
Abstract
Although the two anti-diabetic drugs, dipeptidyl peptidase-4 inhibitors (DPP4is) and glucagon-like peptide-1 (GLP-1) receptor agonists (GLP1RAs), have distinct effects on the dynamics of circulating incretins, little is known of the difference in their consequences on morphology and function of pancreatic islets. We examined these in a mouse model of β cell injury/regeneration. The model mice were generated so as to express diphtheria toxin (DT) receptor and a fluorescent protein (Tomato) specifically in β cells. The mice were treated with a DPP4i (MK-0626) and a GLP1RA (liraglutide), singly or doubly, and the morphology and function of the islets were compared. Prior administration of MK-0626 and/or liraglutide similarly protected β cells from DT-induced cell death, indicating that enhanced GLP-1 signaling can account for the cytoprotection. However, 2-week intervention of MK-0626 and/or liraglutide in DT-injected mice resulted in different islet morphology and function: β cell proliferation and glucose-stimulated insulin secretion (GSIS) were increased by MK-0626 but not by liraglutide; α cell mass was decreased by liraglutide but not by MK-0626. Although liraglutide administration nullified MK-0626-induced β cell proliferation, their co-administration resulted in increased GSIS, decreased α cell mass, and improved glucose tolerance. The pro-proliferative effect of MK-0626 was lost by co-administration of the GLP-1 receptor antagonist exendin-(9-39), indicating that GLP-1 signaling is required for this effect. Comparison of the effects of DPP4is and/or GLP1RAs treatment in a single mouse model shows that the two anti-diabetic drugs have distinct consequences on islet morphology and function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes mellitus (T2DM) is a metabolic disorder characterized by chronic hyperglycemia. Defective insulin secretion from pancreatic β cells and/or defective insulin action in peripheral tissues contributes to its development [1]. Numerous insulin secretagogues (such as sulfonylureas and glinides) and insulin sensitizers (such as biguanides and thiazolidinediones) have been used for the treatment of T2DM [1]. However, several large human clinical trials have revealed that none of these oral anti-diabetic drugs can afford long-lasting, glycemic control [2, 3]. In most patients with T2DM, glycemic control usually deteriorates over time, together with a progressive decline in β cell function [4].
Beta cell mass of T2DM patients has also been shown to be decreased compared with that of normal subjects, as assessed by cadaveric or operatively extracted specimen [5–8]. Notably, β cell mass is known to decline progressively along with duration of the disease [8]. Indeed, such decline begins before the onset of T2DM [6], indicating that decreased β cell mass is not secondary to chronic hyperglycemia. Accordingly, strategies that restore β cell mass as well as function may be a novel treatment for T2DM.
Recently, incretin-based therapies have become widely used in the treatment of T2DM [9]. These drugs are the glucagon-like peptide-1 (GLP-1) receptor agonists (GLP1RAs) and the dipeptidyl peptidase-4 (DPP-4) inhibitors (DPP4is); the former mimic the major incretin GLP-1 and show robust stability in vivo, and the latter inhibit inactivation of various endogenous DPP-4 substrates, including incretins [GLP-1 and glucose-dependent insulinotropic polypeptide (GIP)], neuropeptides, cytokines, and chemokines [9]. Incretins are secreted from enteroendocrine cells in response to meal ingestion and potentiate insulin secretion in a glucose-dose-dependent manner [10]. This strict glucose dependency makes incretins ideal secretagogues applicable in the treatment of T2DM. In addition, incretins have been reported to be capable of inhibiting β cell death and promoting proliferation [10]. Accordingly, both GLP1RAs and DPP4is may prevent progressive decrease in β cells of T2DM patients.
The effects of GLP1RAs and DPP4is on β cell mass as well as function have been studied extensively in experimental animal models. However, the difference in pharmacological consequences of GLP1RAs and DPP4is on islets remains unclear, although the two drugs differ greatly in terms of incretin dynamics [9]. GLP1RAs administration induces continuous and marked increase of GLP-1 in whole systemic circulation, while DPP4is mildly but significantly increase various bioactive peptides (as well as GLP-1) and retain normal temporal (high in postprandial state) and spatial (concentrated in portal vein) distribution of the substrates. Results from differing experimental conditions including animal models, dose and duration of treatments, and outcome measures (i.e., methodologies of β cell mass quantification) are confounding. In the present study, we compared the consequences of treatments with either a GLP1RA, a DPP4i or both together in a single mouse model of β cell injury/regeneration.
Materials and methods
Reagents
Diphtheria toxin (DT) from Corynebacterium diphtheriae was purchased from Merck Millipore (Darmstadt, Germany). A DPP4i, MK-0626, was provided by Merck & Co., Inc. (Kenilworth, NJ, USA) and a GLP1RA, liraglutide, was purchased from Novo Nordisk (Bagsvaerd, Denmark). Exendin-(9-39) was from Abcam (Cambridge, UK). 5-Bromo-2′-deoxyuridine (BrdU) was from Wako (Osaka, Japan). Caspase-3 inhibitor (Ac-DEVD-CHO), LY294002, and PD98059 were from Merck Millipore. Caspase-9 inhibitor (Z-LEHD-FMK) was from R&D Systems (Minneapolis, MN, USA).
