
Abstract
Impaired glucose tolerance and overt diabetes mellitus are becoming increasingly common complications of cystic fibrosis (CF), most probably merely as a result of increased life expectancy. In order to understand the pathophysiology of cystic fibrosis-related diabetes (CFRD), knowledge on the possible expression and cell distribution of the cystic fibrosis transmembrane conductance regulator (CFTR) protein within the endocrine pancreas is required. In this report, we establish the first evidence for expression of CFTR protein in rat pancreatic islets by using independent techniques. First reverse transcriptase-polymerase chain reaction (RT-PCR) amplification showed that CFTR mRNA is present in isolated islets of Langerhans. Furthermore, the analysis of flow cytometry-separated islet cells indicated that the level of CFTR transcripts is significantly higher in the non-β than in β-cell populations. The expression of CFTR protein in rat islet cells was also demonstrated by Western blotting and the level of expression was also found significantly higher in the non-β than in β-cell populations. Last, in situ immunocytochemistry studies with two monoclonal antibodies recognizing different CFTR epitopes indicated that CFTR expression occurs mainly in glucagon-secreting α-cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cystic fibrosis (CF) is the most common lethal autosomal recessive disease in Caucasians with a prevalence of approximately to 1:2,000 [1]. CF is a disorder resulting from a mutation in a single gene—the cftr gene encoding for the cystic fibrosis transmembrane conductance regulator (CFTR) membrane protein—, also called ABCC7, which functions as a cyclic adenosine monophosphate-regulated chloride channel [2, 3]. Over one thousand mutations in the CFTR gene have been reported (CF Genetic Analysis Consortium, http://www.genet.sickkids.on.ca/cftr/) and they are classified in to six categories (class I–VI) according to their consequence on CFTR functionality [4] with respectively, complete lack of synthesis, altered processing, altered regulation, altered conductance, reduced synthesis, and defective stability within the cell membrane. Thus although CF is a monogenic disease, it manifests as a broad clinical spectrum of phenotypes whose severity imperfectly correlates to the residual CFTR activity. The disorder primarily affects salt and fluid transport in epithelia. In the respiratory tract, it results in reduced mucociliary clearance eventually leading to thick dehydrated mucus, chronic and tenacious bacterial infections of the lungs (especially by Pseudomonas aeruginosa and Staphylococcus aureus), and inflammation and loss of pulmonary function. CF disease also includes intestinal obstruction (i.e. meconial ileus in the newborn), male infertility, hepatobiliary complications, chronic sinusitis, nasal polyp formation, and pancreatic insufficiency [5–7]. At birth, CF patients may or may not exhibit multiple symptoms of the disease. The severity and the rate of progression of the disease also vary considerably. Among CF patients, the status of pancreatic function is more directly linked to the CFTR mutations [8] unlike pulmonary involvement which further suggests the influence of environmental factors or modifiers genes to account for the severity of the disease [9–11]. Approximately 85% of CF patients have pancreatic deficiency leading to maldigestion and significant malabsorption. Those patients have a worse prognosis than the remaining “pancreatic sufficient” 15% patients [8–13], with a median survival rate of 29 and 56 years, respectively [14]. The clinical features of pancreatic insufficiency are also variable. Most CF patients present severe fibrosis of the pancreas and exocrine insufficiency. The lesion can begin in utero and can be identified in neonates on the basis of elevated blood concentration of pancreatic enzymes, classically trypsinogen [15]. Pancreatic endocrine dysfunction is also common, and the proportion of the CF population showing cystic fibrosis-related diabetes (CFRD) increases with age. Based on oral glucose tolerance test screening, a CFRD prevalence of 9, 26, 35, and 43% was found in patients aged 5–9, 10–19, 20–30, and >30 years, respectively [16]. CFRD shares features of both type 1 and type 2 diabetes, but these terms do not appear appropriate for CFRD [16]. Fasting hyperglycemia is encountered approximately in 15% of adult CF patients, most often insulin requiring while approximately 25% of adult CF patients have CFRD without fasting hyperglycemia. The expression of CFTR in exocrine pancreas is well demonstrated in intralobular and intercalated duct epithelia [4, 17–19]. In some species, a lower level is also described in acinar cells [20]. Cl−/HCO −3 exchangers (mostly the isoforms SLC26A6 and SLC26A3) [21–23] work in parallel with the CFTR in duct cells to provide a HCO − 3 rich pancreatic secretion. This secretion relies thus critically on epithelial chloride conductance. Its impairment leads to precipitation of proenzymes secreted by acinar cells and obstruction of small ducts with inflammation, necrosis, and fibrosis, and eventually pancreatic atrophy [24, 25]. Endocrine dysfunction in CF has been related to fibrosis and fatty infiltration of the exocrine pancreas, with secondary damage of islets of Langerhans [26, 27]. With these considerations in mind, we examined whether the CFTR protein is expressed in the rat endocrine pancreas. The present observation of CFTR expression within rat islets, mainly in the α-cells, may suggest new hypothesis on the pathogenesis of CFRD.
