
Abstract
Purpose
The definition of growth response in growth hormone (GH)-treated children is controversial. This study aims at: (1) evaluating short-term and long-term efficacy of GH treatment in a cohort of short children with GH deficiency (GHD); (2) assessing and compare various poor response criteria; (3) identifying predictive factors of growth response.
Methods
Our study included 94 children, affected by isolated GHD and treated with GH until they reached final height. Criteria used for calculating the proportion of poor responders to GH for the first year were gain in height (ΔHt) SDS < 0.5 (“Bang criterion”), <0.3 or <0.4 SDS for less-severe and severe GHD, respectively (“Ranke criterion”), height velocity (HV) < mean –1 SDS (“Bakker criterion”); for adult height “Cianfarani criterion” was total ΔHt < 1 SDS.
Results
After 1 year of treatment we defined “poor responders” 55.3% of patients according to Bang criterion, 40.9% according to Bakker criterion and 23.4% according to Ranke criterion. At the end of the treatment, poor responders according to Cianfarani criterion were 22.34%; almost everyone in our population (97.9%) achieved mMid-parental height (MPH). Median final Ht was −1.11 SDS. Our analysis revealed a significant negative association between ΔHt and age at diagnosis.
Conclusions
Bang criterion generated the highest number of poor responders, but had a low negative predictive value (67.5%); Ranke and Cianfarani criteria displayed similar rate of poor response. There is no reliable predictive factor of growth hormone response. However, almost all children treated reached MPH, suggesting good treatment efficacy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Growth hormone deficiency (GHD) is one of the most frequent endocrinological disorder in children with short stature, but we are still limited in making a definitive diagnosis of GHD, due to lack of reliable diagnostic tools and criteria [1]. Growth hormone replacement treatment helps patients to achieve a normal adult height for the general population and for their genetic target [2]. It is clear from reports that individual height response may vary considerably even with individualized treatment regimens, mainly depending on chronological age at the beginning of therapy, severity of the deficiency at GH stimulation tests and stage of pubertal development [3]. Management of children treated with GH should include evaluation of growth response after 1 year, because this seems to be the most important factor in determining the overall success of treatment [4]. If response is poor, further investigation or discontinuation of treatment should be considered [5]. However, despite more than 50 years of experience of GH treatment in children with short stature, there is still some uncertainty about the definition of “poor response”.
A response to GH therapy may be assessed by comparing observed and predicted outcomes. In the 1990s, algorithms aiming at predicting growth during the course of GH treatment were developed for different diagnosis groups (idiopathic short stature—ISS [6], Turner syndrome [7], GHD [8], and small for gestational age—SGA [9]), but they explain only 40–60% of the observed response variability. Consequently, the prediction may be quite far from the actual response in the individual patient. A poor response could also be described using a specific auxological biometric cutoff value. Bakker et al. [10] created diagnosis-, sex-, and age-dependent curves for height velocity (HV) on GH treatment and suggested that patients with a first-year HV < −1 SD of the corresponding category should be labeled as poor responders. Similarly, Ranke suggested that a gain in height SDS < 0.4 in a patient with severe GHD (defined as GH peak < 7 ng/mL at stimulation tests) and a gain <0.3 in patients with less-severe GHD should be considered as a poor response [11]. Lastly, a Swedish study proposed a cutoff of 0.5 SDS for a satisfactory height increase in the first year of GH treatment [12].
Moreover, we still know little about GH treatment effect on adult height and conflicting data exist on long-term studies, with some showing failure to reach the genetic mid-parental target height and others successful target height achievement [13]. No specific cutoff value for GHD to measure final response exists. According to Cianfarani et al. efficacy outcome measure for ISS is a height gain from inclusion to adulthood of at least ≥1 SDS [14].
The primary purpose of the present longitudinal, retrospective, single-center study was to evaluate short-term (1st and 2nd year) and long-term (final or near-final height) efficacy of GH treatment in our population of iGHD children; we then assessed and compared the various criteria commonly used to define poor response to GH therapy in this population. Finally, we analyzed factors predicting final height outcome.
