
Abstract
Thyroid cancer, predominantly of papillary histology (PTC), is a common cancer mostly diagnosed sporadically. Hereditary PTC is encountered in ~ 5% of cases and may present at an earlier age, with greater risks of metastasis and recurrence, compared with sporadic cases. The molecular basis of hereditary PTC is unknown in most cases. In this study, the genetic basis of hereditary PTC in three Brazilian families was investigated. Whole exome sequencing (WES) was carried out for probands in each family, and validated, pathogenic/likely pathogenic sequence variants (P/LPSVs) were genotyped in additional family members to establish their putative pathogenic role. Overall, seven P/LPSVs in seven novel genes were detected: p.D283N*ANXA3, p.Y157S*NTN4, p.G172W*SERPINA1, p.G188S*FKBP10, p.R937C*PLEKHG5, p.L32Q*P2RX5, and p.Q76*SAPCD1. These results indicate that these novel genes are seemingly associated with hereditary PTC, but extension and validation in other PTC families are required.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thyroid cancer is the most common endocrine malignancy, and its incidence has continuously increased worldwide, from age standardized rate (ASR) of 4.6/100,000 in 1974–1977 to 15.8/100,000 in 2012–2016 [1]. In Brazil, an estimated 9610 new cases of thyroid cancer (8040 in females) were expected to be diagnosed in 2018 (http://www1.inca.gov.br/estimativa/2018/sintese-de-resultados-comentarios.asp; accessed 9th September 2019). The most common histological type is papillary thyroid carcinoma (PTC), comprising ~ 85% of all cases, and in most patients, there is no discernible family history of thyroid malignancy. Familial non-medullary thyroid cancer (FNMTC) is reported in ~ 5% of all thyroid cancers, with only a minority associated with specific cancer susceptibility syndromes, such as Cowden syndrome (MIM No. 158350) and Carney complex (MIM No. 160980) with well-defined driver germline mutations in the PTEN and PRKAR1A genes, respectively [2].
Studies that focused on defining the genetic basis of non-syndromic FNMTC have identified germline mutations in several candidate genes including TITF-1/NKX2.1 [3], SRGAP1 [4], FOXE1 [5], HABP2 [6,7,8], and SRRM2 [9]. Moreover, miRNAs [10] and four chromosomal loci (1q21, 6q22, 8p23.1-p22, 8q24) with no known candidate genes [4, 10, 11] were reportedly associated with FNMTC. Notably, the p.G534E*HABP2 variant (rs7080536) has been detected in some FNMTC families, although co-segregation was incomplete in some families, and this sequence variant is also present in controls with an overall population allele frequency of 2.223e-02 (https://gnomad.broadinstitute.org/variant/10-115348046-G-A?dataset=gnomad_r2_1) [6, 8, 12,13,14].
Thus, while some FNMTC are accounted for by known genes and chromosomal loci, the molecular basis for most FNMTC remains elusive. In the present study, we applied whole-exome sequencing (WES) methodology to define germline pathogenic/likely pathogenic sequence variants (P/LPSVs) in three Brazilian families with autosomal dominant FNMTC.
Materials and Methods
Patients’ Identification and Recruitment
The study encompassed individuals from three Brazilian families displaying a likely autosomal dominant inheritance pattern of PTC in at least two generations (Fig. 1). These families were referred to the School of Medicine, Universidade Federal de Minas Gerais. The experimental protocols were approved by the Institutional Review Board at the Universidade Federal de Minas Gerais (ETIC 367/07). All study participants gave written informed consent before participation.

