
Abstract
Purpose
Primary ovarian failure (POF) is characterized by amenorrhea, hypoestrogenism, and elevated gonadotropin levels in women leading to infertility under the age of 40 years. POF is a heterogeneous disease with different causes, and several genes have been associated with the POF phenotype. Thus, Whole-exome sequencing (WES) was performed in a consanguineous family with two sisters affected by POF.
Methods
All exons of both sisters were massively sequenced by WES, and the segregation was confirmed by Sanger sequencing.
Results
The novel homozygous c.1489delT variant in the NOBOX gene was identified in the two sisters with POF. Their parents were heterozygous carriers of this variant and, therefore, consistent with an autosomal recessive mode of inheritance. The c.1489delT NOBOX variant has not been previously reported in any public available databases (1000Genomes, 6500ESP/EVS, ExAC, and gnomAD). Furthermore, this variant was neither present in 387 Brazilian exomes control individuals nor in 200 fertile Brazilian women screened by Sanger sequencing.
Conclusion
We report the first familial case of a novel homozygous NOBOX variant with an autosomal recessive mode of inheritance, thus allowing for a genetic diagnosis of primary ovarian failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Premature ovarian failure (POF), also known as primary ovarian insufficiency (POI), is characterized by the loss of ovarian function before the age of 40 years, thus resulting in amenorrhea, hypoestrogenism, infertility, and elevated gonadotropin levels (follicle-stimulating hormone [FSH] >40 U/L) [1, 2]. POF could be caused by X chromosome abnormalities as well as genetic and environmental conditions. Most POF cases are still idiopathic, and genetic and epigenetic causes are currently being identified to be etiologic causes of POF [1]. POF is a heterogeneous disease with different causes, and several genes have already been associated with POF. Genes affecting ovarian development (SF-1 and FOXL2), gonadotropin production (LH and FSH beta subunit gene defects), gonadotropin signaling (FSHR and LHR), follicle development or maintenance of germ cells (BMP15, NOBOX, and NANOS3), and DNA division and repair (STAG3, MCM8, and MCM9) could contribute to POF [2].
In mammals, folliculogenesis is an intricate process underlying extra ovarian and intraovarian factors. The oocytes and their regulatory factors play a pivotal role in this process. Many transcription factors are implicated in follicle development, maturation, and maintenance of the constituent oocytes. The oocyte-specific homeobox gene encoding the newborn ovary homeobox protein, NOBOX, is expressed in germ cells and primordial oocytes. It belongs to a group of tissue-specific homeobox genes that plays a pivotal role in ovarian development [3]. NOBOX is preferentially expressed in primordial and growing oocytes [3]. In mice and human, NOBOX directly regulates pivotal oocyte-specific factors such as GDF9, OCT4, and KIT-L through its homeodomain and CG-protein region [4]. Despite the crucial role of NOBOX in ovarian development and a relatively higher estimated frequency of heterozygous NOBOX mutations (0–6%), only one homozygous NOBOX mutation has been reported for a patient with POF [4, 5].
Here, we describe a novel homozygous NOBOX variant identified by whole-exome sequencing (WES) as a cause of primary ovarian failure in two sisters.
Patients and methods
Case reports
Institutional review board approval and written informed consent were obtained from all subjects; blood was collected for DNA analysis. This study was approved by the Ethics Committee of the Hospital das Clínicas, São Paulo University School of Medicine, Brazil (protocol number 2015/12837/1.015.223).
From a cohort of 11 families with POF, we identified the NOBOX variant c.1489delT in one family with two affected sisters. The proband (II-3) and her sister (II-4) were born from consanguineous Brazilian parents (Fig. 1). The proband, a 17-year-old woman and her 18-year-old sister had no breast development and menarche. Upon physical examination, the proband and her sister had normal heights (148 and 156 cm, respectively) and body weights (40 and 47 kg, respectively). The stage of breast development for the proband and her sister were characterized by Tanner stages I and III, respectively. Pubic hair was characterized by Tanner III stage for both patients. The karyotypes of both the sisters were 46,XX as determined by an analysis of 50 cells in metaphase.