Results
Immunolocalization of CFTR in rat islets
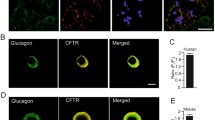
The cellular distribution of CFTR in the pancreas was studied using classical and confocal microscopy on permeabilized pancreatic sections. Immunofluorescence revealed that CFTR protein was observed within peripheral cells of islets of Langerhans. Both CFTR antibodies raised against CFTR [MATG-1104 (Fig 1a, b) and MA1-935 (Fig 1c)] showed a similar distribution. In the rat islet, the peripheral cells are either the α-cells containing glucagon or the δ-cells containing somatostatin. Double immunofluorescence technique was thus performed with each of the CFTR antibodies and either a somatostatin or a glucagon antibody. CFTR as well as somatostatin decorates the periphery of the islets, but they are clearly in two different cell populations since no colocalization of CFTR immunoreactivity (green) with somatostatin (red) was observed (Fig. 2a). On the other hand, a high level of CFTR labeling (Fig. 3a) was observed in the glucagon peripheral cells (Fig. 3b). The subcellular distribution of CFTR and glucagon immunoreactivity in these α pancreatic cells showed qualitative differences. For glucagon, the immunofluorescence labeling was more uniform (Fig. 3b) whereas the CFTR labeling was more reticular (Fig. 3a). The yellow color corresponds to the colocalization of CFTR and glucagon immunoreactivity (Fig. 3c, d). Although all results were consistent with the detection of the genuine CFTR, we cannot exclude in these experiments cross reactions with other related proteins, and the results should therefore be read as CFTR-like immunoreactivity. With respect to this conservative interpretation is the fact that CFTR immunoreactivity was also observed in ductal cells (Fig. 1d), confirming previous observations [17–19].

Immunodetection of CFTR in rat pancreatic section. Two different monoclonal antibodies against CFTR were used, either MATG-1104 (panels a and b) or MA1-935 (panels c and d). Immunohistochemical labeling was performed using avidin–biotin–peroxidase complex with diaminobenzidine (panels a and b) or using fluorescein-labelled secondary antibody (panels c and d). CFTR antibody labels the periphery of islets (panels a, b, and c) as well as diffusely the intercalated ducts of the exocrine pancreas (panels c and d). Scale bar: 100 μM

Co-localization studies of CFTR and somatostatin in rat pancreatic islets. Panel (a) Double labeling with anti-CFTR and anti-somatostatin antibodies demonstrating CFTR (green)- and somatostatin (red)-positive cells. Somatostatin and CFTR immunoreactivity appears to decorate distinct cells at the periphery of the islet. Panel (b) Light transmission microscopy corresponding to the pictures shown in panel (a). Scale bar: 15 μM

Co-localization studies of CFTR and glucagon in rat pancreatic islets. Double labeling with anti-CFTR (panel a) and anti-glucagon antibodies (panel b) demonstrating CFTR (green)- and glucagon (red)-positive cells. These confocal images were captured simultaneously, merged and presented in panels (c) and (d) and appear yellow (merge of green and red) indicating that both antigens are present within the same cells. The magnification in panel (d) indicates that within a single cell, the immunoreactivity does not always coincide, indicating that CFTR and glucagon are probably located in different compartments within the same cell. Scale bar: 10 μm
Immunoblot analysis of CFTR expression in islet cells
The presence of CFTR was also assessed by immunoblot analysis (Fig. 4) of whole-cell extracts (30 μg/lane) of isolated rat islets, of flow cytometry separated β- and non-β-cells, of RIN-m5F β-cells, of INR1-G9 α-cells and of T84 cells (15 μg in this lane). The immunoblot was performed using the monoclonal anti-CFTR antibody MA1-935 raised against the first transmembrane domain. CFTR was detected in all lanes but appeared slightly less abundant in β-cells than in non-β-cells.

Immunoblot analysis of CFTR expression in rat pancreatic islet and α- and β-cell lines extracts. Solubilized proteins from T84 cells (lane 1), total cell lysate of isolated rat islets (lane 2), β-cells (lane 3) and non-β cells (lane 4) providing from flow cytometry separated islet cells, RIN-m5F β-cells (lane 5), INR1-G9 α-cells (lane 6), were fractionated on an SDS/7.5%-PAGE gel and probed with the monoclonal antibody MA1-935 which detected always a band at ∼170 kDa whose intensity was however variable. Each lane corresponded to 30 μg proteins per lane except lane 1 where only 15 μg proteins were deposited.