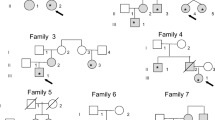
Pedigree status from families A, B, and C with respect to papillary thyroid cancer and the genotypes for each heterozygous mutation identified. Circles and squares represent female and male members, respectively. The proband is indicated by a black arrow. Deceased members are represented by diagonal lines. a Pedigree from family A; b pedigree from family B; c pedigree from family C
DNA Extraction from Peripheral Blood Leukocytes and Paraffin-Embedded Tissue
Genomic DNA was extracted from peripheral blood samples (II.2, III.1, III.2, and III.3, family A; I.2, II.1, and II.2, family B; I.2, II.1, II.2, and III.1, family C—Fig. 1) using standard protocols. DNA sample (II.1, family C) from paraffin-embedded tissue (FFPE) thyroid tumor tissue was extracted according to manufacturer’s instructions (QIAamp DNA FFPE Tissue Kit, Qiagen, Germantown, MD). The quality and quantity of each DNA sample were examined by NanoDrop ND-2000 UV-Vis Spectrophotometer (Thermofisher, Waltham, MA).
Whole-Exome Sequencing
DNA was subjected to whole exome capturing and sequencing using the Roche NimbleGen V2 chip (Madison, WI) or Nextera (San Diego, CA) with the Illumina HiSeq2000 sequencing platform (San Diego, CA). Sanger sequencing was used to validate the candidate variants identified by WES in these affected family members.
Variant Calling and Annotation
For each of the studied samples, raw sequence files were prepared using the Genome Analysis Tool Kit (GATK). Each fastq file was aligned against the human hg19/GRCh37 reference genome and a variant call format (VCF) file generated for each sample. PCR duplicates were removed using Picard (http://picard.sourceforge.net/); reads around known and detected indels were realigned, and base quality was recalibrated using GATK.
All generated VCF files were analyzed using HIPAA-compliant Variantyx Genomic Intelligence platform and Genomic Intelligence diagnostic console (https://www.variantyx.com/) (Variantyx Inc., Framingham, MA, 01701, USA), Genome AnalysisTK 2.3-9 (Theragen Etex Bioinformatics Team), Mendel, MD (https://mendelmd.org/), and Ingenuity Variant Analysis
Sequence variants associated with gain or loss of function, compound heterozygotes, heterozygous ambiguous, haploinsufficient, homozygous or hemizygous, located at a microRNA binding site; frameshift, in-frame indel, or stop codon changes and also assigned “pathogenic/likely pathogenic” by PROVEAN [15], SIFT, Polyphen-2, and CADD [16] algorithms were selected for further analyses.
Genes that harbored P/LPSVs were queried for potential relevance to thyroid cancer tumorigenesis by applying the following criteria: (i) predicted functional consequences using the Ensembl database (https://www.ensembl.org/index.html); (ii) publications relating each gene to cancer, based on PUBMED search of the gene name; (iii) pathway annotation, which includes all pathways in which a given gene product has been involved at Genecards (http://www.genecards.org/), OMIM (https://www.omim.org/), and UniProt (http://www.uniprot.org/) databases; (iv) the occurrence of deleterious or possible deleterious somatic mutations in the COSMIC database (http://cancer.sanger.ac.uk/cosmic); (v) mouse models (when available), gathered from KOMP (https://www.mousephenotype.org/aboutikmc/about-komp); Mouse Genome Informatics (MGI; http://www.informatics.jax.org/) and HomoloGene (http://www.ncbi.nlm.nih.gov/homologene); and (vi) minor allele frequency of less than 1% in gnomAD database (http://genomad.broadinstitute.org; ac). Following these filtering steps, data were manually reviewed to generate final candidate gene list.
WES Sequence Data Validation and Mutation Analysis
Validation of the candidate P/LPSVs on DNA extracted from all consenting individuals was carried out using Sanger sequencing. The same variants were also sequenced in an independent series of 90 ethnically matched elderly controls (≥ 65 years of age) who consented to participate in the study using a previously approved ethics protocol (ETIC 367/07).). In addition, allele frequencies of all pathogenic variants were queried with data derived from the 1000 Genomes Project (1000G), Exome Variant Server (ESP), 10,000 UK Genome (UK10K), The Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/; accessed 6th January 2020) as well as the Brazilian Initiative on Precision Medicine database (http://bipmed.iqm.unicamp.br/genes; accessed 6th January 2020).