Materials and methods
Study population
This study included 94 Caucasian patients (66 males and 28 females), affected by iGHD. Diagnosis was made in accordance with previous version of Note 39 of Italian Drugs Agency (AIFA) for prescribing GH in Italy (used before July 2014), consisting of auxological criteria (a height ≤ −3 SDS OR a height ≤ −2.5 SDS with a pathological growth velocity ≤1 SDS OR a height of more than 1.5 SDS deviation from their mid-parental height (MPH) with a pathological growth velocity ≤−1.5 SDS), GH peak < 10 ng/mL at two stimulation tests and/or symptoms due to GHD (hypoglycemia) [15]. All patients were treated with rhGH with regular follow-up every 6–8 months until they reached final or near-final height, from 1991 to 2014. Criteria for interruption of treatment were HV <3 cm/year, bone age ≥ 13 years (girls) or ≥15 years (boys), complete pubertal development. Reevaluation of GH status in late adolescence or young adulthood was performed in 79 patients (84%). They underwent retesting on average 2.11 years after treatment interruption (2 months–3.6 years). The lowest normal GH cutoff limit in response to insulin tolerance test (ITT) was 6 ng/mL while that to GHRH plus arginine (GHRH + ARG) test was 19.0 μg/L [15].
Patients with GHD secondary to neoplasia, irradiation, or with other chronic diseases (such as diabetes mellitus, coeliac disease, systemic lupus erythematosus) were excluded. Other exclusion criteria were discontinuation of therapy for more than 2 months and unavailable/not sufficient pretreatment data.
Methods
At baseline, 1-year and 2-year follow up visit (±90 days) and at the end of treatment, we recorded age, height, weight, bone age (Greulich & Pyle method [16, 17]), and pubertal stage of the patients [18, 19], together with IGF1 serum measurements.
SDS values for Ht and HV and MPH were calculated using Italian growth reference values with the program “Growth calculator 3” by the Italian Society of Pediatric Endocrinology and Diabetology (ISPED-SIEDP) and available online at www.siedp.it [20]. Birth weight and length were evaluated using Italian growth reference values [21] to identify patients born adequate or small for gestational age (AGA/SGA).
GH peak during GH stimulation tests (one or two tests) was recorded. Patients were divided into two groups according to the results of tests for maximum GH peak level: GH < 5 ng/mL (“severe GHD”), and GH 5–10 ng/mL (“less-severe GHD”) to allow comparison with previous reports [11].
At time of diagnosis a brain MRI was obtained in each patients, except for one who denied consent.
IGF1 values were converted to sex- and age-related percentiles values as reported by Bedogni et al. [22], to set a standard between different assays and time points.
We chose the following criteria and cutoffs for poor responses to GH:
“Bang criterion”: ΔHt 1st year <0.5 SDS [12]
“Ranke criterion”: ΔHt 1st year <0.4 SDS if severe GHD; <0.3 SDS if less-severe GHD. [11]
“Bakker criterion”: HV 1st year (cm/aa) < mean –1 SD [10]
Considering the absence of criteria for efficacy on adult height outcome for GHD, we referred to Cianfarani et al. for ISS (“Cianfarani criterion” for adult height total ΔHt < 1 SDS) [14].
Statistical analyses
Statistical analyses were performed with the program JMP IN®, version 7.0 (SAS Institute Inc., Cary, NC, USA) for Macintosh (Apple Computer Inc., Cupertino, CA, USA) and the program IBM SPSS Statistics 22. Mean ± SD or median [min–max value] are reported where appropriate. A p-value < 0.05 was considered statistically significant. The proportion of poor responders per group was determined as the mean [95% confidence interval]. Comparisons between groups were tested by exploratory analysis using a χ2 test or Fisher’s exact test.
Results
Baseline characteristics
Among the 94 iGHD patients included in our study, 16 (17%) had a birth weight or length <3rd percentile (defined as SGA); 22 (23.4%) were classified as “severe GHD” and 72 (76.6%) as “less-severe GHD”. Brain MRI was predominantly normal (65.6%); in the other cases (34.4%) the main abnormalities were anterior pituitary hypoplasia (14%), thin pituitary stalk (8.6%), ectopic posterior pituitary, or empty sella (11.8%).