Reverse transcriptase-PCR analysis of CFTR mRNA in rat endocrine pancreas
The expression of CFTR in pancreas has been previously studied by immunocytochemistry [17, 20] and by the determination of CFTR messenger RNA by in situ hybridization [18, 19] in mouse, rat, and human. These studies have characterized the expression of CFTR in exocrine pancreatic duct and acinar cells, but never in the endocrine pancreas. Therefore, the more sensitive RT-PCR technique was used in this study to determine the expression of the CFTR gene in isolated islets of Langerhans and in cell lines derived from endocrine pancreas. Based on a partial cDNA sequence from Rattus norvegicus CFTR (Genbank accession no. M89906) spanning a region homologous to human exons 10–16 (80% nucleotide identity), we designed two sets of oligonucleotide primers and used them to amplify by nested PCR, a predicted 401 bp product from reverse transcribed endocrine cell mRNA. CFTR transcripts were readily identified in as little as ∼300 ng of total RNA extract (Fig. 5a) from rat islets isolated by the collagenase technique (see Materials and methods). No PCR product appeared when either reverse transcriptase or the template was omitted. To localize more precisely the CFTR expression in the islets, the RT-PCR analysis was repeated on β and non-β islet cells separated by a method of flow cytometry of dispersed islet cells loaded with a fluorescent calcium indicator, as described previously by Mercan et al. [28]. 9 × 105 β-cells and 1.56 × 105 non-β-cells were obtained from eight rat pancreas (recovery of cells after sorting amounted to 87–90%) and the purified cells were further checked in term of insulin/glucagon contents by radioimmunoassay (data not shown). The RT-PCR (Fig. 5b) was performed with samples of cDNA obtained after reverse-transcription of RNA from approximately 28,000, 84,000 and 168,000 β-cells (Fig. 5b, lanes 3, 4 and 5, respectively), and from ∼5,000, 15,000, and 30,000 non-β-cells (Fig. 5b, lanes 7, 8, and 9, respectively). In three experiments, the 401-bp CFTR band was obtained with cDNA template from a minimum of 168,000 β-cells whereas this CFTR band is already more intense with cDNA from only 5,000 non-β-cells. Taking into account the number of cells used for the preparation of cDNA, the determination of CFTR band intensity by densitometry suggested that the level of CFTR mRNA was 60–80 times higher in non-β-cells than in β-cells. Since the RT-PCR product from β-cell population could result from non-β-cells contamination during the separation, we have repeated the RT-PCR analysis on the RIN-m5F cell line, which derived from rat pancreas insulinoma and is characterized as β-cells producing insulin (Fig. 5c). The results obtained with the cDNA from the non-β-cells separated by flow cytometry (Fig 5c, lanes 7, 8, and 9) were compared with RT-PCR products obtained with reverse-transcripts of mRNA from approximately 40,000, 120,000, and 240,000 RIN-m5F cells (Fig. 5c, lanes 3, 4, and 5, respectively). Three experiments showed the presence of CFTR mRNA in RIN-m5F cells but detectable only when starting from a minimum of 240,000 cells. In agreement with the determination of the 401-bp band intensity and in consideration of the number of cells involved in the preparation of reverse transcripts, the level of CFTR mRNA seems to be ∼90–100 times lower in RIN-m5F cells than in non-β-cells.