Results
Clinical Cases Study
The study included 11 individuals from three Brazilian families (at least three affected members from each family) displaying a likely autosomal dominant inheritance pattern of PTC in at least two successive generations (Fig. 1). Of relevance is the fact that all studied affected patients presented with classical and/or follicular variants of PTC. Table 1 summarizes clinical, histological, and molecular data.
The proband of family A (II.2) was diagnosed with PTC at age 32 years. Her family history (Fig. 1a) included a mother (I.2) diagnosed with metastatic Hurthle Cell carcinoma at age 38 years, two sons (III.1 and III.2) diagnosed with PTC at age 39 and 36 years, respectively, a son who died of leukemia at age 14 years (III.4), and another son aged 36 (III.3) with thyroid nodules who refused biopsy. Pathology report of tumor from II.2 showed infiltrating papillary carcinoma (follicular variant). Immunohistochemistry studies demonstrated positivity for Cytokeratin-7, TTF-1 and thyroglobulin in tumor cells.
The proband (II.1) of family B (Fig. 1b) presented a multifocal PTC at age 43 years. His sister (II.2) and his mother (I.2) were also diagnosed with PTC at age 22 and 70 years, respectively. In addition, his mother (I.2) and another’s sister (II.3) were diagnosed with breast cancer at 73 and 52 years, respectively. Pathology reports of tumors from the proband (II.1) and II.2 showed differentiated infiltrating (classical variant) papillary carcinoma.
The proband (II.1) of Family C (Fig. 1c) was diagnosed with the classical variant of PTC at age 18 years. Her family history included a mother (I.2) and two sisters (II.2 and II.3) who were also diagnosed with PTC at 65, 47, and 45 years of age, respectively. Her niece (III.1) is unaffected. Patients II.2 and II.3 had follicular and classical variants of PTC, respectively.
All patients affected by PTC underwent total thyroidectomy and radioactive iodine treatment. None of these patients had a past history of ionizing radiation exposure.
WES Analysis
Family A
Exome sequencing of the proband (II.2) yielded a total of 49,402 variants in 17,359 genes, with an average read depth of 138× per base and a mean base call quality of 1379. After excluding common SNPs with minor allele frequency (MAF) of ≥ 1%, a total of 488 genes with mutations predicted to have a potentially damaging effect were identified within the coding regions by Ingenuity Variant Analysis. Similar analysis using Mendel, MD yielded a list of 160 genes. Overall, 39 candidate genes were identified by both analyses tools. All 39 genes were queried for any supportive evidence to their putative involvement in thyroid carcinogenesis. Following this selection step, four variants in four genes were selected for validation. Only two mutations in two genes were validated: p.D283N*ANXA3 (MIM No. 106490) and p.Y157S*NTN4 (MIM No. 610401). These two germline variants were also identified in the two other PTC-affected individuals in family A (III.1 and III.2) and in the individual with thyroid nodules (III.3) (Fig. 1a); p.D283N*ANXA3 has been reported in the gnomAD (3.99e−6; 8.59e−4) database. Y157S*NTN4 was not present in any of the 90 healthy ethnically matched controls as well as in the gnomAD and Brazilian Initiative on Precision Medicine databases.
Family B
Exome sequencing of patient II.1 resulted in 49,338 variants in 17,478 genes, with an average read depth of X130−/base and mean call quality of 1472. Ingenuity Variant Analysis identified 112 P/LPSVs (in 91 genes) most likely to be implicated in PTC pathogenesis. Similar analysis using Mendel, MD yielded 227 P/LPSVs in 158 genes. Only two P/LPSVs were shared by both pipelines. Sanger sequencing validated only the c.1087G>T;p.G172W *SERPINA1 (MIM No 107400) gene variant in the proband and all genotyped affected family members (I.2, II.1, and II.2) with PTC. None of the Brazilian population controls harbored the c.1087G>T;p.G172W*SERPINA1 mutation which was not reported in the Brazilian Initiative on Precision Medicine database. The rate of this variant in the gnomAD database was 8.59e−4.