Median age at diagnosis was 11.72 years (range 2.8–15.6). The majority of the patients (64.9%) was prepubertal at diagnosis. Mean age at the onset of puberty (Tanner stage II) was at 13.69 ± 1.5 years (boys) and 12.15 ± 1.49 years (girls). Twenty-two patients entered puberty during 1st year and 13 during 2nd year of treatment; only 26 patients (27.7%) remained prepubertal during the first two years. Bone age was delayed for the majority of our patients (85.1%), and advanced only in two cases (2.1%), who were born AGA and had already entered puberty at baseline.
Mean MPH was 167.8 ± 8.5 cm (−0.79 ± 0.84 SDS). The average ΔMPH was −1.68 ± 0.89 SDS.
Once GHD was diagnosed, treatment with rhGH started at a mean dose of 0.2 ± 0.02 mg/kg/week (0.03 mg/kg die), close to the European recommendation for GH administration [23]. The median dose over the whole period of treatment was 0.186 mg/kg/week (0.146–0.23). Changes in GH dose in individual patients were minimal and exclusively related to weight, IGF1 concentration elevation, or insulin resistance.
Response to treatment
As shown in Table 1, all auxological parameters ameliorated in all patients. In our study the median height after 1 year of treatment was −1.99 SDS (ranging from −3.67 to −0.91 SDS) and resulted in an increase of about 0.49 ± 0.36 SDS from baseline values. Mean HV increased as well from +4.37 ± 2.56 cm/year (−1.91 ± 0.18 SDS) to +8.13 ± 1.96 cm/year (+2.33 ± 0.18 SDS).
Median chronological age at the end of treatment was 16.82 year for boys and 15.4 year for girls, with a range from 13.66 to 19.37 years, thus therapy lasted on average 5.08 ± 2.72 years. Our patients reached a median final height of −1.11 SDS (range from −2.68 to +1.26), with a total height gain of +1.5 ± 0.6 SDS. Difference between height and MPH reduced to −0.17 ± 0.81 SDS (pretreatment value −1.68 ± 0.89).
The patients who remained prepubertal during the first two years of treatment started GH treatment earlier, at a median age of 7.84 ± 3 years and discontinued it after 8.52 ± 2.5 years. Nevertheless, no substantial differences were found in both baseline and final auxological data: prior to treatment their height was −2.69 SDS (range from −3.61 to −1.39) with HV −1.82 ± 2.31 SDS, their final height was −1.02 (range from −2.13 to + 0.38) with a total ∆ Ht + 1.7 ± 0.50 SDS and a ∆MPH −0.22 ± 1.17 SDS (pretreatment value −1.95 ± 1.08 SDS).
Table 2 depicts comparisons in growth response among different categories of patients. In the subgroup with a pathological baseline HV < 3 cm/year (23 of 94 patients, 24.5%), catch up growth was more relevant at the end of the treatment (median height −0.89 SDS, range from −2.6 to + 0.4) than after 1 year (median height −2.35 SDS, range from −3.12 to +1.28 SDS). Interestingly, only four children of this category displayed a permanent GHD due to abnormalities of pituitary region.
Comparison among 1st year-poor responders population according to the three criteria are listed in Table 3. There were no significant differences in sex, birth weight/length, GH peaks, pubertal stage, bone age, and auxological data at diagnosis among these groups.
If we consider only Bang criterion, no substantial differences in baseline features are found between poor and good responders, except for a weak relationship between good response and MRI evidence of hypothalamic/pituitary abnormalities (45.2% vs. 25%, P = 0.12) [Table 4].
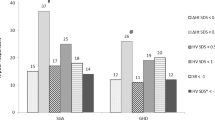
Change in height <0.5 SDS for the 1st year (Bang criterion) was the criterion that generated the highest number of poor responders (55.3%), followed by Bakker criterion (40.9%) and by Ranke criterion (16%) [Fig. 1].

