomes to interact with target cells, we incubated freshly isolated total CD4+T cells for 6 h with BMMSCs-E and DPSCs-E labeled with PKH67 green fluorescent membrane linker dye, and detected them with a fluorescence microscope (Fig. 6d), PKH67 labeled CD4+T cell supporting the occurrence of interaction between exosomes and CD4+T cells.

BMMSCs-E and DPSCs-E increase the apoptosis and inhibit proliferation of CD4+T cells. a CD4+T cells were stained with Ann 647/PI dual-immunofluorescence after addition with BMMSCs-E and DPSCs-E for 72 h, then were detected apoptosis by flow cytometry. b The statistical view of apoptosis rate of CD4+T cells. c The effect of BMMSCs-E and DPSCs-E on the proliferation of CD4+T cells was examined by CCK-8 assay kit after co-culture for 5 days. d PKH67-labeled exosomes were cultured for 6 h with CD4+T cells and analyzed by a fluorescence microscope. Nuclei were stained with DAPI (PKH67 = green; DAPI = blue). Image showed the incorporation of PKH67-labeled BMMSCs-E and DPSCs-E. Comparisons between two groups were performed with paired t test. Values are presented as the mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. All results were obtained at least three independent experiments
Discussion
Generous studies have been done to elucidate the immunomodulatory property of BMMSC, however, at the current stage, clinical trials of cell-based therapy are controversial, and its security is not guaranteed [31]. Many researchers have discovered that cell paracrine product exosomes have functions similar to that of cells, and the study of exosomes in the field of immunology is increasingly extensive [24, 32]. Compared with cells, exosomes are more preservable and have lower risks of tumorigenesis and immune rejection that they can be used as alternative therapies for various immune diseases [24]. BMMSC-derived exosomes have been found to modulate the immune system, both in vivo and in vitro [26,27,28,29,30]. However, it is well known that the trauma of BMMSCs acquisition is inevitable. We considered that DPSC-derived exosomes (DPSCs-E), as a more accessible resource have not been fully studied in the field of immunoregulation. Hence, this article mainly elaborated the function of DPSC-derived exosomes in suppressing inflammatory responses, and to compare that with exosomes derived from BMMSCs.
Massive studies have proved that Th17/Treg imbalance is the driving factor of the occurrence and development of immune diseases, such as RA, SLE, GVHD, MS [33, 34]. Th17/Treg ratio in peripheral blood of patients with immune diseases as RA may elevate abnormally. In addition, the disease symptom was brought under control after adjusting the ratio of Th17/Treg in mice with autoimmune diseases [35, 36]. Recently, a team of researchers investigated subcutaneous injection of BMMSC-derived exosomes in allogeneic skin transplantation mice delayed the occurrence of GVHD, which was concomitant with an increase in the percentage of Treg [27]. Stella Cosenza demonstrated for the first time that MSC-derived exosomes have therapeutic potential in inflammatory arthritis by increasing Treg cells population [26]. In autoimmune uveitis model, treatment with BMMSC-derived exosomes promoted disease recovery through efficiently suppressed the immunological activity of detrimental Th17 and weakened the migration of inflammatory cells [29]. Additionally, Wancheng Chen discovered that the ratio of Th17/Treg was restrained by BMMSC-derived exosomes in vitro [30]. In this article, we found that DPSC-derived exosomes can ameliorate Th17/Treg imbalance in vitro and are more efficacious than BMMSC-derived exosomes. This finding implies the potentiality of DPSC-derived exosomes in the treatment of auto-immune diseases.
It has been known for a long time that transcription factor FOXP3 is required to maintain Treg activity, and transcription factor RORC has been recognized as the main regulator of the Th17 lineage. Besides, more interesting is the fact Treg can redifferentiate into proinflammatory Th17 cells when loses FOXP3 expression [35, 37]. Research indicated defective-FOXP3 mice lose immune tolerance and easily cause autoimmune diseases [38]. Jong Won Hong discovered FOXP3 mRNA levels in the CD4+T cells co-cultured with DPSCs were higher than those in the monocultured CD4+T cells [39]. With our research, we suggested that the expression of FOXP3 increased and RORC decreased in CD4+T cells stimulated by DPSC-derived exosomes and BMMSC-derived exosomes compared with the control group, and DPSC-derived exosomes were more powerful.