Family C
Exome sequencing of PTC-affected patients I.2 (mother) and II.1 (daughter) in family C resulted in 57,436 variants in 15,060 genes. The mean base call quality was 1022 and average read depth was X130/base. Using the above outlined filtering steps, Ingenuity Variant Analysis identified 252 P/LPSVs in 194 genes that were shared by both patients. Similar analysis using Mendel, MD led to a list of 198 variants in 132 genes. Twelve seemingly P/LPSVs were commonly shared by both pipelines. Of these, four heterozygous P/LPSVs were validated by Sanger sequencing: c.666G>A;p.G188S*FKBP10 (MIM No. 607063), c.2815C>T;p.R937C*PLEKHG5 (MIM No. 611101), c.494T>A;p.L32Q*P2RX5 (MIM No. 602836), and c.285C>T;p.Q76*SAPCD1. These four PLSVs were present in all three affected genotyped family members (Fig. 1) as well as in a heterozygous state in a tumor sample from patient II.1. This latter finding implies that there was no allelic loss of the mutant allele in the tumor sample, at least in the region that was genotyped.
None of 90 ethnically matched controls nor did the unaffected case (III.1, family C) carry any of these validated P/LPSVs in these four genes. The rates of these variants in the gnomAD database are 1.77e−5 (FKBP10), 4.12e−03 (P2RX5), and 8.42e−4 (SAPCD1). None were reported in the Brazilian Initiative on Precision Medicine database or in mouse databases.
Discussion
In this study, P/LPSVs in the ANXA3, NTN4, SERPINA1, FKBP10, PLEKHG5, P2RX5, and SAPCD1 genes emerged as candidates for being FNMTC susceptibility genes. Notably, the previously reported FNMTC-associated predisposition genes (AARS, AGK, ALB, ATP13A2, CDH11, CDS2, CIS, CTDSP1, DR6, EDC4, EFCAB8, EIF3A, FGD6, FGFR4, FOXA3, FOXE1, GPR187, HABP2, IDE, ITGGAD, KDSR, KLHL3, LZTR1, MAPKAK3, NAPB, NFRKB, NHLH1, NSMF, PARP4, PDPR, RNF213, SALL4, SRGAP1, SMARCD3, SRRM2, SVIL, TERT, THBS4, TINF2, TITF-1/NKX2.1, ZNF17) [6, 8, 13, 14, 17] were not present in any of the families reported herein and therefore excluded as contributing to PTC phenotype. Moreover, despite the association of breast cancer and PTC in family B, raising the possibility of Cowden syndrome, the PTEN gene as well as the SDH family of genes were not mutated in that family, despite adequate coverage (> 100×).
The ANXA3 gene, located at 4q21.21, encodes calcium-dependent phospholipid-binding proteins—Annexin A3. Abnormal expression of ANXA3 plays a role in different cellular processes, associated with tumorigenesis: cell proliferation, apoptosis, invasion, metastasis, and drug resistance [18]. In vitro and in vivo assays demonstrated increased of thyroid cancer cells proliferation concomitant with reduced ANXA3 expression [18, 19].
Netrin-4 (NTN4) gene, located at 12q22, encodes for NTN4 proteins—basement membrane components [20]. NTN4 may play a role in development and progression of several cancer types such as breast [21], gastric [22], and melanoma [23]. Low NTN4 expression may promote tumor growth, cellular proliferation, and angiogenesis inhibition [21, 24]. NTN4 is markedly downregulated in prostate [25], breast [21], melanoma [23], and cervical cancers [26]. The involvement of NTN4 gene in PTC has not been reported.
