
Abstract
The mechanisms and signaling pathways of the neuroprotective effects of hypercapnia and its combination with hypoxia are not studied sufficiently. The study aims to test the hypothesis of the potentiating effect of hypercapnia on the systems of adaptation to hypoxia, directly associated with A1-adenosine receptors and mitochondrial ATP-dependent K+ -channels (mitoK+ATP-channels). We evaluated the relative number of A1-adenosine receptors and mitoK+ATP-channels in astrocytes obtained from male Wistar rats exposed to various respiratory conditions (15 times of hypoxia and/or hypercapnia). In addition, the relative number of these molecules in astrocytes was evaluated on an in vitro model of chemical hypoxia, as well as in the cerebral cortex after photothrombotic damage. This study indicates an increase in the relative number of A1-adenosine receptors in astrocytes and in cells next to the stroke region of the cerebral cortex in rats exposed to hypoxia and hypercapnic hypoxia, but not hypercapnia alone. Hypercapnia and hypoxia increase the relative number of mitoK+ATP-channels in astrocytes and in cells of the peri-infarct region of the cerebral cortex in rats. In an in vitro study, hypercapnia mitigates the effects of acute chemical hypoxia observed in astrocytes for A1-adenosine receptors and mitoK+ATP-channels. Hypercapnia, unlike hypoxia, does not affect the relative number of A1 receptors to adenosine. At the same time, both hypercapnia and hypoxia increase the relative number of mitoK+ATP-channels, which can potentiate their protective effects with combined exposure.
Graphic Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Intermittent hypoxia is effective in increasing the resistance of organs and tissues to ischemia (Chen et al., 2005; Yang et al., 2009). There is evidence of the effectiveness of hypoxia for the prevention and treatment of cardiovascular disease (Neckár et al., 2002; Shatilo et al., 2008) and ischemic brain damage (Sharp et al., 2004). However, the use of intermittent hypoxia in practice is limited due to long exposures (1–6 h) and long therapeutic courses (at least 7 times) (Neckár et al., 2002; Yang et al., 2009).
In recent years, interest in the therapeutic effects of carbon dioxide has increased significantly (Laffey et al., 2000; Tao et al., 2013). Permissive hypercapnia has been found to have neuroprotective properties (Tao et al., 2013; Zhou et al., 2010). It may be mediated by the inhibition of neuronal apoptosis (Tao et al., 2013; Zhou et al., 2010), antioxidant effect (Zakynthinos et al., 2007), and activation of angiogenesis (Siafakas et al., 2001).
Earlier, we proved that permissive hypercapnia and hypercapnic hypoxia led to a more intensive when compared to intermittent hypoxia increase in the resistance of the body and brain to acute ischemia (Tregub et al., 2013, 2015). However, the mechanisms and signaling pathways underlying the neuroprotective effects of hypercapnia and its combination with hypoxia are studied insufficiently.
Adenosine receptors have long been a target for the development of neuroprotective agents (Coppi et al., 2020). This mainly refers to A1-receptor agonists, which prevail in the central nervous system, reduce glutamate excitotoxicity, and modulate neuroinflammation in various neurological pathologies (Mahmoud et al., 2019; Martí Navia et al., 2020). Studies by Cao et al. (2019) showed that A1-adenosine receptor agonists have a protective potential in hypoxic brain damage by maintaining Ca2+ homeostasis, and Li et al. (2019) found a similar protective effect in an in vitro model.
Maintaining energy homeostasis and mitochondrial permeability is a key link in the protective paradigm of preconditioning, where mitochondrial ATP-sensitive K+ channels are one of the important signaling pathways (Smith et al., 2017). It was shown that the effect of neuroprotection after preconditioning of the brain is mediated by the opening of mitochondrial ATP-sensitive K+ channels with a subsequent increase in the elimination of nitric oxide (Deryagin et al., 2017). Activation of ATP-dependent potassium channels is neuroprotective in in vivo models of focal and global ischemia, and in vitro results suggest that these effects are mediated, not only by neuronal, but also by astrocytic channels (Szeto et al., 2018).