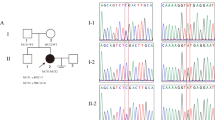
The homozygous pathogenic variant in NOBOX, c.1489delT, was identified in two sisters from Brazil. a Family Pedigree. The black arrow indicates the proband (II-3). Pedigree numbers of individuals are indicated above the symbols. b Electropherogram confirmed that the c.1489delT (red arrow to the nucleotide peak of interest) variant was homozygous in II-3 and II-4, and heterozygous in their parents (I-1 and I-2). WT denotes the wild type allele and MT denotes the c.1489delT variant. An asterisk indicates samples utilized for Whole-exome sequencing (WES). c Schematic cartoon of the NOBOX (ENSG00000106410/NM_001080413.1/NP001073882.3). The ten exons are shown in black and homeodomain in gray box. The black arrow indicates the pathogenic NOBOX variant position found in two Brazilian sisters (p.Cys497Valfs*53)
Basal serum gonadotropin levels were elevated in the proband (FSH = 68 U/L and LH = 32 U/L) and in her affected sister (FSH = 74 U/L and LH = 67 U/L), whereas serum estradiol levels were low in both the sisters.
Adrenal or thyroid autoimmune disorders had been excluded in both patients. Both sisters were treated with conjugated estrogen daily followed by progesterone replacement during the first 12 days of the month, resulting in complete breast development and a normal menstrual cycle. Their final heights at 27 and 26 years of age were 170 and 157 cm, respectively.
The mother of the affected sisters had their children at 25 and 26 years of age, respectively, and had a normal menopause (46 years of age). Moreover, their maternal aunt and their first-degree cousin were also evaluated due to irregular menstrual cycles and delay of puberty. Their physical and hormonal levels were normal for their age.
Genetic analysis
DNA extraction
Genomic DNA was extracted from peripheral blood leukocytes using standard procedures.
WES and data analysis
WES was performed in both sisters using SureSelect Human All Exons Kit (Agilent Technology, Santa Clara, CA, USA) on the Illumina HiSeq2500 (Illumina, San Diego, CA, USA). Reads (FASTQ files) were aligned against the human reference genome GRCh37/hg19 using the Burrows-Wheeler aligner (BWA) [6]. FastQC was used to assess the quality of the raw sequence data (FASTQ files).
The aligned reads were subsequently sorted and marked for duplicated sequences using biobambam2 [7]. Quality control for the sequence alignment data was performed using Qualimap [8]. Variant calling was performed with freebayes [9]. Low quality calls were filtered out using vcflib (https://github.com/vcflib/vcflib). Decomposition of multi-allelic loci and left normalization of indels were performed with vt [10]. The resulting variants were annotated with ANNOVAR [9, 11]. We focused our analysis on rare protein-altering variants (nonsynonymous, indels, nonsense, and splice sites). Variant frequency was analyzed based on different ethnic sub-groups available from the Exome Aggregation Consortium (ExAC), NHLBI Exome Sequencing Project (ESP), Exome Variant Server (EVS), and in the International Genome Sample Resource-1000 Genomes Project [12,13,14]
SNVs were analyzed by nine independent protein pathogenicity predictors: Polyphen-2, SIFT, Mutation Taster, Mutation Assessor, FATHMM, PROVEAN, Radial SVN, and LR. Another aspect of the annotation included base conservation and functional prediction using the Combined Annotation Dependent Depletion database (CADD) and the genomic evolutionary rate profiling score (GERP).
Sanger sequencing
WES variant was confirmed by Sanger sequencing for both subjects. Primers flanking the NOBOX variant (c.1489delT in exon 9, ENSG00000106410/NM_001080413) were used for PCR amplification. All PCR products were sequenced using the BigDye terminator v3.1 followed by automated sequencing with the ABI PRISM 3130XL (Applied Biosystems, Foster City, CA). Moreover, Sanger sequencing was used to evaluate 200 fertile Brazilian women as controls for putative damaging variant that was found from the 2 sisters.