RT-PCR analysis of CFTR expression in rat endocrine pancreatic cells and in the RIN-m5F β-cell line. Panel (a) RT-PCR analysis of CFTR transcripts in extract from full islets of Langerhans. Lane 1: 600 islets were used to extract total RNA with Tri-Pure Reagent, 10 μg of total RNA were reverse-transcribed to cDNA in a volume of 33 μl, 1 μl of this volume was used in first PCR and the second PCR was performed with 1% of the total volume of the first PCR; lane 2: ditto lane 2 except that mRNA was extracted from 65 islets with Micro-Fast Track kit and total mRNA was reverse-transcribed to cDNA; lane 3: negative control (RT-PCR product of total RNA in the absence of reverse transcriptase). Panel (b) RT-PCR analysis of CFTR transcripts in flow cytometry separated β- and non-β-cells. Two samples of total RNA obtained with Tri-Pure Reagent were reverse-transcribed each in a volume of 33 μl. Several quantities of cDNA were used for the first PCR whereas the second PCR was always performed with 1% of the volume of the first PCR as template. Lane 1: 1 kb DNA ladder; lane 2: negative control; lanes 3–5: RT-PCR product of β cells with 1, 3, and 6 μl of cDNA, respectively; lanes 6: negative control; lanes 7–9: RT-PCR product of non-β-cells obtained with 1, 3, and 6 μl of cDNA, respectively. Panel (c): RT-PCR results from RIN-m5F β-cell line compared to flow cytometry separated non-β-cells (as in panel b). Thirty five μg of total RNA was obtained with ∼10 × 106 RIN-m5F cells and 5 μg were reverse-transcribed to cDNA in a volume of 33 μl. Several quantities of cDNA were used for the first PCR whereas the second PCR was performed with 1% of the volume of the first PCR as template. Lane 1, 1 kb DNA ladder; lane 2, negative control; lanes 3, 4, and 5, RT-PCR products of RIN-m5F with 1, 3, and 6 μl of cDNA, respectively; lane 6, negative control; lanes 7, 8, and 9, RT-PCR products of non-β-cells obtained with 1, 3, and 6 μl of cDNA
Discussion
The CFTR protein is well known to be expressed in the exocrine pancreas. On the other hand, CFTR has not been shown in the endocrine pancreas although islet function is also affected in CF patients, this being viewed as secondary to exocrine pancreas inflammation, fibrosis, and scarring. In the present study, we examined the expression of CFTR in Langerhans’ rat islets and related endocrine cell lines and observed that CFTR is constitutively expressed in islet cells, especially in α-cells.
In situ immunolocalization of CFTR using two different monoclonal anti-CFTR antibodies showed its expression mostly restricted to the periphery of the islets. Pancreatic sections double-stained with antibodies against CFTR, glucagon, and somatostatin indicated that the major locus of CFTR expression within the islets is the α-cells and not the δ-cells. The immunoblot analysis further confirmed this CFTR expression in isolated rat islets cell extract as well as in a fraction of non-β-cells separated by flow cytometry and in cultured hamster glucagonoma cells, the INR1-G9 cells. Of importance CFTR expression was also observed in β-cells, separated by flow cytometry, though at a lower level, and in a β-cell line, the RIN-m5F insulin-secreting cells.
Last, the presence of CFTR mRNA was also identified by nested RT-PCR in (i) isolated rat islets, (ii) flow –cytometry-separated islet cells with a much higher level of CFTR mRNA in non-β than in β-cell population, and (iii) a low level of CFTR transcripts in the RIN-m5F β-cell line. It is obviously not possible to decide whether CFTR expression in this β-cell line represents an actual β-cell characteristic or whether it merely results from dedifferentiation, as often observed in cell lines. Thus a low level of CFTR expression appears likely in the β insulin-secreting cells, yet remains questionable. On the other hand, the expression of CFTR in rat endocrine pancreas—mainly within the α-cells—is supported by three independent techniques: immunocytochemistry, immunoblotting, and RT-PCR analysis.
It is quite tempting to speculate that CFTR plays a role in the control of glucagon secretion. Hence mutations of the CFTR protein could be causative of abnormal glucose metabolism and diabetes in CF patients. This could account for the observation that post-mortem examination of CF pancreas [26, 29] failed to show more islet alterations in patients with CFRD than in CF patients without diabetes. Furthermore, although the secretion of islets hormones insulin, glucagon, and pancreatic polypeptide is often diminished in CF patients with pancreatic insufficiency [30], the secretion of somatostatin by islet delta cells is largely preserved or increased in CF patients with diabetes [31], as proinsulin levels in patients with CF who have abnormal glucose tolerance [32, 33]. Taken together, these observations do not support the common theory that CFRD merely results from severe fibrosis-induced islet destruction. Insulin secretion critically depends on membrane depolarization induced by closure of an ATP-dependent K+ channel. CFTR opening should lead to either depolarization or hyperpolarization depending on the electrochemical potential chloride gradient between the intra and extracellular fluid. It has been suggested that the chloride electrochemical potential gradient is high in β-cells due to the activity of a Na-K-2Cl-cotransporter in these cells [34] but low in α-cells due to the presence of a KCl-cotransporter [35]. Thus in α-cells, CFTR opening should induce chloride entry, cell membrane hyperpolarization, hence inhibit glucagon secretion. Thus in α-cells CFTR opening should induce chloride entry, cell membrane hyperpolarization and hence inhibit glucagon secretion. Therefore defective CFTR function in α-cells would be expected to lead to impaired glucagon suppression, a finding reported in CF patients but obviously a common finding in diabetes mellitus. As a matter of fact, glucose intolerance is observed in CF with reduced glucagon suppressibility following an oral glucose load [36]. A very low level of CFTR immunoreactivity was observed in β-cells in pancreatic sections, while the relative proportion of CFTR protein found in β-cells vs. non-β-cells, separated by flow cytometry, was respectively 28% and 72%, as estimated by densitometric scan analysis of the Western blots. Given the problem of islet α vs. β-cells separation, contamination of the β-cell population by non-β-cells should not be excluded. Moreover, the observed expression in derived β-cell lines does not resolve this issue as cell lines often behave differently from in situ islet cells. If present in β-cells, CFTR opening should induce chloride exit and cell membrane depolarization, hence insulin secretion. Interestingly in the distal nephron, the CFTR protein appears to control the permeability of the inwardly rectifying potassium channel (“ROMK”) also inserted within the apical membrane domain [37, 38].