SERPINA1, also known as α1-AntiTrypsin (AAT), located at 14q32.13, is highly expressed in colorectal cancer [27], cutaneous squamous cell carcinoma [28], and papillary thyroid carcinoma [29]. Although the mechanistic role of SERPINA1 in thyroid tumorigenesis is not fully understood, cell invasion and migration in colorectal cancer have been associated to SERPINA1 upregulation of fibronectin [27]. Furthermore, the C-terminal 26-residue peptide of SERPINA1 presents a mitogenic action, stimulating malignant cell proliferation [28].
The suppressor APC domain containing 1 (SAPCD1) gene, located to 6p21.33, is associated with DNA repair [30]. Expression of SAPCD1 is somatically decreased in breast cancer, and a deleterious variant (p.Q76*) has been reported to be associated with lung cancer predisposition [31].
The PLEKHG5 (also known as TECH or Syx) gene localizes to 1p36.31, a region frequently associated with somatic tumor gene rearrangements [32]. PLEKHG5 encodes for a guanine nucleotide exchange factor that selectively couples RhoA activation at leading cell edges with Dia1 signaling and ROCK suppression. PLEKHG5 leads to migration of human cells, a feature suggestive of its involvement in malignant transformation [33]. Indeed, p.R937C*PLEKHG5 somatic mutation has been detected in urinary tract carcinoma (COSMIC database; http://cancer.sanger.ac.uk/cosmic).
The FKBP10 gene, located at 17q21.2 encodes for the FK506 binding protein 10 [34]. Ge et al. [35] reported that FKBP10 is present at higher levels in renal cancer cells compared with non-tumoral cells, inducing cell cycle progression, cell proliferation, migration, and invasion. FKBP10 expression is decreased in epithelial ovarian carcinomas [36]. Notably, 191 non-synonymous somatic FKBP10 point mutations are noted in the COSMIC database but not the p.G188S mutation.
The P2RX5 gene localizes to 17p13.2 and encodes an ion channel receptor for adenosine 5′-triphosphate (ATP) and adenosine, which acts as extracellular purinergic signaling molecules [37]. Overexpression of P2RX5 is reported in high grade bladder cancer56, basal cell and squamous carcinomas [38], and colon cancer [39]. It was suggested that activated P2RX5 receptors may be associated with growth inhibition of cancer cells, switching the cell cycle from proliferation into a state of differentiation [37]. Presently, mutations in this gene have not been shown to contribute to cancer formation and/or predisposition.
The association of papillary carcinoma and Hurthle Cell carcinoma in only one patient from one single family (I:2, family A) may represent a rare co-occurrence only of a sporadic Hurthle Cell case in a FNMTC.
Although germline mutations in more than one gene were detected in each family, it seems unlikely that the inheritance pattern is multigenic, as these families were selected based on the unusual clustering of thyroid cancer in more than one generation a pattern suggestive of autosomal dominant FNMTC. The inherent inability to single out one gene variant stems in part from the size of the families and the lack of unaffected individuals who could be genotyped. Furthermore, there have been no published functional analyses or animal models that examined the putative role of these genes in FNMTC predisposition.
The list of genes described in this study should be considered tentative at best, as there are no functional studies or animal models that prove the direct involvement of these mutations in thyroid tumorigenesis. Moreover, the list for genes that have been reported in FNMTC is long and exhaustive [3,4,5,6,7,8,9,10,11,12,13,14, 17, 19, 29]. No single gene seems to account for more than one or a handful of families. One can speculate as to the reasons for this genetic reality in FNMTC predisposition: inclusion of phenocopies in the analyses, oligogenic inheritance, population genetic-based differences, to name a few [40,41,42,43]. This feature of multiple genes in inherited cancer syndrome is similar to inherited prostate cancer where no single gene accounts for more than 5% of all inherited cases [42].
Conclusion
In conclusion, our results suggest that FKBP10, PLEKHG5, P2RX5, SAPCD1, ANXA3, NTN4, and SERPINA1 may contribute to susceptibility to non-medullary thyroid cancer. The validation of these results and affirming the possible contribution of the genes reported herein to other FNMTC families as well as the exact pathogenic mechanism await further studies.