Under the exposure to intermittent hypoxia, adenosine and adenosine receptors (mainly the A-subtype) are involved in the development of the brain’s resistance to ischemia (Heurteaux et al., 1995). Adenosine leads to the activation of mitoK+ATP-channels (Kulinskiĭ et al., 2006) and reduces transmission of synaptic excitation (Ilie et al., 2006). Moreover, activation of adenosine A1 receptors fully reproduces the effect of preconditioning (Björklund et al., 2008; Yellon & Downey, 2003). Additionally, mitoK+ATP-channels are the end effectors of the signaling pathway of hypoxic preconditioning (Mayanagi et al., 2007). Activation of these channels causes a protective effect similar to ischemic preconditioning, and their inhibition, on the contrary, enhances ischemic damage (Ahmet et al., 2004). Herewith, carbon dioxide can cause the induction of Ca2+-activated and ATP-dependent membrane potassium channels (Lindauer et al., 2003). Therefore, the assessment of the role of A1-adenosine receptors and mitoK+ATP-channels in the mechanism of development of resistance to ischemia after the combined exposure to hypercapnia and hypoxia is of great interest.
In one study, we found that A1-adenosine receptors and mitoK+ATP-channels are involved in increasing resistance to acute oxygen deprivation after the combined exposure to hypercapnia and hypoxia (Tregub et al., 2014). Moreover, adenosine receptors were mainly affected by hypoxia, without the hypercapnic component. However, due to the lack of experimental data, it was not possible to draw reliable conclusions about the role of hypercapnia in the activation of A1-adenosine receptors and mitoK+ATP-channels.
We suggest that hypercapnia alone and/or in combination with hypoxia can activate signaling pathways of adaptation to ischemia/hypoxia associated with A1-adenosine receptors and mitoK+ATP-channels, since carbon dioxide has an effect on the factors of adaptation to hypoxia that are directly associated with these molecules. Testing this hypothesis was the goal of this study.
Establishing the role of A1-adenosine receptors and mitoK+ATP-channels in the molecular mechanisms of the neuroprotective efficacy of hypercapnia and hypercapnic hypoxia opens prospects for reducing the labor intensity and increasing the effects through the use of pharmacological modulators of ischemic and hypoxic preconditioning.
Materials and Methods
Animals
The studies were performed using in vivo and in vitro models on 60 male Wistar rats (Institute of Cytology and Genetics of the Siberian branch of the Russian Academy of Medical Sciences, Novosibirsk, Russia), with an average weight of 250–300 g. Animal experiments were approved by the bioethical board of the local ethical committee of the Krasnoyarsk State Medical University named after prof. V.F. Voino-Yasenetsky and conducted in compliance with the principles of the European Convention for the Protection of Vertebrate Animals. The animals were randomized using SPSS 11.5 (SPSS Inc, Chicago, IL). The rats have been kept in cages at controlled room temperature (~ 23 °C) and natural lighting. The rats had free access to food and water. Before and after the experiments, the animals were weighed.
Groups and Experimental Design
The study consisted of three series of experiments, with each series including four groups of samples/animals as shown in the experimental design (Online Resource 2). The first and second series were performed in in vitro models, and the third series was performed in an in vivo model. In each series of experiments, the groups differed in the partial oxygen pressure (PO2) and partial carbon dioxide pressure (PCO2), depending on the in vitro/in vivo model used:
-
NbH (normobaric hypoxia): in vivo—PO2 ≈ 90 mmHg and PCO2 ≈ 1 mmHg; the rest was N2; in vitro—PO2 ≈ 35 mmHg and PCO2 ≈ 35 mmHg; the rest was N2;
-
PermH (permissive hypercapnia): PO2 ≈ 150 mmHg; PCO2 ≈ 50 mmHg; the rest was N2;
-
HyperH (hypercapnic hypoxia): in vivo—PO2 ≈ 90 mmHg and PCO2 ≈ 50 mmHg; the rest was N2; in vitro—PO2 ≈ 35 mmHg and PCO2 ≈ 50 mmHg; the rest was N2;
-
Con (control group): in vivo—PO2 ≈ 150 mmHg and PCO2 ≈ 1 mmHg; the rest is N2; in vitro—PO2 ≈ 150 mmHg and PCO2 ≈ 35 mmHg; the rest is N2. Animals in this group underwent all experimental procedures, but without exposure to hypoxia and/or hypercapnia.