Animals and in vivo experiments
We developed a mouse model expressing DT receptor (DTR) and a fluorescent protein (Tomato) specifically in β cells using toxin receptor-mediated cell ablation system [11]. Rosa26 DTR/+ mice and Rosa26 tdTomato/+ were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Rat insulin promoter-Cre (Rip-Cre) transgenic mice [12] were crossbred with Rosa26 DTR/+ mice and Rosa26 tdTomato/+ mice to generate Rip-Cre; Rosa26 DTR/tdTomato mice (Fig. 1a). Mice were housed in a climate-controlled room with a temperature of 23 ± 3 °C, humidity of 55 ± 15 %, and a 12 h light/12 h dark cycle and fed standard laboratory chow (CRF-1; Charles River Japan, Yokohama, Japan) ad libitum. All animal experiments were approved by the Animal Care Committee of Chiba University.

Pancreatic β cell injury/regeneration mice. a Scheme of Rip-Cre;Rosa26 DTR/tdTomato mice. b Immunofluorescence of insulin (green), tomato (red), and Hoechst (blue) in pancreatic islets of Rip-Cre;Rosa26 DTR/tdTomato mice. c Insulin and tomato staining of islets in mice at 6 and 14 days after DT injection. d Quantification of β and tomato-positive cell mass in mice at 6 and 14 days after DT injection (n = 4). e Immunofluorescence of insulin (green), Ki67 (red) and Hoechst (blue) in islets of mice at 6 and 14 days after DT injection. An arrowhead indicates a Ki67-positive β cell and enlarged image is also shown. f Quantification of Ki67-positive cells in β cells of mice at 6 and 14 days after DT injection (n = 3). The cells of thirty random islets were analyzed. g Blood glucose levels after 1 g/kg oral glucose in mice at 14 days after DT injection (n = 4). Scale bar 50 μm. *P < 0.05 and **P < 0.01 versus DT-untreated mice
For β cell ablation, mice were singly intraperitoneally injected with 20 μg/kg DT (10:00–17:00), and pancreata were excised after 6 or 14 days and subjected to immunohistochemical analyses. For administration of MK-0626, mice were placed on a normal chow diet (D10001; Research Diets Inc., New Brunswick, NJ, USA) containing 0.0025 % MK-0626. Daily intake of MK-0626 calculated from daily chow intake (2.9 g/mouse/day) was estimated to be approximately 3.8 mg/kg, which exceeds the dose (~3 mg/kg) reported to increase plasma active GLP-1 concentrations significantly in ob/ob mice [13]. For administration of liraglutide, mice were injected subcutaneously with 100 μg/kg liraglutide twice daily (10:00 and 22:00) [14] to raise its concentration continuously to a supra-physiological level. For exendin-(9-39) administration, an Alzet osmotic pump (model 1002, for 14 days, 0.25 μl/h; Alza, Cupertino, CA, USA) containing 77 nmol/100 μl of exendin-(9-39) was implanted subcutaneously to deliver at an estimated rate of 276 nmol/kg/day [15]. For BrdU incorporation analysis, mice were labeled continuously with 1 mg/ml BrdU dissolved in drinking water for 14 days. Blood glucose levels at indicated time points were measured using Glutestmint (Sanwa Chemical Co., Nagoya, Japan). For oral glucose tolerance tests (OGTTs), mice were fasted for 16 h and then administered 1 g/kg glucose. For assessment of insulin secretory capacity in vivo, serum insulin levels in fasted (14 h) or refed (3 h) mice were measured using an ELISA kit (Morinaga Institute of Biological Science, Yokohama, Japan).
Immunohistochemistry
Extracted pancreata were fixed and embedded in paraffin, and 4 μm sections were cut. For quantitative analysis of each cell mass, we examined all sections, spaced 160 μm apart from each other, prepared from the whole pancreas. Eleven to 14 sections of each pancreas were stained with hematoxylin–eosin (HE) or immunostained as previously described [16]. Primary antibodies used were anti-RFP (1:500) (BML, Tokyo, Japan), anti-insulin (1:200) (Life Technologies, Gaithersburg, MD, USA), and anti-glucagon (1:400) (Cell Signaling Technology, Danvers, MA, USA). Secondary antibodies used were immunoglobulins/HRP (1:200) (Dako, Glostrup, Denmark), and the bound antibodies were visualized by DAB. Ratios of Tomato-positive area-, insulin-positive area-, and glucagon-positive area-to-total pancreas area in all sections were calculated through the 4× objective using BZ-II Analyzer software (Keyence, Osaka, Japan). Total masses of tomato-positive, β and α cells were determined by multiplying the ratios of positive-area-to-total pancreas area by total weight of the pancreas.
For immunofluorescence staining, primary antibodies used were anti-RFP, anti-insulin, anti-glucagon (1:200), anti-somatostatin (1:200) (Merck Millipore), anti-pancreatic polypeptide (PP) (1:100), anti-chromogranin A (1:50) (Merck Millipore), anti-Ki67 (1:50) (Abcam), and anti-BrdU (1:150) (Abcam). Secondary antibodies used were anti-IgG antibodies conjugated with Alexa Fluor 405, 488 and 555 (Life Technologies). TUNEL staining was performed using an ApopTag in situ apoptosis detection kit (Merck Millipore). The nuclei were counterstained with Hoechst 33342 (Sigma, St. Louis, MO, USA). The slides were analyzed in an FV10i confocal microscope (Olympus, Tokyo, Japan). Thirty randomly selected islets (amounting to 3000–7000 cells in total) per each mouse were subjected to quantification.