References
Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, eds (2016) SEER Cancer Statistics Review, 1975–2016, National Cancer Institute. Bethesda, MD. Available from https://seer.cancer.gov/csr/1975_2016/.
Giordano TJ. Genomic hallmarks of thyroid neoplasia (2018) Annu Rev Pathol 13:141–162.
Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, Leon TY, Cherny SS, Tsai SY, Lo CY, Khoo US, Tam PK, Garcia-Barceló MM (2009) A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2.1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst 101:162–175.
He H, Nagy R, Liyanarachchi S, Jiao H, Li W, Suster S, Kere J, de la Chapelle A (2009) A susceptibility locus for papillary thyroid carcinoma on chromosome 8q24. Cancer Research 69:625–631.
Pereira JS, da Silva JG, Tomaz RA, Pinto AE, Bugalho MJ, Leite V, Cavaco BM (2015) Identification of a novel germline FOXE1 variant in patients with familial non-medullary thyroid carcinoma (FNMTC). Endocrine 49:204–214.
Gara SK, Jia L, Merino MJ, Agarwal SK, Zhang L, Cam M, Patel D, Kebebew E (2015) Germline HABP2 mutation causing familial nonmedullary thyroid cancer. N Engl J Med 373:448–455.
Tomsic J, He H, de la Chapelle A (2015) HABP2 Mutation and nonmedullary thyroid cancer. N Engl J Med 373:2086.
Tomsic J, Fultz R, Liyanarachchi S, He H, Senter L, de la Chapelle A (2016) HABP2 G534E variant in papillary thyroid carcinoma. PLoS One 11:e0146315.
Tomsic J, He H, Akagi K, Liyanarachchi S, Pan Q, Bertani B, Nagy R, Symer DE, Blencowe BJ, de la Chapelle A (2015) A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci Rep 5:10566.
Cavaco BM, Batista PF, Sobrinho LG, Leite V (2008) Mapping a new familial thyroid epithelial neoplasia susceptibility locus to chromosome 8p23.1-p22 by high-density single-nucleotide polymorphism genome-wide linkage analysis. J Clin Endocrinol Metab 93:4426–4430.
Suh I, Filetti S, Vriens MR, Guerrero MA, Tumino S, Wong M, Shen WT, Kebebew E, Duh QY, Clark OH (2009) Distinct loci on chromosome 1q21 and 6q22 predispose to familial nonmedullary thyroid cancer: a SNP array-based linkage analysis of 38 families. Surgery 146:1073–1080.
Sahasrabudhe R, Stultz J, Williamson J, Lott P, Estrada A, Bohorquez M, Palles C, Polanco-Echeverry G, Jaeger E, Martin L, Magdalena Echeverry M, Tomlinson I, Carvajal-Carmona LG; TCUKIN (2016) The HABP2 G534E variant is an unlikely cause of familial non-medullary thyroid cancer. J Clin Endocrinol Metab 10:1098–1103.
de Mello LEB, Araujo AN, Alves CX, de Paiva FJP, Brandão-Neto J, Cerutti JM (2017) The G534E variant in HABP2 is not associated with increased risk of familial nonmedullary thyroid cancer in Brazilian kindreds. Clin Endocrinol (Oxf) 87:113–114.
Pinheiro M, Drigo SA, Tonhosolo R, Andrade SCS, Marchi FA, Jurisica I, Kowalski LP, Achatz MI, Rogatto SR (2017) HABP2 p.G534E variant in patients with family history of thyroid and breast cancer. Oncotarget 8:40896–40905.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP (2012)Predicting the functional effect of amino acid substitutions and indels. PLoS One 7:e46688.
Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X (2015) Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 24:2125–2137.
Wang Y, Liyanarachchi S, Miller KE, Nieminen TT, Comiskey DF Jr, Li W, Brock P, Symer DE, Akagi K, DeLap KE, He H, Koboldt DC, de la Chapelle A (2019) Identification of rare variants predisposing to thyroid cancer. Thyroid 29:946–955.