In summary, regardless of any speculation on the physiopathological role of CFTR in islet cells, the present study unambiguously demonstrates that at least in the rat, α islet cells strongly express the CFTR protein.
Materials and methods
Immunochemistry
Simple immunohistochemical and double immunofluorescence staining were performed on pancreatic sections of Wistar rats by using antiserum raised against CFTR, glucagon, and somatostatin. All experimental procedures were conducted in agreement with the protocol approved by the Ethical and Animal Welfare Committee of the Université Libre de Bruxelles. Rats were killed by carbon dioxide inhalation. Pancreas were dissected free, fixed for 24 h at 4°C in freshly prepared 4% paraformaldehyde (PAF) solution in 0.1 M phosphate-buffered saline (PBS) at pH 7.4 and cryoprotected in graded solutions of sucrose in PBS (10, 20, and 30%; one day each) covered with OCT compound and frozen. Using a Leitz cryostat, 15-μm sections were cut, then mounted onto poly-l-lysine-coated slides and stored at −20°C until used for immunochemistry. Further pancreas samples were fixed for 24 h in formalin 10% diluted in PBS (v/v), dehydrated, embedded in paraffin, cut in tissue sections of 5 or 15-μm thickness using a conventional microtome (2035 Reichert-Jung, Biocut, Germany) and placed onto Superfrost slides (International Medical Products, Brussels, Belgium).
Two mouse monoclonal antibodies directed to CFTR were used. The first one was raised against the first extracellular loop of human CFTR, in a position where it is highly homologous to mouse and rat (MA1-935 from Affinity Bioreagents, Golden, CO, USA); the second one was R domain specific (MATG 1104 from Transgene S.A.,Strasbourg, France). A rabbit polyclonal antibody to pancreatic glucagon was from Novocastra, Newcastle upon Tyne, UK). The anti-somatostatin antibody was a kind gift of Prof. V. Leclercq-Meyer (Laboratoire de Médecine Expérimentale, Université Libre de Bruxelles, Brussels, Belgium).
Single immunohistochemical labeling ABC-DAB
Immunodetection was conducted following a standard ABC-DAB technique. Paraffin sections were deparaffinized in toluol and rehydrated in graded ethanol and propanolol solutions. Slides were treated for 30 min with H2O2 0.3% (w/v) in CH3OH to inhibit endogenous peroxidase, rinsed in PBS and incubated for 1 h at room temperature with the blocking solution, 5% (v/v) normal horse serum (S-2000, Vector Laboratories, Amsterdam, Netherlands) diluted in PBS. After several rinses, non-specific staining related to endogenous biotin was blocked by an avidin–biotin blocking kit (SP-2001, Vector Laboratories). After an overnight incubation with the anti-CFTR MAI-935 antibody (1:1,000), the sections were rinsed and incubated with a horse anti-mouse antibody conjugated to biotin (Vector Laboratories), at a dilution of 1/200. Controls were performed by omitting the primary antibody. The slides were then rinsed with PBS and the avidin–biotin–peroxidase complex (Vectastain® Elite® ABC kit, Vector Laboratories, the Netherlands) was applied. Peroxidase was revealed after 5 min incubation with diaminobenzidine (DAB substrate kit for peroxidase, SK-4100, Vector Laboratories, the Netherlands). The slides were finally dehydrated and permanently mounted with DePeX mounting medium (Gurr®, BDH, UK). The staining pattern was visualized using a Zeiss Axioplan (Carl Zeiss, Hamburg, Germany) and images were recorded with an Axiocam (Carl Zeiss).
Simple and double immunofluorescence labeling
Pancreas cryosections were incubated for 1 h at room temperature with 1:20 normal serum (goat or sheep normal serum depending of the origin of the secondary antiserum) in Tris-buffered Saline Solution (TBS). Incubation with primary antibodies was performed overnight in presence of blocking serum 1:20 diluted in TBS. Antibodies against CFTR were used at two different concentrations (1:100–1:250). After washing in TBS (three times) the sections were incubated for 1 h in the dark with both fluorescein-labeled secondary antibody and Texas-Red-labeled secondary antibody at 1:30 dilution. The preparation were again washed three times in PBS for 10 min, rinsed with water, and mounted with a drop of Gelvatol solution containing 100 mg/ml Dabco reagent. Pancreatic sections were observed under a Zeiss Axiovert fluorescence microscope and some images were obtained using a Laser-Scanning Confocal Microscope (MRC 1000, BioRad, Nazareth Eke, Belgium) using an argon-krypton laser and equipped with Laser-Sharp software (BioRad). Images were further analyzed using the J-Image software (National Institutes of Health, Bethesda, USA).