Islet isolation and insulin release
Isolated pancreatic islets were prepared from mice by collagenase digestion technique as previously described [17], with slight modifications. Isolated islets were incubated at 37 °C in RPMI 1640 medium containing 10 % FBS and 5.5 mM glucose for 4 h. Islets were preincubated at 37 °C in Krebs–Ringer bicarbonate buffer supplemented with 0.2 % BSA and 10 mM HEPES (incubation medium) containing 2.8 mM glucose for 30 min and then incubated in the incubation medium containing 2.8 or 16.7 mM glucose for 60 min. Aliquots of supernatant of the incubation medium were subjected to ELISA (Mesacup insulin test; MBL, Tokyo, Japan).
In vitro experiments using MIN6 cells overexpressing DTR
A pancreatic β cell line, MIN6 cells [18], was cultured in Dulbecco’s modified Eagle medium containing 10 % FBS and 71 μmol/l 2-mercaptoethanol (Sigma) at 37 °C. MIN6 cells stably overexpressing DTR (MIN6-DTR) were generated using the retrovirus vector, the MSCV-IRES-EGFP vector, as previously reported [19]. After transduction, EGFP-positive MIN6 cells were subcloned by limiting dilution.
MIN6-DTR were seeded in 96-well-plates (1.5 × 104 cells/well) at 2 days before the experiment and treated with DT in the presence or absence of MK-0626, liraglutide, caspase-3 inhibitor, or caspase-9 inhibitor for 24 h. Effects of LY294002 and PD98059 were evaluated in MIN6-DTR by adding either of them at 30 min prior to the initiation of 24 h treatment with DT plus liraglutide. Cell viability was determined with Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan). Experiments using the same protocol were repeated twice or three times to ascertain reproducibility.
To evaluate the anti-apoptotic effect of liraglutide, MIN6-DTR was incubated with DT and/or liraglutide for 16 h. To confirm the efficacy of LY294002 and PD98059, inhibition of Akt phosphorylation or Erk1/2 phosphorylation was assessed in cells pretreated for 5 min with LY294002 or PD98059, followed by add-on incubation with liraglutide for 3 min. Cells were lysed and subjected to immunoblotting, as previously described [20]. Primary antibody used was anti-cleaved caspase-3, anti-β actin, anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-Erk1/2, and anti-Erk1/2 (Cell Signaling Technology). Secondary antibody used was HRP-conjugated anti-rabbit IgG (GE Healthcare, Buckinghamshire, UK).
Statistical analysis
Data are expressed as mean ± SEM. Statistical significance of difference was evaluated by Student’s t test or by analysis of variance followed by the Dunnett multiple comparison test. P < 0.05 was considered significant.
Results
Pancreatic β cell injury/regeneration mice
The model mice were established by introducing DTR and a red fluorescent protein (Tomato) in β cells (Rip-Cre;Rosa26 DTR/tdTomato mice) (Fig. 1a). Tomato protein was expressed in about 80 % of insulin-positive cells (Fig. 1b), in accord with the Cre recombinase efficiency in β cells of Rip-Cre mice [21]. Single DT injection to the mice eliminated most Tomato-positive cells at day 6 (Fig. 1c), which resulted in a marked decrease in Tomato-positive cell mass to only 7 % of that in DT-untreated mice (Fig. 1d), suggesting co-expression of DTR in Tomato-positive β cells. Quantitative measurement revealed that β cell mass, i.e., the sum of Tomato-positive and -negative cell mass, decreased to 34 % by DT administration at day 6. The β cell mass then increased up to 55 % at day 14, but Tomato-positive cell mass did not (Fig. 1d), indicating that an increase in Tomato-negative β cell mass exclusively contributes to the increase in total β cell mass. We also detected β cell proliferation in these mice, frequently at 6 days after DT ablation, but rarely at day 14, as assessed by Ki67 immunostaining (Fig. 1e, f), indicating a vigorous but transient regeneration of β cells in the mice. After β cell ablation, the mice retained normoglycemia (Tables 1, 2) but exhibited significantly impaired glucose tolerance as assessed by OGTTs (Fig. 1g).
Cytoprotective effects of DPP4is and/or GLP1RAs against β cell injury
To analyze cytoprotective effects of DPP4is and/or GLP1RAs on β cells, 7-week-old mice were administered MK-0626 and/or liraglutide for 7 days, followed by a single administration of DT. Six days after β cell ablation, pancreata were excised and subjected to immunohistochemical examination (Fig. 2a). MK-0626 administration did not affect body weight, pancreas weight, and blood glucose levels (Table 1). On the other hand, liraglutide administration induced a mild but significant decrease in body weight, irrespective of co-administration of MK0626.

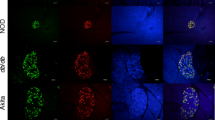
Cytoprotective effects of DPP4is and/or GLP1RAs against β cell injury. a Experimental protocol to examine cytoprotective effects of DPP4is and/or GLP1RAs on β cells. b Tomato and insulin staining of islets in mice treated with MK-0626, liraglutide, or MK-0626 plus liraglutide at 6 days after DT injection. c, d Tomato-positive cell mass c and β cell mass d in the treated mice at 6 days after DT injection (n = 3–4). e TUNEL staining with insulin in islets of the treated mice at 4 days after DT injection. f Quantification of apoptotic cells in β cells of the treated mice at 4 days after DT injection (n = 3–4). The cells of thirty random islets were analyzed. Scale bar 50 μm. *P < 0.05; **P < 0.01
Immunohistochemical analysis revealed that the number of Tomato-positive cells that survived after β cell ablation was significantly increased by MK-0626 and also by liraglutide, but was not further increased by co-administration of MK-0626 plus liraglutide (Fig. 2b, c). Although treatment with MK-0626 and/or liraglutide almost doubled the number of surviving Tomato-positive β cells (Fig. 2c), the increase (~0.05 mg) was too small to show statistically significant difference in total (Tomato-positive plus-negative) β cell mass (0.4–0.5 mg) (Fig. 2d).