Du R, Liu B, Zhou L, Wang D, He X, Xu X, Zhang L, Niu C, Liu S (2018) Downregulation of annexin A3 inhibits tumor metastasis and decreases drug resistance in breast cancer. Cell Death Dis 9:126.
Jung EJ, Moon HG, Park ST, Cho BI, Lee SM, Jeong CY, Ju YT, Jeong SH, Lee YJ, Choi SK, Ha WS, Lee JS, Kang KR, Hong SC (2010) Decreased annexin A3 expression correlates with tumor progression in papillary thyroid cancer. Proteomics Clin Appl 4:528–537.
Lambert E, Coissieux MM, Laudet V, Mehlen P (2012) Netrin-4 acts as a pro-angiogenic factor during zebrafish development. J Biol Chem 287:3987–3999.
Xu X, Yan Q, Wang Y, Dong X (2017) NTN4 is associated with breast cancer metastasis via regulation of EMT-related biomarkers. Oncol Rep 37:449–457.
Lv B, Song C, Wu L, Zhang Q, Hou D, Chen P, Yu S, Wang Z, Chu Y, Zhang J, Yang D, Liu J (2014) Netrin-4 as a biomarker promotes cell proliferation and invasion in gastric cancer. Oncotarget 6:9794–9806.
Jayachandran A, Prithviraj P, Lo PH, Walkiewicz M, Anaka M, Woods BL, Tan B, Behren A, Cebon J, McKeown SJ (2016) Identifying and targeting determinants of melanoma cellular invasion. Oncotarget 7:41186–41202.
Eveno C, Contreres JO, Hainaud P, Nemeth J, Dupuy E, Pocard M (2013) Netrin-4 overexpression suppresses primary and metastatic colorectal tumor progression. Oncol Rep 29:73–78.
Latil A, Chêne L, Cochant-Priollet B, Mangin P, Fournier G, Berthon P, Cussenot O (2003) Quantification of expression of netrins, slits and their receptors in human prostate tumors. Int J Cancer 103:306–315.
Zhang J, Zheng F, Yu G, Yin Y, Lu Q (2013) miR-196a targets netrin 4 and regulates cell proliferation and migration of cervical cancer cells. Biochem Biophys Res Commun 440:582–588.
Kwon CH, Park HJ, Choi JH, Lee JR, Kim HK, Jo HJ, Kim HS, Oh N, Song GA, Park DY (2015) Snail and serpinA1 promote tumor progression and predict prognosis in colorectal cancer. Oncotarget 6:20312–20326.
Farshchian M, Kivisaari A, Ala-Aho R, Riihilä P, Kallajoki M, Grénman R, Peltonen J, Pihlajaniemi T, Heljasvaara R, Kähäri VM (2011) Serpin peptidase inhibitor clade A member 1 (SerpinA1) is a novel biomarker for progression of cutaneous squamous cell carcinoma. Am J Pathol 179:1110–1119.
Vierlinger K, Mansfeld MH, Koperek O, Nöhammer C, Kaserer K, Leisch F (2011) Identification of SERPINA1 as single marker for papillary thyroid carcinoma through microarray meta analysis and quantification of its discriminatory power in independent validation. BMC Med Genomics 4:30.
Matta J, Morales L, Dutil J, Bayona M, Alvarez C, Suarez E (2013) Differential expression of DNA repair genes in Hispanic women with breast cancer. Mol Cancer Biol 1:54.