Isolation of islets of Langerhans
Pancreatic islets were isolated by the collagenase technique [39] from eight fed female Wistar rats. Briefly, the minced tissue is mixed with collagenase powder (7 mg/pancreas; Boehringer Mannheim) during 15 min at 37°C, and passed through a 400-μm mesh nylon filter (Nytal) to discard large exocrine aggregates. The islets were then placed in iced Hank’s solution and isolated under control of dissecting microscope, to minimize exocrine contamination. The islets were centrifuged for 30 min at 200g, and the pellet resuspended in a Ca2+-free HEPES-buffered Earle’s dissociation medium (NaCl 124 mM, MgSO4 0.8 mM, NaH2PO4 1 mM, NaHCO3 14.4 mM, HEPES 10 mM; pH 7.3) containing bovine serum albumin (2.5 mg/ml), EGTA (1 mM), and d-glucose (5.6 mM).
Separation of β and non-β islet cells
The isolation of pancreatic β- and non-β-cells is performed by flow cytometry of dispersed cells labeled with the calcium-sensitive fluorochrome fluo-3 [28]. Briefly, further digestion of islets was conducted in the presence of trypsin (0.8 μg/ml) and DNAse (0.3 μg/ml) for 15 min at 30°C, the digestion being stopped when about 60% of the islet cells were present as single cells. The suspension of cells was then passed through a 70-μm mesh nylon filter (Falcon products, In Vitrogen, Merelbeke, Belgium) and carefully layered on the surface of a 1.045 density Earle-HEPES-Percoll solution (Amersham Pharmacia Biotech Benelux, Roosendaal, Netherlands). After 15 min centrifugation (300g) at 6°C, the supernatant was discarded, the cell pellet being resuspended in 5 ml of dissociation medium (see above). For loading with fluo-3, an aliquot of a 1 mM stock solution of fluo-3 AM (Molecular Probes, In Vitrogen, Merelbeke, Belgium) prepared in DMSO was mixed by vortexing with an equal volume of 25% (w/v) solution of the dispersing agent Plutonic F-127 (Molecular Probes), 4 μl of this mixture then added to 1 ml of the cell suspension. The cells were then incubated for 30 min at 20°C. Thereafter, the cells were separated by centrifugation from the incubation medium, washed in a dye-free medium, resuspended in 5 ml of Earle HEPES buffer (see above) now containing 1.8 mM CaCl2, 16.7 mM d-glucose, and 5 mg/ml bovine serum albumin. Cell sorting was performed on a FACSTAR PLUS flow cytometer (Becton Dickinson, San Jose, CA, USA) equipped with an Enterprise Argon-ion laser (Coherent, Palo Alto, CA, USA). Data collection and processing were accomplished with Lysis II software. The 488 nm blue line of the laser was used for excitation at 50 mW output power; emission was measured through a 530/30 bandpass filter. Two populations of cells with significant different fluorescence were observed and gated for sorting and their specificity was checked by measuring insulin and glucagon in cell extracts, as described elsewhere [40].
Cell cultures
Cell culture media, l-glutamine, fetal calf serum, and antibiotics were purchased from Gibco BRL (Merelbeke, Belgium). The RIN-m5F beta cell line (ATCC nr: CRL-11605) is derived from a rat pancreas insulinoma. Cells were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS). The INR1-G9 hamster glucagonoma alpha cells [41] (a generous gift of Prof. H.-C. Fehmann, University of Marburg, Germany) were cultured in RPMI 1640 medium containing 11 mM d-glucose, supplemented with 10% FCS. The T84 cell line (ATCC nr: CCL-248), derived from a human colonic carcinoma and expressing CFTR protein, was cultured in DMEM supplemented with 5% FCS. The COS-1 cells (ATCC n°: CRL-1650) transfected transiently with human CFTR cDNA [42] were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS.