We then investigated whether increased survival of Tomato-positive cells by MK-0626 and/or liraglutide is attributable to suppression of apoptotic β cell death by DT administration. The number of apoptotic cells was suppressed similarly by MK-0626 treatment and liraglutide treatment, but no further suppression was observed by their co-treatment (Fig. 2e, f), in accord with the results of the mass of Tomato-positive cells surviving after DT ablation. These results indicate that potentiation of GLP-1, but not that of other DPP-4 substrates, plays a major role in suppressing DT-induced apoptosis by MK-0626 in these mice.
Molecular mechanisms of cytoprotection of GLP-1 in β cells
The mechanism of β cell cytoprotection by GLP-1 was examined in MIN6-DTR in vitro. DT administration reduced cell viability in a dose-dependent manner (Fig. 3a), while liraglutide co-treatment significantly increased viability (Fig. 3b). Since DPP-4 is found to be expressed in MIN6 cells as well as in pancreatic islets (data not shown), DPP4is may exert cytoprotection through inhibiting DPP-4 released from or expressed in MIN6 cells. However, this possibility was not supported by the experiment of MK-0626 administration to MIN6-DTR, since MK0626 failed to rescue MIN6-DTR from DT ablation (Fig. 3c). Considering that DT-induced cell death might be mediated by activated caspase signaling [22], we attempted to protect MIN6-DTR from DT by inhibitors of caspase-3 and -9; both of these inhibitors were found to increase the cell viability impaired by DT (Fig. 3d, e). Cleaved caspase-3, a critical player in apoptosis [23], was induced by DT but was completely inhibited by co-administration of liraglutide (Fig. 3f).

Molecular mechanisms of cytoprotection of GLP-1 in β cells. a Cell viability of MIN6-DTR incubated with various concentrations of DT for 24 h. b Cell viability of MIN6-DTR incubated with various concentrations of liraglutide and 10 μg/ml DT for 24 h. c Cell viability of MIN6-DTR incubated with various concentrations of MK-0626 and 10 μg/ml DT for 24 h. d, e Cell viability of MIN6-DTR incubated with various concentrations of caspase-3 inhibitor d or caspase-9 inhibitor e and 10 μg/ml DT for 24 h. f Western blot analysis for cleaved caspase-3 (upper panel) and β actin (lower panel) of MIN6-DTR incubated with 10 μg/ml DT and/or 100 nM liraglutide for 16 h. g Western blot analysis for phospho-Akt (upper panel) and Akt (lower panel) of MIN6-DTR preincubated with 5 μM LY294002 for 5 min and then incubated with 100 nM liraglutide in the presence of LY294002 for 3 min. h Western blot analysis for phospho-Erk1/2 (upper panel) and Erk (lower panel) of MIN6-DTR preincubated with 10 μM PD98059 for 5 min and then incubated with 100 nM liraglutide in the presence of PD98059 for 3 min. i Cell viability of MIN6-DTR preincubated with 2 μM LY294002 and/or 10 μM PD098059 for 30 min and then incubated with 10 μg/ml DT and 10 nM liraglutide in the presence of LY294002 and/or PD098059 for 24 h. Data are mean ± SEM of four or five observations in the same experiment. Experiments using the same protocol were repeated twice or three times to ascertain reproducibility. *P < 0.05; **P < 0.01
We also examined whether the cytoprotective effect of liraglutide is mediated by phosphatidylinositol 3 (PI3) kinase and mitogen-activated protein (MAP) kinase signaling [10]. Phosphorylation of Akt and Erk by liraglutide was suppressed by inhibitors of PI3 kinase (LY294002) and MEK (PD098059), respectively (Fig. 3g, h), and both inhibitors attenuated cytoprotection by liraglutide, although their co-treatment evoked no additional decline in cell viability (Fig. 3i).
Effects of DPP4is and/or GLP1RAs on β cell proliferation during regeneration
Potentiating effects of DPP4is and/or GLP1RAs on β cell proliferation were evaluated by quantifying changes in β cell mass and BrdU incorporation during 14 days of regeneration. After an injection of DT, 8-week-old mice were administered MK-0626 and/or liraglutide for 14 days before pancreas excision (Fig. 4a). With or without DT injection, treatments with MK-0626, liraglutide, or MK-0626 plus liraglutide did not affect body weight, pancreas weight, or blood glucose levels (Table 2). MK-0626 treatment induced a significantly larger increase in mass of β cells (mostly Tomato-negative cells) after DT ablation compared with untreated group (Fig. 4b, c). By contrast, β cell mass was not increased by treatment with either liraglutide alone or MK-0626 plus liraglutide. Quantification of cell division by continuous labeling with BrdU revealed that β cell proliferation was increased significantly by MK-0626, but not by liraglutide or MK-0626 plus liraglutide (Fig. 4d, e). These results indicate that MK-0626 but not liraglutide increases β cell mass by promoting β cell proliferation. In addition, co-administration of liraglutide nullified the potentiating effect of MK-0626.