In the first experimental series, the respiratory effects in rats (n = 20) were performed in a special chamber for 15 days, 30 min per day. 24 h after completion of exposure courses, 5 rats from each group were subjected to euthanasia by decapitation. The hippocampus was removed to obtain a primary tissue culture of central nervous system progenitor cells and neurospheres (Kuvacheva et al. 2015). Neurospheres were used for further differentiation into astrocytes and neurons. After differentiation, immunostaining of adenosine A1 receptors and mitochondrial ATP-dependent potassium channels was performed in cells cultured in normal conditions and in chemical hypoxia.
In the second series of experiments, astrocytes obtained from the primary tissue culture of progenitor cells were cultured in the hypoxic chamber under conditions of normobaric hypoxia (PO2 ≈ 35 mmHg; PCO2 ≈ 35 mmHg; rest—N2) and hypoxia in combination with hypercapnia (PO2 ≈ 35 mmHg; PCO2 ≈ 50 mmHg; the rest—N2) for 24 h. In 6 h, they were subjected to chemical hypoxia followed by immunostaining of adenosine A1 receptors and mitochondrial ATP-dependent potassium channels. As a control for this series, cells cultured in standard conditions were used: temperature—37 °C, relative humidity—80%, PO2 ≈ 150 mmHg and PCO2 ≈ 35 mmHg; the rest was N2.
In the third experimental series, respiratory exposures were performed in rats (n = 40) in a special chamber for 15 days, 30 min daily. On the next day after completion of the course of the exposure, focal ischemic brain damage was modeled in all animals by photochemical thrombosis (Barth & Mody, 2011; Pevsner et al., 2001). 72 h after the surgery, perfusion fixation was carried out followed by decapitation and extraction of the brain.
Respiratory Exposure
To conduct respiratory exposure, the flow chamber described earlier was used (Tregub et al., 2015). The experimental groups of rats breathed a gas mixture corresponding to the experimental group. The control group was placed in the chamber under similar conditions, but instead of the gas mixture, atmospheric air was pumped by the compressor. The gas composition in the chamber was controlled by a “Microlux O2 + CO2” gas analyzer (Microlux Ltd., Russia).
Surgical Procedures and Photochemical Thrombosis
On the day before the surgery inducing focal ischemic damage, all animals did not receive food but had free access to water. Each rat was anesthetized by intraperitoneal administration of chloral hydrate (400 mg/kg). A sterile incision was made in the left inguinal region. A 4% solution of pink rose bengal (Sigma-Aldrich, Germany) was injected into the left femoral vein in 0.9% NaCl solution at a dose of 40 mg/kg.
Ischemic damage to the cerebral cortex was performed by transcranial photochemical thrombosis (Barth & Mody, 2011; Pevsner et al., 2001). A 10-min illumination of the scalped bones of the skull was performed with a green laser (532 nm, 20 mW). A section of the right parietal bone, 2 mm in diameter, located in the middle between the bregma and the lambdoid suture, and 2 mm lateral to the sagittal suture was illuminated on the skull.
Before decapitation, the animals underwent transcardial perfusion (jet injection of 500 ml of PBS followed by the perfusion with 250 ml of 4% PBS-buffered paraformaldehyde). After perfusion, the brain was removed and kept for 2 days in a 4% solution of PBS-buffered paraformaldehyde and 2 days in a 30% sucrose solution. Next, the brain was partitioned on a vibratome (thickness of the frontal sections was 60 μm). The sections were individually transferred to a 30% sucrose solution in 24-well plates for storage or analysis.