Immunoblot analysis of CFTR expression
Cell cultures (RIN-m5F, INR1-G9, and T84) were washed with ice-cold PBS and lysed in RIPA buffer (25 mM Tris, 150 mM NaCl, 0.5% deoxycholate, 0.1% SDS, 0.1% Triton X-100, pH 8.0). Nuclei and unbroken cells were removed by centrifugation (15,000g for 15 min at 4°C). Protein concentration of each sample was determined using the Biorad protein assay kit (Biorad). Soluble proteins (35 μg) were denaturated with Laemmli sample buffer and subjected to SDS-polyacrylamide gel electrophoresis on 7.5% separating gels. After electroblotting, Hybond polyvinylidene difluoride membranes (Amersham Pharmacia Biotech Benelux), were saturated with 5% non-fat dried milk in TBS (Tris 0.02 M, pH 7.5, NaCl 0.15 M) containing 0.1% Tween 20 (TBS-T) and then incubated for 1 h with a CFTR monoclonal antibody (MA1-935), raised against residues 103–117, and then incubated with the appropriate peroxidase-labeled secondary antibody (Amersham Pharmacia Biotech Benelux). Proteins were detected by exposure to enhanced chemiluminescence (ECL+Plus Western Blotting kit, Amersham Pharmacia Biotech Benelux) according to the manufacturer’s instructions.
RT-PCR analysis of CFTR mRNA
PCR was performed with a Perkin-Elmer apparatus using the Taq/Pwo polymerase from Roche Diagnostics Belgium (Vilvoorde, Belgium). Total RNA was extracted from rat pancreatic cells with Tri-Pure reagent (Roche Diagnostics Belgium). RNA was subsequently converted into complementary DNA using First-Strand cDNA synthesis kit (Amersham Pharmacia Biotech Benelux). Samples of cDNA were used as template for PCR amplification of CFTR with two sets of primers (M69-M70 and M71-M72) used successively for nested PCR. Assuming rat CFTR gene is interrupted by intronic DNA at the same positions as in human CFTR gene, probes M69 (antisense) and M70 (sense) encompass the last 275 base pairs (bp) of exon 13, all of exon 14 and the first 122 bp of exon 5. Probes M71 (sense) and M72 (antisense) encompass the last 157 bp of exon 13, all of exon 14 and the first 77 bp of exon 15. Oligodeoxyribonucleotides were synthesized on an Applied Biosystems synthesizer model 380A via the solid-phase phosphoramide method. The primer sequences were as follows:
primer set 1: 5′-GCAAAGTGTCAGCCACTCCCACG-3′ (M69) and 5′-GTTCCAGACTCTGAACATGGAGAGG-3′ (M70); primer set 2: 5′-GCTCAGTCTCCAGCAGTCTCCAGAGGACC-3′ (M71) and 5′-GCTGGAACTGGTGATGACCACAACATAGG-3′ (M72).
For the first and second sets of CFTR primers, the PCR reactions were carried out following a classical cycle. DNA fragments were separated on a 1.5% agarose gel containing 0.1 μg/ml ethidium bromide. Predicted fragment lengths were 565 bp for primer set 1 and 401 bp for primer set 2.
References
M.J. Welsh, L.-C. Tsui, T.F. Boat, A.L. Beaudet, in The Metabolic Basis of Inherited Disease, ed. by C.L. Scriver, A.L. Beaudet, W.S. Sly, D. Valle (McGraw Hill, New York, 1995), pp. 3799–3876
J.R. Riordan, J.M. Rommens, B. Kerem, et al., Science 245, 1066–1073 (1989)
C.E. Bear, C.H. Li, N. Kartner, et al., Cell 68, 809–818 (1992)
I. Crawford, P.C. Maloney, P.L. Zeitlin, et al., Proc. Natl. Acad. Sci. USA 88, 9262–9266 (1991)
F.S. Collins, Science 256, 774–779 (1992)
M.J. Welsh, A.E. Smith, Cell 73, 1251–1254 (1993)
S.M. Rowe, S. Miller, E.J. Sorscher, N. Engl. J. Med. 352, 1992–2001 (2005)
P. Kristidis, D. Bozon, M. Corey, et al., Am. J. Hum. Genet. 50, 1178–1184 (1992)
E. Kerem, M. Corey, B.S. Kerem, et al., N. Engl. J. Med. 323, 1517–1522 (1990)
R.K. Rowntree, A. Harris, Ann. Hum. Genet. 67, 471–485 (2003)
J. Clain, J. Lehmann-Che, E. Girodon, et al., Hum. Genet. 116, 454–460 (2005)