Effects of DPP4is and/or GLP1RAs on β cell proliferation during regeneration. a Experimental protocol to examine potentiating effects of DPP4ia and/or GLP1RAs on β cell proliferation. b Insulin and glucagon staining of islets in mice treated with MK-0626, liraglutide, or MK-0626 plus liraglutide at 14 days after DT injection. Scale bar 50 μm. c β cell mass in the treated mice at 14 days after DT injection (n = 4–5). Left graph DT-uninjected group; right graph DT-injected group. d Immunofluorescence of insulin (green), BrdU (red), and Hoechst (blue) in islets of the treated mice at 14 days after DT injection. e Quantification of BrdU-positive cells in β cells of the treated mice at 14 days after DT injection (n = 4–5). The cells of thirty random islets were analyzed. f α cell mass in the treated mice at 14 days after DT injection (n = 4–5). g Quantification of apoptotic cells in α cells of liraglutide-treated mice at 14 days (n = 3). The cells of thirty random islets were analyzed. h Immunofluorescence of chromogranin A (green), insulin (red), and glucagon/somatostatin/PP (blue) in islets of liraglutide-treated mice at 14 days. Arrowheads indicate cells positive only for chromogranin A. i Quantification of glucagon-positive and glucagon/somatostatin/PP-positive cells in islet cells of liraglutide-treated mice at 14 days (n = 4). The cells of thirty random islets were analyzed. Scale bar 15 μm. *P < 0.05; **P < 0.01
We next examined involvement of GLP-1 signaling on the effect of MK-0626. Co-administration of exendin-(9-39), a selective GLP-1 receptor antagonist, significantly suppressed the pro-proliferative effect of MK0626 judging from a lack of both β cell mass increase and BrdU incorporation (Fig. 4c, e), which indicates the involvement of GLP-1 signaling.
We also found that liraglutide treatment evoked a significant reduction in glucagon-positive α cells (Fig. 4b, f). Neither preceding DT administration nor co-administration of MK-0626 affected the α cell reduction by liraglutide. The mechanism of the reduction of α cell mass was evaluated by quantifying apoptotic α cells; the number of TUNEL-positive α cells was not increased by liraglutide treatment (Fig. 4g). On the other hand, we detected several endocrine cells stained negative to either insulin, glucagon, somatostatin, or PP in liraglutide-treated mice (Fig. 4h), suggesting that a loss of glucagon expression of α cells may explain the reduction of α cell number. We also compared the numbers of glucagon-positive cells and glucagon/somatostatin/PP-positive (positive for any of the three) cells. The number of glucagon-positive cells and that of glucagon/somatostatin/PP-positive cells were significantly decreased by liraglutide treatment (5.8 and 6.6 %, respectively), suggesting that pancreatic α cells are unlikely to transdifferentiate to other types of non-β endocrine cells.
Effects of DPP4is and/or GLP1RAs on islet function in vitro and glucose tolerance in vivo
We then evaluated the effects of MK-0626 and/or liraglutide on insulin secretory function of β cells and glucose tolerance in vivo. Insulin secretion of size-matched isolated islets under basal glucose level (G2.8) was not different among all groups (Fig. 5a), indicating basal insulin secretory capacity of β cells was not affected by these pharmacological interventions. High glucose-stimulated insulin secretion (GSIS) of islets isolated from MK-0626-treated mice was significantly increased, indicating that MK-0626 improves insulin secretory capacity of β cells as well as their volume. By contrast, liraglutide treatment failed to increase secretory capacity. Co-treatment of liraglutide did not attenuate the improvement in GSIS by MK-0626. In OGTTs, there was no significant difference in glucose excursion among the groups in DT-untreated mice (Fig. 5b). By contrast, in DT-treated mice, blood glucose levels of mice treated with MK-0626 plus liraglutide were significantly lower than those of the other three groups. Insulin secretory response to glucose was reported to be barely seen in Rip-Cre mice [24]; there was no significant increase in serum insulin levels in all our experimental groups (data not shown). To evaluate insulin secretory capacity under physiological conditions in vivo, serum insulin levels in fasted and refed conditions were measured in mice treated with MK-0626 and/or liraglutide. Refeeding substantially increased serum insulin levels in all animal groups (Fig. 5c). The insulin levels in refed mice treated with MK-0626 plus liraglutide were significantly higher than those of untreated mice, suggesting that concomitant administration might improve insulin secretory capacity in vivo.