Obtaining the Initial Culture of the Neurospheres and the Primary Culture of Neurons and Astrocytes
The hippocampus was separated and removed from the brain in a 2% glucose solution in PBS. The resulting fragments were transferred to a fresh 2% glucose solution in PBS, followed by removal of the supernatant after passive sedimentation. After sedimentation, the tissue was resuspended in 1 ml of the medium from the NeuroCult Proliferation Kit (Mouse & Rat) (StemCell, Canada) and triturated to a homogeneous cell suspension. Further, the supernatant obtained after sedimentation was centrifuged at 150g for 5 min.
Progenitor cells isolated from the brain, with a density of 6–12 million viable cells/ml, were plated in T-25 cm2 culture vials in 10 ml of NeuroCult Proliferation Kit (Mouse & Rat) culture medium (StemCell, Canada). Subsequently, the cells were incubated in a CO2 incubator at 5% CO2 and a temperature of 37 °C.
Differentiation of the Neurospheres
After 24-h incubation, establishment of freely floating neurospheres (a conglomerate of cells proliferating as spheroids) was observed. Neurospheres were used to differentiate the cells into astrocytes after 4–5 days of culture.
To differentiate the neurospheres, differentiation factors were added to the culture medium. The commercial Astrocyte Medium kit media (Cat. No. 1821, ScienCell, USA) were used as nutrient media. After 7 days, an immunocytochemical assessment of the purity of the obtained cultures was carried out. GFAP was used as an astrocyte marker. Then astrocytes were transferred to 24-well plates for further experiments.
In Vitro Chemical Hypoxia Model
Astrocytes were incubated with sodium iodoacetate for 30 min at 37 °C in a CO2 incubator. The concentration of sodium iodoacetate in the medium was 50 μM. At the end of the incubation, the cells were washed and the culture medium was completely replaced, and cells were further cultured under standard conditions for 24 h.
In Vitro Modeling of Hypoxia and Hypercapnia
Modeling of hypoxic and hypercapnic-hypoxic effects in a culture of astrocytes isolated from progenitor cells of rats of the control group was carried out in the HypoxyLab chamber (Oxford Optronix Ltd, UK) (PO2 ≈ 35 mmHg; PCO2 ≈ 50 mmHg; the rest is N2), at 37 °C and a humidity level of 80% for 24 h.
Immunocytochemistry
The immunocytochemical assessment of A1-adenosine receptors and Kir6.2 (a subunit of the mitochondrial ATP-dependent potassium channel) relative number in astrocytes was performed by double indirect immunofluorescence. Preliminarily, the cells were prefixed with 2% paraformaldehyde solution in the culture medium for 5 min. Subsequent cell fixation was carried out with a 4% paraformaldehyde solution for 15 min.
After the fixation, permeabilization with 0.1% Triton X-100 solution (10 min at room temperature) and blocking with 10% BSA in PBS (30 min at 37°) have been performed. After BSA was removed and washed 3 times with PBS, the primary antibody solution was added to astrocytes for 2 h at 37 °C. After removing the primary antibodies and washing 3 times with PBS, a solution of secondary antibodies was added to the astrocytes for 1 h at 37 °C. To contrast the nuclei, a DAPI solution was used.
We used polyclonal rabbit antibodies to A1-adenosine receptors (Thermo Fisher, Cat. No. PA1-041A) at a dilution of 1:500 and monoclonal rabbit antibodies to Kir6.2 (Thermo Fisher, Cat. No. PA5-75331) at a dilution of 1:200 as primary antibodies. Polyclonal goat anti-rabbit antibodies conjugated with AlexaFlour555 at a dilution of 1:500 (Thermo Fisher, Cat. No. A-21428) were used as secondary antibodies.
Fluorescence microscopy was carried out on a ZOE cell analysis system (Bio-Rad, USA) including a count of the number of immunopositive cells relative to the total number of cells. Microphotographs were processed using ImageJ 1.41 (Scion Inc., USA).