Proportion of poor responders after 1 year (according to Bang, Ranke, or Bakker criteria) and at the end of GH treatment (according to Cianfarani criterion) in 94 iGHD children
At the end of the treatment, 21 patients (22.34%) had a height gain <1 SDS (Cianfarani criterion) [Table 5]. These poor responder patients had a lower MPH and showed a less satisfactory 1st year outcome, especially regarding HV (+0.27 SDS vs. +2.85 SDS) than final good responders. Age at diagnosis, baseline height, and therapy duration were found to be correlated to good response (P < 0.05).
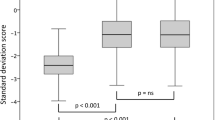
Only 32.7% of those that were considered poor responders after 1 year of therapy according to Bang criterion, actually still belonged to this category considering Cianfarani criterion for final height. Moreover, all subjects in our population (97.9%), except for two, achieved or exceeded mid-parental target height (MPH ± 2 SDS). Figure 2 compares final heights of poor responders according to different criteria and target heights: adult height SDS was similar in all groups.

Pattern of growth in the cohort according to different response categories. The upper and the lower limits of each rectangle indicate the 75th and the 25th height SDS percentiles, respectively; the lines inside the rectangles indicate median values. The vertical lines extending from the rectangles represent maximum and minimum height SDS values
Seventy nine patients (84%) were retested using the ITT or GHRH + ARG test after discontinuation of therapy. Sixty one patients (77%) showed normalization of GH secretion, whereas GH treatment was restarted in 18 patients due to pathological retesting results. Patients with permanent GHD showed better height outcome (mean 1st-year ΔHt 0.67 ± 0.38 SDS and total ΔHt 1.81 ± 0.6 SDS).
Predictive factors
Multiple regression analysis showed that both short-term and long-term outcomes correlated significantly with age at diagnosis (P = 0.0011 e P = 0.0004, respectively) and pretreatment height (both P < 0.0001, Rsquare = 0.99). Statistically significant relationships for adult height were age at diagnosis (P = 0.0004), treatment duration (P = 0.0004) and Ht SDS at diagnosis (P < 0.0001). The association between height gain during 1st year of treatment and final response was not statistical significant (p = 0.2871). Moreover, there was no difference in growth response between patients who were born SGA (16/94) or AGA (P = 0.525) nor between patients with a “severe GHD” and “less severe GHD” (P = 0.30) for the first year of treatment; in contrast, final height gain was significantly higher in the “severe GHD” group (P = 0.0041). We found that males presented a poorer 1st year response than females (P = 0.0062): this may be explained by age and sex-dependent differences in pubertal development.
Discussion
Our study supports GH short-term and long-term efficacy for the treatment of GHD, as height and HV after 1 year of treatment increased in all our patients. These results are consistent with those reported by KIGS showing a mean HV of 9.1 cm/year [24]. At the end of the treatment all patients appeared to have reached a height close to (in some cases over) their target height, except for two (2.1%), in which ΔMPH was −3.19 and −2.08 SDS. Median final or near-final height reached by our patients are generally better than some previously reported, in which mean final height was −2.3 SDS and < 2.0 SDS below the MPH [25]. Those data, however, referred to children treated with extractive human growth hormone, administered at a lower dose and not on a daily basis regimen. Other studies reported good outcomes, similar to ours: height of −0.9 SDS, close to target height, for Reiter et al. [13], −1.5 SDS for Cutfield et al. [26], −0.7 SDS in the American trial Genentech [27]. In the Genentech trial, however, most of the included patients failed to achieve full genetic height potential, despite the use of a 40% higher dose in USA than in Europe. Previous data regarding impact of GH dose on adult height are discordant: some studies suggested that a GH treatment at an early age and with high dose leads to a greater catch up growth. In our analysis, instead, no relationships were found between growth response and mean GH dose. Mean GH dose used in our patients was in the range permitted by European Medicines Agency [23], so we cannot exclude an advantage in terms of height gain by using higher dosage.