Results
The mean coverage depth of the targeted regions in our exome sequencing data was 87.8–96.0×, with at least 96.46% of the sequenced bases covered more than 10-fold. We applied the following exome filter: total variants called in this family → homozygous variants only in both affected sisters → variants that were absent or with minor allele frequency less than 0.01% in the population databases (1000Genomes, ExAC, and ESP6500) → coding or splicing variants only → variants absent from the Brazilian exomes database. After these filters, only a single homozygous NOBOX variant was identified in both sisters with POF (Table 1). The c.1489delT NOBOX variant is located in exon 9, the second last exon, resulting in a cysteine to valine substitution at that position 497 as well as a frameshift and downstream premature stop codon (NM_001080413 [NOBOX_i001]:c.1489delT:p.Cys497Valfs*53) as predicted by the Mutalyzer analysis. Sanger sequencing confirmed that both sisters were homozygous for the c.1489delT NOBOX variant whereas the parents were heterozygous. Furthermore, their maternal aunt and first-degree cousin were heterozygous for this variant. Moreover, this variant was not present in the 400 fertile Brazilian women that were analyzed as well as the 386 exomes sequences from the Brazilian control database (unpublished data) and gnomAD browser.
Discussion
NOBOX plays an important role in early folliculogenesis. Though female NOBOX +/- mice were fertile, female NOBOX knockout mice were infertile and also presented the majority of their oocytes and follicular growth beyond primordial follicle stage as well as an accelerating loss of oocytes [15]. Moreover, NOBOX deficiency in mice resulted in the faulty development of germ cell cysts during embryonic development, thus leading to abnormal follicles formation [16].
The first reported case of a heterozygous missense NOBOX mutation associated with POF emerged in 2007 [17]. The p.Arg355His mutation disrupted the homeodomain of NOBOX, thus affecting its binding to DNA, and the functional study showed a dominant negative effect. Despite that, no family segregation was performed. Moreover, this affected woman had a mild phenotype presenting normal puberty, menarche was at 11 years, had two healthy children and entered menopause at 32 years of age. She was the only child of a woman who conceived her at 26 years of age and who entered menopause at 42 years of age [17].
Several cohorts were analyzed and different heterozygous variants were identified in women with POF [18,19,20,21]. Heterozygous mutations in NOBOX were reported in high frequency (6.2%) in 178 patients with POF of Caucasian and African ancestry [18]. One nonsense and four missense mutations in NOBOX were described, and these heterozygous mutations were verified to compromise the ability of the proteins to bind to the GDF9 promotor [18]. Moreover, the patients exhibited a heterogeneous phenotype, since primary amenorrhea until fertile woman. The heterozygous loss-of-function NOBOX mutation (p.Arg303*), localized in the conserved homeodomain, was reported in a patient who became spontaneously pregnant few months after POF diagnosis [18]. Furthermore, three novel and two previously described mutations in NOBOX were found in a heterozygous state in a cohort of 213 patients with POF. They have confirmed the functional effects of four heterozygous missense mutations, and concluded that no variants exhibited dominant negative effect [19]. Interestingly, the POF patients carrying the same mutations presented different phenotypes and one of them carried two different mutations, which were not segregated in the parents [19].
In a cohort of 125 Tunisian women diagnosed with POF, screening of NOBOX revealed three known heterozygous missense mutations in eight patients. They also reported a homozygous NOBOX variant (p.Arg117Trp), already described as deleterious heterozygous variant in POF woman [20]. Moreover, 107 European women with idiopathic POF were screened, and genetic analyses revealed five novel heterozygous NOBOX variants. They reported heterozygous mutations in the NOBOX homeobox domain and into c-terminus domain [21]. However, in a cohort comprised of 200 Chinese women with POF, NOBOX mutations were not identified as a common explanation for POI in that population, though only the homeodomain domain was screened [22].