Immunohistochemistry
An immunohistochemical study was performed on free-floating sections of the brain. A blocking solution with 10% BSA in PBS was added to the sections for 60 min at 37 °C. After removal of BSA and washing twice with PBS solution, a primary antibody solution was added to the sections, incubating on a shaker overnight at + 4 °C. After the primary antibodies were removed and washed twice with PBS solution, a secondary antibody solution was added to the sections for 2 h at room temperature. Primary and secondary antibodies were used as indicated in “Immunocytochemistry” section.
After incubation with secondary antibodies and washing three times, the sections were transferred onto glass slides with the addition of fluoromount and DAPI. Counting the number of immunopositive cells in the peri-infarction region was performed in at least 5 fields of view using an FV10i-W confocal microscope (Olympus, Japan).
Statistical Analysis
The size of the total sample and of each group was calculated from previous studies on a similar model using the quantitative scale method (Dell et al., 2002). Statistical analysis was performed with the SPSS 11.5 program (SPSS Inc, Chicago, IL). The hypothesis of distribution normality was tested by the Shapiro–Wilk test. Due to the fact that some of the data did not correspond to the normal distribution, univariate Kruskal–Wallis analysis of variance was performed to compare the differences between groups. In the analysis, the dependent variables were the percentage of A1-adenosine receptors in astrocytes, the percentage of A1-adenosine receptors in the peri-stroke region of the cerebral cortex, the percentage of mitoK+ATP-channels in astrocytes, and the percentage of mitoK+ATP-channels in the peri-stroke region of the cerebral cortex.
Quantitative data are presented as median (Me), lower quartile (25%) and upper quartile (75%). The differences were considered significant if the probability of error (p) was < 0.05.
Results
Relative Number of A1-Adenosine Receptors in Astrocytes In Vitro
The relative number of A1-adenosine receptors in astrocytes isolated from rat hippocampus after 15 respiratory effects of hypoxia and/or hypercapnia in vivo (Fig. 1) differed between groups both during cell growth under normal conditions and after chemical hypoxia modeling.

The relative number of A1-adenosine receptors in astrocytes isolated from rat progenitor cells subjected to respiratory exposure in vivo: A, B the percentage of immunopositive cells in relation to the total number of cells under normal and hypoxic conditions. Data are presented as Me (25; 75%). *Differences compared to control (p < 0.05); **differences compared to the control (p < 0.001); #differences compared to the NbH group (p < 0.001); &differences compared to the PermH group (p < 0.001). C, D A1-adenosine receptors in astrocytes under normal and hypoxic conditions. Magnification ×175. Blue color—DAPI; Red color—primary antibodies to A1-adenosine receptors associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia, PermH permissive hypercapnia, HyperH hypercapnic hypoxia
Moreover, under normoxic conditions (Fig. 1A), a decrease in the relative number of A1 receptors for adenosine was found in the PermH group compared to the control group by 26% (p < 0.05), and in the NbH and HyperH groups, there was an increase in the relative number of 2 and 2.5 times, respectively (p < 0.001).
After chemical hypoxia (Fig. 1B), the control group (p < 0.001) and the PermH group (p < 0.05) had an increased relative number of adenosine A1 receptors in more than twofold compared to normoxic conditions. In the NbH group, on the contrary, there was a twofold decrease in their relative number relative to the control group (p < 0.001). Moreover, the number of A1 receptors in the HyperH group did not change under conditions of chemical hypoxia, remaining comparable to the control group.
The relative number of A1-adenosine receptors in astrocytes after a 24-h exposure to in vitro hypoxia and/or hypercapnia (Fig. 2) differed between groups both during cell growth under normal toxic conditions and after chemical hypoxia modeling.