Extensive analysis was undertaken to find out any associations between total height increment and various baseline parameters. The most influential variables with negative relationship were height gain (after 1 year and at the end of treatment) and chronological age at diagnosis; strong positive relationship were final ΔHt and duration of therapy. These results emphasize the importance of initiating GH treatment as soon as possible and of providing therapy over a long period. Several clinical trials and postmarketing studies consistently reported better responses when treatment was started at an early age [13, 28]. Brain MRI was negative for hypothalamic/pituitary abnormalities in the majority of our patients, but it was not significantly associated with growth response, in contrast with previous literature that showed better response in children with pathological findings of hypothalamic/pituitary region [29, 30]. Neither birth weight/length nor MPH was relevant in predicting adult height, in line with the report by Dahlgren [31]. No statistically significant association existed between growth response and GH maximum peak at GH stimulation tests in the first year of treatment. This result reflects, at least in part, the well-known inaccuracies and limits in the diagnosis of GHD [12, 32]. Difficulties in distinguishing less-severe GHD from ISS or costitutional delay of growth and puberty (CDGP) have already been discussed by Kriström et al. [3] and Loche et al. [33]. A reduction of diagnostic cutoff of the GH stimulation tests could better recognize “real” GHD patients, as suggested by Bereket [34]. The revision of Note 39 of AIFA about regulation of rhGH prescription in Italy can be considered an effort in that sense.
We assessed IGF1 levels for every patient at baseline and during follow-up, but we decided to exclude these data from our analysis, because they were determined in different laboratories and with different assays so that a valid standardization could not be performed. It is known that IGF1 levels are related to height gain and HV increase during the first year and could ameliorate prediction of growth response [35].
The present study reports, for the first time, how 1st year response cutoffs compare with end-of-treatment response criteria in the same population of iGHD patients. In our study 55.3% of patients were defined as “poor responders” after 1 year of treatment using Bang criterion, 40.9% using Bakker criterion, and 23.4% using Ranke criterion. These percentages are a little higher, but similar in the order, than the ones reported by Bang et al. in the analysis on 456 North European children treated with GH [5]. No substantial differences emerged between these three different criteria in terms of sex, birth weight/length, or GH stimulation tests at diagnosis. The cutoff for 1st year ΔHt SDS of 0.3 (Ranke criterion) is probably too close to the normal growth variation during childhood. On the other hand, the Bakker criterion needs specific growth charts and may result uneasy in everyday clinical practice; moreover, sometimes it is not possible to measure 1st year HV because of unavailability or inaccuracy of pretreatment data, as happened for 33 patients in our cohort.
At the end of the treatment, poor responders according to Cianfarani criterion were 21 (22.34%). Main differences between poor and good responders can be seen especially in the first year: this again underlines that the 1st year response is the most important predictor of final outcome. Comparing 1st year and end-of-treatment response criteria, it can be noticed that 67.3% of poor responders according to Bang did not meet Cianfarani criterion and achieved a satisfactory final outcome (Final ∆ Ht > 1 SDS). To summarize, Bang criterion presented relatively good positive predictive value (90.5% of the 1st year-good responders reached a satisfactory final height), but a low negative predictive value (67.3%). Our data showed that almost every subject in our population (97.9%) reached his MPH. Only 2 patients of 94 (2.1%) showed a final height lower than target height and resulted “real poor responders” at the end of treatment, also according to Cianfarani criterion: in these cases, a genetic cause of short stature in addition to GHD may be undiagnosed. In agreement with Reiter [13], we ascertained that a satisfactory height response to GH treatment should lead to catch-up toward the target height. The difference between near-adult height SDS and MPH SDS is perhaps the best indication of whether an individual has achieved his/her genetic height potential.
One limitation of the present study is that we did not perform sex hormone priming in any patient; moreover, our population displayed relatively high median chronological age at diagnosis (11.82 years old, ranging from 2.82 to 15.62) in our population could mislead the interpretation of results. This problem, however, is common in studies about iGHD children, because in this category CDGP or familial short stature are often included and patients come later to the pediatrician’s attention. This bias may confuse data interpretation: for this reason, we underlined that most patients (64.9%, 61/94) were prepubertal at diagnosis. Males entered puberty (Tanner stage II) at 13.69 ± 1.5 years and girls at 12.15 ± 1.49 years, later than the rest of Italian population [36]. This late onset of puberty was described also in the study by Ranke on Pharmacia International Growth Database [37]. In our population there were probably cases of overlapping GHD in CDGP, even if it is difficult to identify and quantify them, as previously mentioned.