Liu Y, Lusk CM, Cho MH, Silverman EK, Qiao D, Zhang R, Scheurer ME, Kheradmand F, Wheeler DA, Tsavachidis S, Armstrong G, Zhu D, Wistuba II, Chow CB, Behrens C, Pikielny CW, Neslund-Dudas C, Pinney SM, Anderson M, Kupert E, Bailey-Wilson J, Gaba C, Mandal D, You M, de Andrade M, Yang P, Field JK, Liloglou T, Davies M, Lissowska J, Swiatkowska B, Zaridze D, Mukeriya A, Janout V, Holcatova I, Mates D, Milosavljevic S, Scelo G, Brennan P, McKay J, Liu G, Hung RJ, Christiani DC, Schwartz AG, Amos CI, Spitz MR (2018) Rare variants in known susceptibility loci and their contribution to risk of lung cancer. J Thorac Oncol 13:1483–1495.
Garnaas MK, Moodie KL, Liu ML, Samant GV, Li K, Marx R, Baraban JM, Horowitz A, Ramchandran R (2008) Syx, a RhoA guanine exchange factor, is essential for angiogenesis in vivo. Circ Res 103:710–716.
Dachsel JC, Ngok SP, Lewis-Tuffin LJ, Kourtidis A, Geyer R, Johnston L, Feathers R, Anastasiadis PZ (2013) The Rho guanine nucleotide exchange factor Syx regulates the balance of dia and ROCK activities to promote polarized-cancer-cell migration. Mol Cell Biol 33:4909–4918.
Romano S, D'Angelillo A, Romano MF (2015) Pleiotropic roles in cancer biology for multifaceted proteins FKBPs. Biochim Biophys Acta 1850:2061–2068.
Ge Y, Xu A, Zhang M, Xiong H, Fang L, Zhang X, Liu C, Wu S (2016) FK506 binding protein 10 is overexpressed and promotes renal cell carcinoma. Urol Int 98:169–176.
Quinn MC, Wojnarowicz PM, Pickett A, Provencher DM, Mes-Masson AM, Davis EC, Tonin PN (2013) FKBP10/FKBP65 expression in high-grade ovarian serous carcinoma and its association with patient outcome. Int J Oncol 42:912–920.
Di Virgilio F, Adinolfi E (2017) Extracellular purines, purinergic receptors and tumor growth. Oncogene 36:293–303.
Greig AV, Linge C, Healy V, Lim P, Clayton E, Rustin MH, McGrouther DA, Burnstock G (2013) Expression of purinergic receptors in non-melanoma skin cancers and their functional roles in A431 cells. J Invest Dermatol 121:315–327.
Gao P, He M, Zhang C, Geng C (2018) Integrated analysis of gene expression signatures associated with colon cancer from three datasets. Gene 654:95–102.
Bauer AJ. Clinical behavior and genetics of nonsyndromic, familial nonmedullary thyroid cancer. Front Horm Res. 2013;41:141–148. doi: https://doi.org/10.1159/000345674
Kousi M, Katsanis N. Genetic modifiers and oligogenic inheritance. Cold Spring Harb Perspect Med. 2015;5(6). pii: a017145. doi: https://doi.org/10.1101/cshperspect.a017145.
Peiling Yang S, Ngeow J. Familial non-medullary thyroid cancer: unraveling the genetic maze. Endocr Relat Cancer. 2016 Dec;23(12):R577-R595
Dudbridge F, Brown SJ, Ward L, Wilson SG, Walsh JP. How many cases of disease in a pedigree imply familial disease? Ann Hum Genet. 2018 Mar;82(2):109–113. doi: https://doi.org/10.1111/ahg.12222.
Acknowledgments
We thank all family members for their support.
Funding
This work was partially funded by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq No. 405053/2013-4) and CAPES, Brazil
Author information
Authors and Affiliations
Contributions
Marta Sarquis and Debora C. Moraes contributed equally to this manuscript.
Corresponding author
Ethics declarations
Ethics Statement
The experimental protocols were approved by the Institutional Review Board at the Universidade Federal de Minas Gerais (ETIC 367/07).
Disclosure Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sarquis, M., Moraes, D.C., Bastos-Rodrigues, L. et al. Germline Mutations in Familial Papillary Thyroid Cancer. Endocr Pathol 31, 14–20 (2020). https://doi.org/10.1007/s12022-020-09607-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12022-020-09607-4