Effector cytokines of Th17 and Treg have been increasingly recognized as key players in anaphylaxis, autoimmunity and inflammation [40]. Some researchers figured out that interdiction of IL-17 attenuates the severity of CIA, and IL-17-deficient mice are not susceptible to CIA [41, 42]. While IL-10 produced by Treg cells resulting in the amelioration of severity of CIA mice compared with wild-type mice [43]. Previous studies also found that lymphocytes stimulated by BMMSC-derived exosomes secreted fewer pro-inflammatory factors and more anti-inflammatory factors [30]. Our experimental results confirmed that DPSC-derived exosomes treatment significantly suppressed the concentration of IL-17 and TNF-α, while promoted the production of IL-10 and TGF-β. What’s more, we found that the role of DPSC-derived exosomes is stronger than that of BMMSC-derived exosomes. These results also demonstrated the superiority of DPSC-derived exosomes in regulating the immune system.
It is reported that the imbalance of proliferation and apoptosis of lymphocytes is also a key factor in the pathogenesis of autoimmune diseases. Researchers found that the apoptosis of CD4+T cells in CIA mice was obviously inhibited and the proliferation was markedly increased. Once this phenomenon was improved, the symptoms of the disease weakened [44]. Andrea Del Fattore found that BMMSC-derived extracellular vesicles could induce T cell apoptosis [45], and then another researcher discovered that BMMSC-derived exosomes could promote the apoptosis of both PBMC and CD3+T cells [30]. These appearances indicated that BMMSCs exert the function in immunity through paracrine microvesicles. In this paper, we pleasantly surprised to find that DPSC-derived exosomes had a stronger property of promoting the apoptosis of CD4+T cells than BMMSC-derived exosomes. Previous studies also shown that BMMSCs could inhibit T cells proliferation [31]; however, Gouveia found that BMMSC-derived extracellular vesicles lost this capacity [46]. Moreover, Wancheng Chen detected that BMMSC-derived exosomes did not affect the proliferation of PBMC and CD3+T cells consistently [30]. Interestingly enough, exosomes derived from BMMSCs and DPSCs had similar ability to inhibit the proliferation of CD4+T cells in our study, albeit slightly. However, the special underlying mechanism of exosomes inhibiting of CD4+T cell proliferation remains to be defined.
In general, this article clarified that DPSC-derived exosomes have stronger immunomodulatory capacity than BMMSC-derived exosomes. However, whether proteins, microRNAs or lncRNAs in exosomes take effects and exactly how they operate under this process need to be researched and discussed deeply. In order to increase the possibility of clinical application, we will carry out animal experiments at the next phase. Altogether, DPSC-derived exosomes are promising to be a novel hope in cell-free therapy for various autoimmune diseases.
References
Friedenstein AJ, Petrakova KV, Kurolesova AI, Frolova GP. Heterotopic of bone marrow. Analysis of precursor cells for osteogenic and hematopoietic tissues. Transplantation. 1968;6(2):230–47.
Romanov YA, Balashova EE, Volgina NE, Kabaeva NV, Dugina TN, Sukhikh GT. Isolation of multipotent mesenchymal stromal cells from cryopreserved human umbilical cord tissue. Bull Exp Biol Med. 2016;160(4):530–4.
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci U S A. 2003;100(10):5807–12.
Yañez R, Lamana ML, García-Castro J, Colmenero I, Ramírez M, Bueren JA. Adipose tissue-derived mesenchymal stem cells have in vivo immunosuppressive properties applicable for the control of the graft-versus-host disease. Stem Cells. 2006;24(11):2582–91.
Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19(3):180–92.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
REB F, Mazurek MS, Soos A, REB SCAF, Mazurek MS, Soos A, et al. Mesenchymal stromal/stem cells in regenerative medicine and tissue engineering. Stem Cells Int. 2018;2018:8031718.
Shi Y, Wang Y, Li Q, Liu K, Hou J, Shao C, et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol. 2018;14(8):493–507.
Swart JF, de Roock S, Hofhuis FM, Rozemuller H, van den Broek T, Moerer P, et al. Mesenchymal stem cell therapy in proteoglycan induced arthritis. Ann Rheum Dis. 2015;74(4):769–77.
Woodworth TG, Furst DE. Safety and feasibility of umbilical cord mesenchymal stem cells in treatment-refractory systemic lupus erythematosus nephritis: time for a double-blind placebocontrolled trial to determine efficacy. Arthritis Res Ther. 2014;16(4):113.