In conclusion, our study does not provide evidence that one criterion is preferable than another. Bang criterion generated the highest number of poor responders, but had a low negative predictive value; Ranke and Cianfarani criteria displayed similar rate of poor response. GH treatment should aim at achieving genetically determined height potential: a catch-up growth to MPH may be the best indication of its efficacy. The decision to start (and continue) therapy with GH should mostly rely on clinical and auxological parameters, not only on a numerical cutoff; we suggest to evaluate each patient in his whole personal and familiar context. Moreover, most of the patients have pubertal delay and a potential for spontaneous catch up growth; this also underlines the difficulties related to a clear diagnosis of GHD. Administration of rhGH should start as soon as possible in order to optimize efficacy of treatment. There is still a need to define poor/good response and to investigate predictors of outcome in order to obtain greater cost-effectiveness and increased opportunities for clinical benefit.
References
S. Cianfarani, E. Inzaghi, The challenge of growth hormone deficiency diagnosis and treatment during the transition from puberty into adulthood. Front Endocrinol. 4, 34 (2013)
J.C. Carel, E. Ecosse, M. Nicolino, M. Tauber, J. Leger, S. Cabrol, I. Bastié-Sigeac, J.L. Chaussain, J. Coste, Adult height after long term treatment with recombinant growth hormone for idiopathic isolated growth hormone deficiency: observational follow up study of the French population based registry. BMJ 325.7355, 70 (2002)
B. Kriström, A.S. Aronson, J. Dahlgren, J. Gustafsson, M. Halldin, S.A. Ivarsson, N.O. Nilsson, J. Svensson, T. Tuvemo, K. Albertsson-Wikland, Growth hormone (GH) dosing during catch-up growth guided by individual responsiveness decreases growth response variability in prepubertal children with GH deficiency or idiopathic short stature. J. Clin. Endocrinol. Metab. 94.2, 483–490 (2009)
M.A. de Ridder, T. Stijnen, A.C. Hokken-Koelega, Prediction of adult height in growth-hormone-treated children with growth hormone deficiency. J. Clin. Endocrinol. Metab. 92.3, 925–931 (2007)
P. Bang, S.F. Ahmed, J. Argente, P. Backeljauw, M. Bettendorf, G. Bona, R. Coutant, R.G. Rosenfeld, M.J. Walenkamp, M.O. Savage, Identification and management of poor response to growth‐promoting therapy in children with short stature. Clin. endocrinol. 77.2, 169–181 (2012)
M.B. Ranke, A. Lindberg, D.A. Price, F. Darendeliler, K. Albertsson-Wikland, P. Wilton, E.O. Reiter, KIGS International Board, Age at growth hormone therapy start and first-year responsiveness to growth hormone are major determinants of height outcome in idiopathic short stature. Horm. Res 68.2, 53–62 (2007)
M.B. Ranke, A. Lindberg, P. Chatelain, P. Wilton, W. Cutfield, K. Albertsson-Wikland, D.A. Price, KIGS International Board. Kabi International Growth Study, Prediction of long-term response to recombinant human growth hormone in Turner syndrome: development and validation of mathematical models. J. Clin. Endocrinol. Metab. 85.11, 4212–4218 (2000)
M.B. Ranke, A. Lindberg, P. Chatelain, P. Wilton, W. Cutfield, K. Albertsson-Wikland, D.A. Price, Derivation and validation of a mathematical model for predicting the response to exogenous recombinant human growth hormone (GH) in prepubertal children with idiopathic GH deficiency. J. Clin. Endocrinol. Metab. 84.4, 1174–1183 (1999)