The second homozygous NOBOX mutation was recently identified in one patient from a cohort of 96 Chinese patients with POF using whole-exome sequencing [5]. They revealed a homozygous truncating variant (c.567delG;p.Thr190Hisfs*13), which showed a severe defect in transcriptional activation of GDF9, a well-known target of NOBOX. The patient had primary amenorrhea and her unaffected mother was not screened to verify the variant status [5]. In our study, the novel homozygous c.1489delT NOBOX variant was identified in two sisters with POF born from consanguineous parents. The heterozygous mother had normal ovarian function and generated six children demonstrating that some heterozygous NOBOX variants are not cause of POF phenotype. The family reported here and a previously described Chinese patient [5] presented homozygous loss-of-function NOBOX variants suggesting an autosomal recessive inheritance pattern for NOBOX gene in POF.
Further evidence to support the pathogenicity of the c.1489delT variant is its absence in any public available databases (1000Genomes, 6500ESP/EVS, ExAC, and gnomAD). Moreover, the c.1498delT NOBOX variant is neither present in 387 Brazilian exomes controls (unpublished data) nor in 200 fertile Brazilian women screened by Sanger sequencing. According to guidelines from the American College of Medical Genetics and Genomics and Association for Molecular Pathology, supporting evidence that the present variant may be pathogenic include the fact that this variant was absent in all available databases, it cosegregated with the disease in affected and unaffected family members, and the fact that the null variants (such as nonsense or frameshift) of this gene is known to contribute to the mechanism for POF [23].
The published data suggested that heterozygous variants might cause ovarian defects due to haploinsufficiency of NOBOX and lead to POF. Although different groups demonstrated this effect, they are not in agreement with animal models for which heterozygous NOBOX +/- is fertile as well as the WT mice [15, 16]. Furthermore, all POF cohorts have shown a heterogeneous phenotype such as mild and severe phenotypes due to the same mutation with no clear explanation. Interestingly, all heterozygous NOBOX mutations previously described were found using Sanger sequencing in contrast with the two homozygous variants, which were identified by massive parallel sequencing. The Whole-exome sequencing was able to exclude others genes that could explain the POF phenotype in the proband mentioned in this study, whereas Sanger sequencing could not rule out causative variants in other genes in previously described patients with heterozygous NOBOX mutations.
According to protein database, the NOBOX protein contains a conserved homeodomain region involved in the transcriptional regulation of key eukaryotic development processes. This homeobox domain region is located at between 272 and 363 amino acids position at the end of exon 4 and middle of exon 6 (see cartoon Fig. 1C) (NCBI reference sequence/ENSG00000106410/NP_001073882.3). Despite the NOBOX homeobox domain being conserved among species and seeming to be a crucial to NOBOX transcriptional activity, heterozygous missense and loss-of-function mutations located outside of this region and nearby to c-terminus site (p.Lys371Thr, p.Arg449*, p.Asp452Asn), were also functionally confirmed to lead POF phenotype [19, 21]. Ferrari and collaborators [21] have shown that NOBOX variants p.Arg449* and p.Asp452Asn, identified in POF patients, had an aberrant activity in vitro. Their inability to sustain gene expression, with the likely deleterious effects of protein instability and degradation, reinforced that C-terminus NOBOX region can also lead to POF phenotype [21]. Considering that the pathogenic variant p.Cys497Valfs*53 is located nearby this c-terminus region, which was also confirmed to be involved in transcriptional activity, the deleterious effect in the protein predicted by in silico tool, and the perfect autosomal recessive inheritance segregation, we suggest that this variant led to POF phenotype in our two patients.
Finally, human disorders may have two types of inheritance with the same phenotype as for NR5A1 defects. A heterozygous loss-of-function NR5A1 mutation (p.Gly35Glu) was described in a patient with adrenal failure and complete 46,XY sex-reversal, indicating that haploinsufficiency of this gene is sufficient to cause a severe clinical phenotype. In an infant with a similar clinical phenotype, the homozygous NR5A1 mutation (p.Arg92Gln) was identified, but three relatives (parents and sister) were phenotypically normal despite being heterozygous for this mutation [24, 25]. Therefore we could not exclude that POF disease caused by NOBOX mutations may also have autosomal dominant and recessive inheritance pattern.