Effects of DPP4is and/or GLP1RAs on islet function in vitro and glucose tolerance in vivo. a GSIS of islets isolated from mice treated with MK-0626, liraglutide, or MK-0626 plus liraglutide at 14 days after DT injection (n = 6–7). Isolated islets were incubated in RPMI medium containing 10 % FBS and 5.5 mM glucose for 4 h. Islets were preincubated in the incubation medium (described in “Materials and methods” section) containing 2.8 mM glucose for 30 min and then incubated in the incubation medium containing 2.8 or 16.7 mM glucose for 60 min. Fold increases are shown in inset. b Blood glucose levels after 1 g/kg oral glucose loading to the mice at 14 days after DT injection (n = 4). Left graph DT-uninjected group; right graph DT-injected group. c Serum insulin levels after 14 h-fasting and 3 h-refeeding at 14 days after DT injection (n = 4). *P < 0.05; † P < 0.05 and ‡ P < 0.01 versus untreated; §§ P < 0.01 versus MK-0626-treated; # P < 0.05 versus liraglutide-treated
Discussion
Quantification of β cell mass, its temporal changes, and the effectiveness of therapeutic intervention have been studied in animal models of β cell regeneration. These models include naturally occurring rodent diabetes models, β cell ablation models using streptozotocin or alloxan, partially pancreatectomized animal models, and genetically engineered mouse models [25]. In the present study, we generated a novel mouse model, Rip-Cre;Rosa26 DTR/tdTomato mice. In these mice, since we can induce cell death specifically in ~80 % β cells (i.e., β cells expressing DTR plus Tomato) at any desired time point, the precise time-course of subsequent regeneration can be observed. In fact, we detected vigorous β cell regeneration after cell ablation with DT. Our present study revealed that increased β cell mass in the mice was implemented by an increase in Tomato-negative β cell mass. These results indicate that β cell regeneration is mediated by proliferation of the β cells that remain unaffected by DT ablation, and not those formed by neogenesis or transdifferentiation from non-β cells. In the case of neogenesis and transdifferentiation, β cells generated de novo start to express Tomato and DTR for the first time in their cell lineage history, at the moment of differentiation to insulin-expressing β cells, as previously reported in similar mouse models specialized for cell lineage tracing experiments [26, 27]. In Rip-Cre;Rosa26 DTR/tdTomato mice, therefore, cytotoxic events and cell regeneration can be independently analyzed by focusing on Tomato-positive cells and -negative cells, respectively. In addition, a lack of hyperglycemia during regeneration enables assessment of direct actions of anti-diabetic drugs on β cells independently of their anti-hyperglycemic effect. Using this mouse model, we compared the effects of GLP1RA (liraglutide) and DPP4i (MK-0626) by quantifying several types of cells and analyzing β cell function.
Regarding β cell protection from DT ablation, MK-0626 and liraglutide have a similar effect without any additive or synergistic effect on each other, indicating that enhanced GLP-1 signaling accounts for this effect, although the possibility of involvement of DPP-4 substrates other than GLP-1 cannot be eliminated. In addition, in vitro study using MIN6-DTR revealed that β cell cytoprotection by GLP-1 is elicited via suppression of caspase-3/-9 signaling in PI3 kinase- and MAP kinase-dependent manners. Considering that activated caspase-3 signaling is involved in β cell apoptosis in islets of animals and humans with T2DM [28, 29], β cell injury by DT may well reflect the pathophysiology of T2DM.
In the present study, we found that β cell proliferation was accelerated by MK-0626, but not by liraglutide in Rip-Cre;Rosa26 DTR/tdTomato mice, which differs from several previous reports [10]. Since liraglutide increased proliferation of MIN6 cells in vitro (data not shown), the lack of increase in β cell proliferation by liraglutide in vivo may be a consequence secondary to its action on the cells other than β cells. In recent study of non-diabetic mice, GLP1RA treatment was reported to lower β cell proliferation and result in reduced β cell mass [14]. Accordingly, the difference in glycemic levels during proliferation may explain the distinct effect of GLP1RAs. The present study shows that MK-0626 and liraglutide have distinct consequences in β cell proliferation. However, since co-treatment with exendin-(9-39) nullified the potentiation of β cell proliferation by MK-0626, GLP-1 signaling is considered to be required for the pro-proliferative effect of MK-0626.
Since α cell mass was significantly decreased by liraglutide both with and without DT administration, reduction in α cell mass is likely to be the event independent to β cell regeneration. In addition, α cell mass was not affected by co-administration of MK-0626, indicating that the effect of liraglutide on α cell mass dominates that of MK-0626. It has been reported recently that β cell ablation failed to induce hyperglycemia in glucagon receptor deficient mice [30], suggesting that insulin might lose its physiological importance under glucagon deficiency. In the present study, β cell ablation in our mice triggered active β cell proliferation, which terminated after β cell recovery. In addition, ectopic islet transplantation to our mice markedly suppressed β cell proliferation (unpublished data), as previously reported [31]. These findings suggest that β cell proliferation is likely to be induced when more β cells are required. Taking these findings together, the reduction in α cell mass by liraglutide may lessen the need for β cell proliferation in our mice.
Considering (1) the lack of increase in apoptotic α cells by liraglutide treatment, (2) the very low frequency of cell proliferation under static condition (data not shown), (3) no transdifferentiation to δ or to PP cells, and (4) the existence of hormone negative endocrine cells, the reduction in α cells by liraglutide treatment may well be due to a loss of glucagon expression in α cells. Recently, dedifferentiation of endocrine cells has been reported in diabetic conditions [31]. Thus, α cells may lose glucagon expression under potent GLP-1 signaling.
We found that GSIS of islets isolated from MK-0626 treated mice was significantly improved. Since insulin secretion under basal glucose level was not affected by MK-0626, the improved GSIS is unlikely to be caused by increased β cell mass. Unexpectedly, liraglutide treatment was found not to improve GSIS in our experiments, and improvement in GSIS by MK-0626 also was observed in co-treatment with liraglutide. Therefore, although the mechanism of improved GSIS by MK-0626 is not clarified in the present study, substrates of DPP-4 other than GLP-1, such as GIP [32] and pituitary adenylate cyclase-activating polypeptide [33], may be involved in the favorable effects of MK-0626 on GSIS.