Wu Y, Cao Y, Li X, Xu L, Wang Z, Liu P, et al. Cotransplantation of haploidentical hematopoietic and umbilical cord mesenchymal stem cells for severe aplastic anemia: successful engraftment and mild GVHD. Stem Cell Res. 2014;12(1):132–8.
Dahbour S, Jamali F, Alhattab D, Al-Radaideh A, Ababneh O, Al-Ryalat N, et al. Mesenchymal stem cells and conditioned media in the treatment of multiple sclerosis patients: clinical, ophthalmological and radiological assessments of safety and efficacy. CNS Neurosci Ther. 2017;23(11):866–74.
Xu K, Xiao J, Zheng K, Feng X, Zhang J, Song D, et al. MiR-21/STAT3 signal is involved in odontoblast differentiation of human dental pulp stem cells mediated by TNF-α. Cell Rep. 2018;20(2):107–16.
Feng X, Xing J, Feng G, Sang A, Shen B, Xu Y, et al. Age-dependent impaired neurogenic differentiation capacity of dental stem cell is associated with Wnt/β-catenin signaling. Cell Mol Neurobiol. 2013;33(8):1023–31.
Zhao Y, Wang L, Jin Y, Shi S. Fas ligand regulates the immunomodulatory properties of dental pulp stem cells. J Dent Res. 2012;91(10):948–54.
Sonoda S, Yamaza H, Ma L, Tanaka Y, Tomoda E, Aijima R, et al. Interferon-gamma improves impaired dentinogenic and immunosuppressive functions of irreversible pulpitis-derived human dental pulp stem cells. Sci Rep. 2016;6:19286.
Tomic S, Djokic J, Vasilijic S, Vucevic D, Todorovic V, Supic G, et al. Immunomodulatory properties of mesenchymal stem cells derived from dental pulp and dental follicle are susceptible to activation by toll-like receptor agonists. Stem Cells Dev. 2011;20(4):695–708.
Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79.
Makino Y, Yamaza H, Akiyama K, Ma L, Hoshino Y, Nonaka K, et al. Immune therapeutic potential of stem cells from human supernumerary teeth. J Dent Res. 2013;92(7):609–15.
Yamaza T, Kentaro A, Chen C, Liu Y, Shi Y, Gronthos S, et al. Immunomodulatory properties of stem cells from human exfoliated deciduous teeth. Stem Cell Res Ther. 2010;1(1):5.
Huang GT, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res. 2009;88(9):792–806.
Kay AG, Long G, Tyler G, Stefan A, Broadfoot SJ, Piccinini AM, et al. Mesenchymal stem cell-conditioned medium reduces disease severity and immune responses in inflammatory arthritis. Sci Rep. 2017;7(1):18019.
Jeong JO, Han JW, Kim JM, Cho HJ, Park C, Lee N, et al. Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ Res. 2011;108(11):1340–7.
Yu B, Zhang X, Li X. Exosomes derived from mesenchymal stem cells. Int J Mol Sci. 2014;15:4142–57.
Sarvar DP, Shamsasenjan K, Akbarzadehlaleh P. Mesenchymal stem cell-derived exosomes: new opportunity in cell free therapy. Adv Pharm Bull. 2016;6(3):293–9.
Yu J, He H, Tang C, Zhang G, Li Y, Wang R, et al. Differentiation potential of STRO-1+ dental pulp stem cells changes during cell passaging. BMC Cell Biol. 2010;11:32.
Zhang B, Yeo RWY, Lai RC, Sim EWK, Chin KC, Lim SK. Mesenchymal stromal cell exosome-enhanced regulatory T-cell production through an antigen-presenting cell-mediated pathway. Cytotherapy. 2018;20(5):687–96.
Perez-Hernandez J, Redon J, Cortes R. Extracellular vesicles as therapeutic agents in systemic lupus erythematosus. Int J Mol Sci. 2017;18(4):E717.
Harrell CR, Simovic Markovic B, Fellabaum C, Arsenijevic A, Djonov V, Arsenijevic N, et al. Therapeutic potential of mesenchymal stem cell-derived exosomes in the treatment of eye diseases. Adv Exp Med Biol. 2018;1089:47–57.