In conclusion, we report the first familial case of a homozygous NOBOX variant, thus indicating an autosomal recessive mode of inheritance. This finding reveals a novel NOBOX variant associated with POF, therefore contributing to the genetic diagnosis of this disorder.
References
A.N. Shelling, Premature ovarian failure. Reprod. Rev. 140, 633–641 (2010). https://doi.org/10.1530/REP-09-0567
E.J. Tucker, S.R. Grover, A. Bachelot, P. Touraine, A.H. Sinclair, Premature ovarian insufficiency: new perspectives on genetic cause and phenotypic spectrum. Endocr. Rev. 37(6), 609–635 (2016). https://doi.org/10.1210/er.2016-1047
N. Suzumori, C. Yan, M.M. Matzuk, A. Rajkovic, Nobox is a homeobox-encoding gene preferentially expressed in promordial and growing oocytes. Mech. Dev. 111(1–2), 137–141 (2002). https://doi.org/10.1016/S0925-4773(01)00620-7
R. Rossetti, I. Ferrari, M. Bonomi, L. Persani, Genetics of primary ovarian insufficiency. Clin. Genet. 91(2), 183–198 (2017). https://doi.org/10.1111/cge.12921
L. Li, B. Wang, W. Zhang, B. Chen, M. Luo, J. Wang, X. Wang, Y. Cao, K. Kee, A homozygous NOBOX truncating variant causes defective transcriptional activation and leads to primary ovarian insufficiency. Hum. Reprod. 32(1), 248–255 (2017). https://doi.org/10.1093/humrep/dew271
H. Li, R. Durbin, Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26(5), 589–595 (2010). https://doi.org/10.1093/bioinformatics/btp698
G. Tischler, S. Leonard, Biobambam: tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 9, 13 (2014). https://github.com/gt1/biobambam2/releases
K. Okonechnikov, A. Conesa, F. García-Alcalde, Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32(2), 292–294 (2016). https://doi.org/10.1093/bioinformatics/btv566
A. Garrison, G. Marth, Haplotype-based variant detection from short-read sequencing, (2012). https://arxiv.org/abs/1207.3907
J.M. Alvarez-Castro, R.C. Yang, R.C. Multiallelic, models of genetic effects and variance decomposition in non-equilibrium populations. Genetics 139(9), 1119–1134 (2011). https://doi.org/10.1007/s10709-011-9614-9
K. Wang, M. Li, H. Hakonarson, ANNOVAR: functional annotation of genetic variants from high- throughput sequencing data. Nucl. Acid. Res. 38(16), e164 (2010). https://doi.org/10.1093/nar/gkq603
M. Lek, K.J. Karczewski, E.V. MiniKel et al. Exome aggregation consortium: analysis of protein-coding genetic variation in 60,706 humans. Nature 536(7616), 285–291 (2016). https://doi.org/10.1038/nature15393
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (URL:http://evs.gs.washington.edu/EVS/)
The 1000 Genomes Project Consortium : a global reference for human genetic variation. Nature 526, 68–74 (2015). https://doi.org/10.1038/nature15393
A. Rajkovic, S.A. Pangas, D. Ballow, N. Suzumori, M.M. Matzuk, NOBOX deficiency disrupts early folliculogenesis and oocytes-specific gene expression. Science 305(5687), 1157–1159 (2004). https://doi.org/10.1126/science.1099755
A. Lechowska, S. Bilinski, Y. Choi, Y. Shin, M. Kloc, A. Rajkovic, Premature ovarian failure in nobox-deficient mice is caused by defects in somatic cell invasion and germ cell cyst breakdown. J. Assist. Reprod. Genet. 28(7), 583–589 (2011). https://doi.org/10.1007/s10815-011-9553-5
Y. Qin, Y. Choi, H. Zhao, J.L. Simpson, Z.I. Chen, A. Rajkovic, NOBOX homeobox mutation causes premature ovarian failure. Am. J. Hum. Genet. 81(3), 576–581 (2007). https://doi.org/10.1086/519496