OGTTs showed that glucose tolerance was improved only by co-administration of MK-0626 and liraglutide. Serum insulin levels in fasted and refed mice treated with MK-0626 plus liraglutide were significantly higher than those of untreated mice. The trends of the insulin levels on refeeding among the four animal groups were similar to those during OGTT (data not shown). This raises the possibility that residual MK-0626 and liraglutide remaining in the body after 16 h (wash-out time for MK-0626) food deprivation and 24 h (wash-out time for liraglutide) interval from the last injection of liraglutide could have influenced glucose tolerance during OGTT. This might also explain the difference in insulin secretory function between in vitro and in vivo experiments. Our present study shows that MK-0626 increased β cell mass by accelerating β cell proliferation and improving insulin secretory function, while liraglutide reduces α cell mass. Improved secretory function by MK-0626 and reduced α cell mass by liraglutide was well retained in co-administered mice. Accordingly, α cell mass reduction by liraglutide may also contribute to improved glucose tolerance in conjunction with the beneficial effect of MK-0626 on β cells. Since beneficial effects of combination therapy have been reported in patients with T2DM [34], combination therapy with DPP4is and GLP-1RAs may well be a useful approach to obtain better glycemic control.
References
F. Ismail-Beigi, Clinical practice. Glycemic management of type 2 diabetes mellitus. N. Engl. J. Med. 366, 1319–1327 (2012). doi:10.1056/NEJMcp1013127
UK Prospective Diabetes Study (UKPDS) Group, Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352, 854–865 (1998)
S.E. Kahn, S.M. Haffner, M.A. Heise, W.H. Herman, R.R. Holman, N.P. Jones, B.G. Kravitz, J.M. Lachin, M.C. O’Neill, B. Zinman, G. Viberti, Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 355, 2427–2443 (2006). doi:10.1056/NEJMoa066224
UK Prospective Diabetes Study Group, UK prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes 44, 1249–1258 (1995)
H. Sakuraba, H. Mizukami, N. Yagihashi, R. Wada, C. Hanyu, S. Yagihashi, Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 45, 85–96 (2002). doi:10.1007/s001250200009
A.E. Butler, J. Janson, S. Bonner-Weir, R. Ritzel, R.A. Rizza, P.C. Butler, Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 (2003)
K.H. Yoon, S.H. Ko, J.H. Cho, J.M. Lee, Y.B. Ahn, K.H. Song, S.J. Yoo, M.I. Kang, B.Y. Cha, K.W. Lee, H.Y. Son, S.K. Kang, H.S. Kim, I.K. Lee, S. Bonner-Weir, Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J. Clin. Endocrinol. Metab. 88, 2300–2308 (2003). doi:10.1210/jc.2002-020735
J. Rahier, Y. Guiot, R.M. Goebbels, C. Sempoux, J.C. Henquin, Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 10(Suppl 4), 32–42 (2008). doi:10.1111/j.1463-1326.2008.00969.x
M.A. Nauck, Incretin-based therapies for type 2 diabetes mellitus: properties, functions, and clinical implications. Am. J. Med. 124, S3–S18 (2011). doi:10.1016/j.amjmed.2010.11.002
J.E. Campbell, D.J. Drucker, Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 17, 819–837 (2013). doi:10.1016/j.cmet.2013.04.008
M. Saito, T. Iwawaki, C. Taya, H. Yonekawa, M. Noda, Y. Inui, E. Mekada, Y. Kimata, A. Tsuru, K. Kohno, Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 19, 746–750 (2001)
K. Kaneko, K. Ueki, N. Takahashi, S. Hashimoto, M. Okamoto, M. Awazawa, Y. Okazaki, M. Ohsugi, K. Inabe, T. Umehara, M. Yoshida, M. Kakei, T. Kitamura, J. Luo, R.N. Kulkarni, C.R. Kahn, H. Kasai, L.C. Cantley, T. Kadowaki, Class IA phosphatidylinositol 3-kinase in pancreatic beta cells controls insulin secretion by multiple mechanisms. Cell Metab. 12, 619–632 (2010). doi:10.1016/j.cmet.2010.11.005
T. Ohyama, K. Sato, Y. Yamazaki, H. Hashizume, N. Horiguchi, S. Kakizaki, M. Mori, M. Kusano, M. Yamada, MK-0626, a selective DPP-4 inhibitor, attenuates hepatic steatosis in ob/ob mice. World J. Gastroenterol. 20, 16227–16235 (2014). doi:10.3748/wjg.v20.i43.16227
J.H. Ellenbroek, H.A. Tons, N. de Graaf, M.A. Hanegraaf, T.J. Rabelink, F. Carlotti, E.J. de Koning, Glucagon-like peptide-1 receptor agonist treatment reduces beta cell mass in normoglycaemic mice. Diabetologia 56, 1980–1986 (2013). doi:10.1007/s00125-013-2957-2
D.D. De Leon, S. Deng, R. Madani, R.S. Ahima, D.J. Drucker, D.A. Stoffers, Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes 52, 365–371 (2003)
E. Mukai, T. Ohta, H. Kawamura, E.Y. Lee, A. Morita, T. Sasase, K. Miyajima, N. Inagaki, T. Iwanaga, T. Miki, Enhanced vascular endothelial growth factor signaling in islets contributes to beta cell injury and consequential diabetes in spontaneously diabetic Torii rats. Diabetes Res. Clin. Pract. 106, 303–311 (2014). doi:10.1016/j.diabres.2014.08.023