M.B. Ranke, A. Lindberg, C.T. Cowell, K.A. Wikland, E.O. Reiter, P. Wilton, D.A. Price, KIGS International Board, Prediction of response to growth hormone treatment in short children born small for gestational age: analysis of data from KIGS (Pharmacia International Growth Database). J. Clin. Endocrinol. Metab. 88.1, 125–131 (2003)
B. Bakker, J. Frane, H. Anhalt, B. Lippe, R.G. Rosenfeld, Height velocity targets from the national cooperative growth study for first-year growth hormone responses in short children. J. Clin. Endocrinol. Metab. 93.2, 352–357 (2008)
M.B. Ranke, A. Lindberg, KIGS International Board, Observed and predicted growth responses in prepubertal children with growth disorders: guidance of growth hormone treatment by empirical variables. J. Clin. Endocrinol. Metab. 95.3, 1229–1237 (2010)
P. Bang, R. Bjerknes, J. Dahlgren, L. Dunkel, J. Gustafsson, A. Juul, B. Kriström, P. Tapanainen, V. Aberg, A comparison of different definitions of growth response in short prepubertal children treated with growth hormone. Horm. Res. Paediatr. 75.5, 335–345 (2011)
E.O. Reiter, D.A. Price, P. Wilton, K. Albertsson-Wikland, M.B. Ranke, Effect of growth hormone (GH) treatment on the near-final height of 1258 patients with idiopathic GH deficiency: analysis of a large international database. J. Clin. Endocrinol. Metab. 91.6, 2047–2054 (2006)
A. Deodati, S. Cianfarani, Impact of growth hormone therapy on adult height of children with idiopathic short stature: systematic review. BMJ 342, c7157 (2011)
Determinazione dell’Agenzia Italiana del farmaco—Modifica alla Nota AIFA 39, 26/11/2009: Gazzetta Ufficiale, Suppl. n. 229 09/12/2009
Determinazione n. 616/2014 modifica alla Nota AIFA 39. Ormone della crescita (somatotropina)—di cui alla determinazione del 29 luglio 2010: Gazzetta Ufficiale, Suppl. n. 270 06/06/2014
W.W. Greulich, S.I. Pyle, Radiographic atlas of skeletal development of hand and wrist (Stanford University Press, London, 1959)
W.A. Marshall, J.M. Tanner, Variations in pattern of pubertal changes in girls. Arch. Dis. Child. 44(235), 291–303 (1969)
W.A. Marshall, J.M. Tanner, Variations in the pattern of pubertal changes in boys. Arch. Dis. Child. 45(239), 13–23 (1970)
Growth Calculator 3. http://www.siedp.it/pagina/151/growth+calculator+3. Accessed 26 June 2014
E. Bertino, E. Spada, L. Occhi, A. Coscia, F. Giuliani, L. Gagliardi, G. Gilli, G. Bona, C. Fabris, M. De Curtis, S. Milani, Neonatal anthropometric charts: the Italian neonatal study compared with other European studies. J. Pediatr. Gastroenterol. Nutr. 51(3), 353–361 (2010)
G. Bedogni, G. Giannone, M. Maghnie, C. Giacomozzi, N. Di Iorgi, S. Pedicelli, E. Peschiaroli, G. Melioli, M. Muraca, M. Cappa, S. Cianfarani, Serum insulin-like growth factor-I (IGF-I) reference ranges for chemiluminescence assay in childhood and adolescence. Data from a population of in- and out-patients. Growth Horm. IGF Res. 22(3–4), 134–138 (2012)
EMA (European Medicines Agency), 27 February 2012 EMA/110423/2012, Patient Health Protection, Assessment report for Somatropin-containing medicinal products
M.B. Ranke, A. Lindberg, K. Albertsson-Wikland, P. Wilton, D.A. Price, E.O. Reiter, Increased response, but lower responsiveness, to growth hormone (GH) in very young children (aged 0–3 years) with idiopathic GH Deficiency: analysis of data from KIGS. J. Clin. Endocrinol. Metab. 90(4), 1966–1971 (2005)
E.C. Burns, J.M. Tanner, M.A. Preece, N. Cameron, Final height and pubertal development in 55 children with idiopathic growth hormone deficiency, treated for between 2 and 15 years with human growth hormone. Eur. J. Pediatr. 137(2), 155–164 (1981)