B. Kerem, J.M. Rommens, J.A. Buchanan, et al., Science 245, 1073–1080 (1989)
M. Wilschanski, P.R. Durie, J. Roy. Soc. Med. 91(Suppl 34), 40–49 (1998)
S.C. FitzSimmons, Cystic Fibrosis Foundation Patient Registry 1995: Annual Data Report (Cystic Fibrosis Foundation, Bethesda, MD, 1996)
E. Lebenthal, A. Lerner, D.D.K. Rolston, in The Pancreas: Biology, Pathobiology, and Disease, ed. by V.L.W. Go, E.P. Dimagno, J.D. Gardner, E. Lebenthal, H.A. Lebenthal, G.A. Sheele (Raven Press Ltd, New York, 1993), pp. 1041–1081
A. Moran, L. Doherty, X. Wang, W. Thomas, J. Pediatr. 133, 10–17 (1998)
C.R. Marino, L.M. Matovcik, F.S. Gorelick, J.A. Cohn, J. Clin. Invest. 88, 712–716 (1991)
A.E. Trezise, M. Buchwald, Nature 353, 434–437 (1991)
T.V. Strong, K. Boehm, F.S. Collins, J. Clin. Invest. 93, 347–354 (1994)
W. Zeng, M.G. Lee, M. Yan, et al., Am. J. Physiol. 273, C442–C455 (1997)
H. Shumaker, H. Amlal, R. Frizzell, C.D. Ulrich, M. Soleimani, Am. J. Physiol. 276, C16–C25 (1999)
D.B. Mount, M.F. Romero, Pflugers Arch. 447, 710–721 (2004)
H. Ishiguro, W. Namkung, A. Yamamoto, et al., Am. J. Physiol. 292, G447–G455 (2007)
H. Kopelman, P. Durie, K. Gaskin, Z. Weizman, G. Forstner, N. Engl. J. Med. 312, 329–334 (1985)
G.A. Scheele, S.I. Fukuoka, H.F. Kern, S.D. Freedman, Pancreas 12, 1–9 (1996)
A. Iannucci, K. Mukai, D. Johnson, B. Burke, Hum. Pathol. 15, 278–284 (1984)
M. Lohr, P. Goertchen, H. Nizze, et al., Virchows Arch. A 414, 179–185 (1989)
D. Mercan, I. Conget, E. Raspé, V. Leclercq-Meyer, W.J. Malaisse, Endocrine 2, 597–600 (1994)
L.E. Kopito, H. Shwachman, Pediatr. Res. 10, 742–749 (1976)
W.M. Moran, R.L. Hudson, S.G. Schultz, Am. J. Physiol. 251, G155–G159 (1986)
L.R. Meacham, L.P. McKean, C.N. Buchanan, et al., Pediatric Pulm. 6(suppl), 33 (1991)
S.G. Hartling, S. Garne, C. Binder, et al., Diabetes Res. 7, 165–169 (1988)
I. Hamdi, M. Green, J.M. Shneerson, C.R. Palmer, C.N. Hales, Clin. Endocrinol. 39, 21–26 (1993)
A. Majid, T. Speake, L. Best, P.D. Brown, Pflugers Arch. 442, 570–576 (2001)
S.L. Davies, E. Roussa, R.P. Le, et al., Biochim. Biophys. Acta 1667, 7–14 (2004)
A. Hinds, A.G. Sheehan, H. Machida, H.G. Parsons, Diabetes Res. 18, 69–78 (1991)
N. Inagaki, T. Gonoi, J.P. Clement, et al., Neuron 16, 1011–1017 (1996)
M. Lu, Q. Leng, M.E. Egan, et al., J. Clin. Invest. 116, 797–807 (2006)
F. Malaisse-Lagae, W.J. Malaisse, in Methods in Diabetes Research Vol. I, part B, ed. by J. Larner S.L. Pohl (Wiley, New York, 1984), pp. 147–152
V. Leclercq-Meyer, J. Marchand, M.C. Woussen-Colle, M.-H. Giroix, W.J. Malaisse, Endocrinology 116, 1168–1174 (1985)
R. Takaki, J. Ono, M. Nakamura, et al., In Vitro Cell Dev. Biol. 22, 120–126 (1986)
J.F. Pollet, G.J. Van, S.E. Van, et al., Biochim. Biophys. Acta 1500, 59–69 (2000)
Acknowledgments
We thank Jacqueline Van Geffel for assistance in the RT-PCR and Prof. Alex Bollen for helpful discussion. We also thank Prof. H.-C. Fehmann (from Philipps-University of Marburg, Marburg, Germany) for providing INR1-G9 alpha cell line and Dr A. Pavirani from Transgène (Strasbourg, France) for providing the MATG 1104 antibody used in this study. This work was supported by the “Fonds Alphonse et Jean Forton”, the “Fondation Jean Brachet” and the “Fonds de la Recherche Scientifique Médicale” (FRSM).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Boom, A., Lybaert, P., Pollet, JF. et al. Expression and localization of cystic fibrosis transmembrane conductance regulator in the rat endocrine pancreas. Endocr 32, 197–205 (2007). https://doi.org/10.1007/s12020-007-9026-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-007-9026-x