T. Miki, K. Nagashima, F. Tashiro, K. Kotake, H. Yoshitomi, A. Tamamoto, T. Gonoi, T. Iwanaga, J. Miyazaki, S. Seino, Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. USA 95, 10402–10406 (1998)
J. Miyazaki, K. Araki, E. Yamato, H. Ikegami, T. Asano, Y. Shibasaki, Y. Oka, K. Yamamura, Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 (1990). doi:10.1210/endo-127-1-126
A. Iwama, H. Oguro, M. Negishi, Y. Kato, Y. Morita, H. Tsukui, H. Ema, T. Kamijo, Y. Katoh-Fukui, H. Koseki, M. van Lohuizen, H. Nakauchi, Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21, 843–851 (2004). doi:10.1016/j.immuni.2004.11.004
E. Mukai, S. Fujimoto, H. Sato, C. Oneyama, R. Kominato, Y. Sato, M. Sasaki, Y. Nishi, M. Okada, N. Inagaki, Exendin-4 suppresses SRC activation and reactive oxygen species production in diabetic Goto-Kakizaki rat islets in an Epac-dependent manner. Diabetes 60, 218–226 (2011). doi:10.2337/db10-0021
C. Postic, M. Shiota, K.D. Niswender, T.L. Jetton, Y. Chen, J.M. Moates, K.D. Shelton, J. Lindner, A.D. Cherrington, M.A. Magnuson, Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315 (1999)
N. Komatsu, T. Oda, T. Muramatsu, Involvement of both caspase-like proteases and serine proteases in apoptotic cell death induced by ricin, modeccin, diphtheria toxin, and pseudomonas toxin. J. Biochem. 124, 1038–1044 (1998)
D.R. McIlwain, T. Berger, T.W. Mak, Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 5, a008656 (2013). doi:10.1101/cshperspect.a008656
J.Y. Lee, M. Ristow, X. Lin, M.F. White, M.A. Magnuson, L. Hennighausen, RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 281, 2649–2653 (2006)
K. Oyama, K. Minami, K. Ishizaki, M. Fuse, T. Miki, S. Seino, Spontaneous recovery from hyperglycemia by regeneration of pancreatic beta-cells in Kir6.2G132S transgenic mice. Diabetes 55, 1930–1938 (2006). doi:10.2337/db05-1459
Y. Dor, J. Brown, O.I. Martinez, D.A. Melton, Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41–46 (2004). doi:10.1038/nature02520
T. Nir, D.A. Melton, Y. Dor, Recovery from diabetes in mice by beta cell regeneration. J. Clin. Invest. 117, 2553–2561 (2007)
Q. Wang, P.L. Brubaker, Glucagon-like peptide-1 treatment delays the onset of diabetes in 8 week-old db/db mice. Diabetologia 45, 1263–1273 (2002). doi:10.1007/s00125-002-0828-3
T. Tomita, Immunocytochemical localisation of caspase-3 in pancreatic islets from type 2 diabetic subjects. Pathology 42, 432–437 (2010). doi:10.3109/00313025.2010.493863
Y. Lee, E.D. Berglund, M.Y. Wang, X. Fu, X. Yu, M.J. Charron, S.C. Burgess, R.H. Unger, Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc. Natl. Acad. Sci. USA 109, 14972–14976 (2012). doi:10.1073/pnas.1205983109
C. Talchai, S. Xuan, H.V. Lin, L. Sussel, D. Accili, Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150, 1223–1234 (2012). doi:10.1016/j.cell.2012.07.029
D. Yabe, Y. Seino, Two incretin hormones GLP-1 and GIP: comparison of their actions in insulin secretion and beta cell preservation. Prog. Biophys. Mol. Biol. 107, 248–256 (2011). doi:10.1016/j.pbiomolbio.2011.07.010
Y. Sakurai, N. Shintani, A. Hayata, H. Hashimoto, A. Baba, Trophic effects of PACAP on pancreatic islets: a mini-review. J. Mol. Neurosci. 43, 3–7 (2011). doi:10.1007/s12031-010-9424-z
R. Violante, J.H. Oliveira, K.H. Yoon, V.A. Reed, M.B. Yu, O.P. Bachmann, J. Ludemann, J.Y. Chan, A randomized non-inferiority study comparing the addition of exenatide twice daily to sitagliptin or switching from sitagliptin to exenatide twice daily in patients with type 2 diabetes experiencing inadequate glycaemic control on metformin and sitagliptin. Diabet. Med. 29, e417–e424 (2012). doi:10.1111/j.1464-5491.2012.03624.x
Acknowledgments
This study was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, a grant (the 3rd Lilly Incretin Basic Research Grant) from Japan Diabetes Foundation, and a research grant from MSD K.K.
Author contribution
All authors contributed to conception and design, acquisition of data or analysis and interpretation of data, and to drafting the article or revising it critically for intellectual content. All authors gave final approval of the version to be published. The opinions expressed in this paper are those of the authors and do not necessarily represent those of MSD K.K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest associated with this manuscript.
Ethical standards
All animal experiments were approved by the Animal Care Committee of Chiba University.
Additional information
Asuka Morita and Eri Mukai have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Morita, A., Mukai, E., Hiratsuka, A. et al. Distinct effects of dipeptidyl peptidase-4 inhibitor and glucagon-like peptide-1 receptor agonist on islet morphology and function. Endocrine 51, 429–439 (2016). https://doi.org/10.1007/s12020-015-0733-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-015-0733-4