W. Cutfield, A. Lindberg, K. Albertsson Wikland, P. Chatelain, M.B. Ranke, P. Wilton, Final height in idiopathic growth hormone deficiency: the KIGS experience. KIGS International Board. Acta Paediatr. Suppl. 88(428), 72–75 (1999)
S.L. Blethen, J. Baptista, J. Kuntze, T. Foley, S. LaFranchi, A. Johanson, Adult height in growth hormone (GH)-deficient children treated with biosynthetic GH. The Genentech Growth Study Group. J. Clin. Endocrinol. Metab. 82(2), 418–420 (1997)
J. Ross, P.A. Lee, R. Gut, J. Germak, Factors influencing the one- and two-year growth response in children treated with growth hormone: analysis from an observational study. Int J. Pediatr. Endocrinol. 2010, 494656 (2010)
M. Bozzola, F. Mengarda, P. Sartirana, L. Tatò, J.L. Chaussain, Long-term follow-up evaluation of magnetic resonance imaging in the prognosis of permanent GH deficiency. Eur. J. Endocrinol. 143(4), 493–496 (2000)
M. Bozzola, C. Adamsbaum, I. Biscaldi, M. Zecca, M. Cisternino, E. Genovese, I. Richard, G. Kalifa, J.L. Chaussain, Role of magnetic resonance imaging in the diagnosis and prognosis of growth hormone deficiency. Clin. Endocrinol. 45(1), 21–26 (1996)
J. Dahlgren, B. Kriström, A. Niklasson, A.F. Nierop, S. Rosberg, K. Albertsson-Wikland, Models predicting the growth response to growth hormone treatment in short children independent of GH status, birth size and gestational age. BMC Med Inf. Decis. Mak. 7, 40 (2007)
T. Stanley, Diagnosis of growth hormone deficiency in childhood. Curr. Opin. Endocrinol. Diabetes Obes. 19(1), 47–52 (2012)
S. Loche, C. Bizzarri, M. Maghnie, A. Faedda, C. Tzialla, M. Autelli, M.R. Casini, M. Cappa, Results of early reevaluation of growth hormone secretion in short children with apparent growth hormone deficiency. J. Pediatr. 140(4), 445–449 (2002)
A. Bereket, Diagnosis of growth hormone deficiency: the role of growth hormone (GH), insulin- like growth factor (IGF-I) and IGF-binding protein (IGFBP-3). J. Clin. Res Pediatr. Endocrinol. 1(1), 23–35 (2009)
B. Kriström, C. Jansson, S. Rosberg, K. Albertsson-Wikland, Growth response to growth hormone (GH) treatment relates to serum insulin-like growth factor I (IGF-I) and IGF-binding protein-3 in short children with various GH secretion capacities. Swedish Study Group for Growth Hormone Treatment. J. Clin. Endocrinol. Metab. 82(9), 2889–2898 (1997)
F. Rigon, L. Bianchin, S. Bernasconi, G. Bona, M. Bozzola, F. Buzi, A. Cicognani, C.De Sanctis, V.De Sanctis, G. Radetti, L. Tatò, G. Tonini, E. Perissinotto, Update on age at menarche in Italy: toward the leveling off of the secular trend. J. Adolesc. Health 46(3), 238–244 (2010)
M.B. Ranke, D.A. Price, K. Albertsson-Wikland, M. Maes, A. Lindberg, Factors determining pubertal growth and final height in growth hormone treatment of idiopathic growth hormone deficiency. Analysis of 195 Patients of the Kabi Pharmacia International Growth Study. Horm. Res. 48(2), 62–71 (1997)
Funding
C.P. was supported by unrestricted grant from Pfizer Italia SRL (Pfizer Reference Number WI210391).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The study protocol has been approved by the San Raffaele Ethics Committee on human research. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pozzobon, G., Partenope, C., Mora, S. et al. Growth hormone therapy in children: predictive factors and short-term and long-term response criteria. Endocrine 66, 614–621 (2019). https://doi.org/10.1007/s12020-019-02057-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-019-02057-x