Chen W, Huang Y, Han J, Yu L, Li Y, Lu Z, et al. Immunomodulatory effects of mesenchymal stromal cells-derived exosome. Immunol Res. 2016;64(4):831–40.
Castro-Manrreza ME, Montesinos JJ. Immunoregulation by mesenchymal stem cells: biological aspects and clinical applications. J Immunol Res. 2015;2015(2):394917.
Tan L, Wu H, Liu Y, Zhao M, Li D, Lu Q. Recent advances of exosomes in immune modulation and autoimmune diseases. Autoimmunity. 2016;49(6):357–65.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441(7090):235–8.
Romano M, Tung SL, Smyth LA, Lombardi G. Treg therapy in transplantation: a general overview. Transpl Int. 2016;30(8):745–53.
Van Hamburg JP, Tas SW. Molecular mechanisms underpinning T helper 17 cell heterogeneity and functions in rheumatoid arthritis. J Autoimmun. 2018;87:69–81.
Nistala K, Wedderburn LR. Th17 and regulatory T cells: rebalancing pro- and anti-inflammatory forces in autoimmune arthritis. Rheumatology (Oxford). 2009;48(6):602–6.
Sun L, Fu J, Zhou Y. Metabolism controls the balance of Th17/T-regulatory cells. Front Immunol. 2017;8:1632.
Sakaguchi S. Naturally arising CD4+ regulatory T cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531–62.
Hong JW, Lim JH, Chung CJ, Kang TJ, Kim TY, Kim YS, et al. Immune tolerance of human dental pulp-derived mesenchymal stem cells mediated by CD4+CD25+FoxP3+ regulatory T-cells and induced by TGF-β1 and IL-10. Yonsei Med J. 2017;58(5):1031–9.
Kondo Y, Yokosawa M, Kaneko S, Furuyama K, Segawa S, Tsuboi H, et al. Review: transcriptional regulation of CD4+ T cell differentiation in experimentally induced arthritis and rheumatoid arthritis. Arthritis Rheum. 2018;70(5):653–61.
Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171(11):6173–7.
Iwanami K, Matsumoto I, Tanaka-Watanabe Y, Inoue A, Mihara M, Ohsugi Y, et al. Crucial role of the interleukin-6/interleukin-17 cytokine axis in the induction of arthritis by glucose-6-phosphate isomerase. Arthritis Rheum. 2008;58(3):754–63.
Kondo Y, Yao Z, Tahara M, Iizuka M, Yokosawa M, Kaneko S, et al. Involvement of RORgammat-overexpressing T cells in the development of autoimmune arthritis in mice. Arthritis Res Ther. 2015;17:105.
Chen SY, Wu CL, Lai MD, Lin CC, Yo YT, Jou IM, et al. Amelioration of rat collagen-induced arthritis through CD4+ T cells apoptosis and synovial interleukin-17 reduction by indoleamine 2,3-dioxygenase gene therapy. Hum Gene Ther. 2011;22(2):145–54.
Del Fattore A, Luciano R, Pascucci L, Goffredo BM, Giorda E, Scapaticci M, et al. Immunoregulatory effects of mesenchymal stem cell-derived extracellular vesicles on T lymphocytes. Cell Transplant. 2015;24(12):2615–27.
Gouveia de Andrade AV, Bertolino G, Riewaldt J, Bieback K, Karbanová J, Odendahl M, et al. Extracellular vesicles secreted by bone marrow-and adipose tissue-derived mesenchymal stromal cells fail to suppress lymphocyte proliferation. Stem Cells Dev. 2015;24(11):1374–6.
Acknowledgments
We appreciate Dan Huang, Jingwen Xiao, and Yihua Song for collecting samples and providing method of extracting DPSCs and appreciate Junling Yang for her assistance with technical help in flow cytometry. We also thank Xingyu Li for helping us buy experimental reagents.
Funding
This work was supported by National Natural Science Foundation of China (81871278; 81671616; 81471603), Science and Technology Projects of Jiangsu Province (BE2018671), the project of “333 National Science Foundation” of Jiangsu Province (BRA2016527), Science and Technology Projects of Nantong City (MS1201712-2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committees and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ji, L., Bao, L., Gu, Z. et al. Comparison of immunomodulatory properties of exosomes derived from bone marrow mesenchymal stem cells and dental pulp stem cells. Immunol Res 67, 432–442 (2019). https://doi.org/10.1007/s12026-019-09088-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-019-09088-6