J. Bouilly, A. Bachelot, I. Broutin, P. Touraine, N. Binart, Novel NOBOX loss-of-function mutations accountfor 6.2% of cases in a large primary ovarian insufficiency cohort. Hum. Mutat. 32(10), 1108–1113 (2011). https://doi.org/10.1002/humu.21543
J. Bouilly, F. Roucher-Boulez, A. Gompel, H. Bry-Gauillard, K. Azibi, C. Beldjord, C. Dodé, J. Bouligand, A.G. Mantel, A.C. Hécart, B. Delemer, J. Young, N. Binart, New NOBOX mutations identified in a large cohort of women with primary ovarian insufficiency decrease KIT-L expression. J. Clin. Endocrinol. Metab. 100(3), 994–1001 (2015). https://doi.org/10.1210/jc.2014-2761
N. Bouali, B. Francou, J. Bouligand, B. Lakhal, I. Malek, M. Kammoun, J. Warszawski, S. Mougou, A. Saad, A. Guiochon-Mantel, NOBOX is a strong autosomal candidate gene in Tunisian patients with primary ovarian insufficiency. Clin. Genet. 89(5), 608–613 (2016). https://doi.org/10.1111/cge.12750
I. Ferrari, J. Bouilly, I. Beau, F. Guizzardi, A. Ferlin, M. Pollazzon, M. Salerno, N. Binart, L. Persani, R. Rossetti, Impaired protein stability and nuclear localization of NOBOX variants associated with premature ovarian insufficiency. Hum. Mol. Genet. 25(23), 5223–5233 (2016). https://doi.org/10.1093/hmg/ddw342
Y. Qin, Y. Shi, Y. Zhao, S.A. Carson, J.I. Simpson, Z.J. Chen, Mutation analysis of NOBOX homeodomain in chinese women with premature ovarian failure. Fertil. Steril. 91(Suppl.4), 1507–1509 (2009). https://doi.org/10.1016/j.fertnstert.2008.08.020
S. Richards, N. Aziz, S. Bale, D. Bick, S. Das, J. Gastier-Foster, W.W. Grody, M. Hegde, E. Lyon, E. Spector, K. Voelkerding, H.L. Rehm, ACMG Laboratory Quality Assurance Committee, standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology.Genet. Med. 17(5), 405–424 (2015) https://doi.org/10.1038/gim.2015.30
J.C. Achermann, M. Ito, M. Ito, P.C. Hindmarsh, J.L. Jameson, A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat. Genet. 22(2), 125–126 (1999). https://doi.org/10.1038/9626
J.C. Achermann, G. Ozisik, M. Ito, U.A. Orun, K. Harmanci, B. Gurakan, J.L. Jameson, Gonadal determination and adrenal development are regulated by the orphan nuclear receptor steroidogenic factor-1 in dose-dependent manner. J. Clin. Endocrinol. Metab. 87(4), 1829–1833 (2002). https://doi.org/10.1210/jcem.87.4.8376.
Acknowledgements
The authors thank the patients and their families for participating in this study. They are also grateful to the LIM/42 team for all technical assistance.
Funding
This work was supported by: Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 2014/14231-0 (to M.M.F.); Fundação de Amparo à Pesquisa do Estado de São Paulo Grant 2013/02162-8, Nucleo de Estudos e Terapia Celular e Molecular (NETCEM) and Conselho Nacional de Desenvolvimento Científico e Tecnológico Grant 303002/2016-6 (to B.B.M.); Fundação de Amparo à Pesquisa do Estado de São Paulo 2014/50137-5 (to SELA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Hospital das Clínicas, University of São Paulo School of Medicine, Brazil (protocol number 2015/12837/1.015.223).
Informed consent
Institutional review board approval and written informed consent were obtained from all individual participants included in the study before the collection of blood for DNA analysis.
Rights and permissions
About this article

Cite this article
França, M.M., Funari, M.F.A., Lerario, A.M. et al. A novel homozygous 1-bp deletion in the NOBOX gene in two Brazilian sisters with primary ovarian failure. Endocrine 58, 442–447 (2017). https://doi.org/10.1007/s12020-017-1459-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-017-1459-2