The relative number of A1-adenosine receptors in astrocytes subjected to hypoxia and hypercapnia in vitro: A, B the percentage of immunopositive cells in relation to the total number of cells under normal and hypoxic conditions. Data are presented as Me (25; 75%). *Differences compared to control (p < 0.05); **differences compared to control (p < 0.001); #differences compared to the NbH group (p < 0.001). C, D A1-adenosine receptors in astrocytes under normal and hypoxic conditions. Magnification ×175. Blue color—DAPI; Red color—primary antibodies to A1-adenosine receptors associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia; HyperH hypercapnic hypoxia
An increase in the relative number of A1-adenosine receptors under normoxic conditions (Fig. 2A) was found in the NbH group, exceeding control values by 3 times (p < 0.001). After modeling chemical hypoxia (Fig. 2B) in the astrocytes of the control group (p < 0.001), the relative number of A1-adenosine receptors increased by 2.3 times, in the HyperH group by 20% (p < 0.01), and, in contrast, in 2.7 times (p < 0.01) in relation to normoxic conditions. Moreover, the values in the experimental groups remained below the corresponding control level (p < 0.01), without differences.
The Number of A1-Adenosine Receptors in the Peri-infarct Zone
The number of A1-adenosine receptors in the near-stroke region of the cerebral cortex (Fig. 3) in the NbH group was twice as high as in the control (p < 0.001). In the HyperH group, this value was higher by 45%, relative to the control values (p < 0.05). Moreover, exposure in the PermH group reduced the number of A1-adenosine receptors by almost half (p < 0.05).

The number of A1-adenosine receptors in the peri-infarct region of the cerebral cortex (A). Microphotographs of a peri-infarct region of the cerebral cortex (B). Data are presented as Me (25; 75%). **Differences compared to control (p < 0.001); *differences compared to control (p < 0.05); #differences compared to the NbH group (p < 0.05); ##differences compared to the NbH group (p < 0.001); &differences compared to the PermH group (p < 0.001). Magnification ×250. Confocal microscopy. Blue color—DAPI; red—primary antibodies to A1-adenosine receptors associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia; PermH permissive hypercapnia; HyperH hypercapnic hypoxia
The Relative Number of mitoK+ ATP-Channels in Astrocytes In Vitro
The relative number of mitoK+ATP-channels in astrocytes isolated from rat hippocampus after 15 respiratory effects of hypoxia and/or hypercapnia in vivo (Fig. 4A) was increased under normoxic conditions in all experimental groups (p < 0.001). Moreover, in the HyperH group the relative number of mitoK+ATP-channels increased by almost 7 times, in the PermH group by 2.5 times, and in the NbH group by 1.8 times.

The relative number of mitoK+ATP-channels in astrocytes isolated from rat progenitor cells subjected to respiratory exposure in vivo: A, B percentage of immunopositive cells in relation to the total number of cells under normal and hypoxic conditions. Data are presented as Me (25; 75%). *Differences compared to control (p < 0.05); **differences compared to control (p < 0.001); #differences compared to the NbH group (p < 0.001); &differences compared to the PermH group (p < 0.001). C, D mitoK+ATP-channels in astrocytes under normal and hypoxic conditions. Magnification ×175. Blue color—DAPI; red color—primary antibodies to Kir6.2 (a subunit of mitoK+ATP-channel) associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia; PermH permissive hypercapnia; HyperH hypercapnic hypoxia
The exposure to chemical hypoxia (Fig. 4B) caused a decrease in the relative number of mitoK+ATP-channels, compared with the corresponding control. Minimal values were observed in astrocytes from the PermH group, where there was a decrease in the relative number of mitoK+ATP-channels by 5.4 times (p < 0.001). Moreover, groups with the hypoxic component had a decrease of 24% (p < 0.05 for NbH; p < 0.001 for HyperH).
Notably, in the control and NbH groups, adding sodium iodoacetate increased the relative number of mitoK+ATP-channels in relation to normoxic conditions (p < 0.001), while the opposite effect occurred in the groups PermH (p < 0.001) and HyperH (p < 0.05).
The relative number of mitoK+ATP-channels in astrocytes after a 24-h exposure to hypoxia and/or hypercapnia in vitro under normoxic conditions (Fig. 5A) increased in the NbH group by 4.2 times, and in the HyperH group by 3.2 times (p < 0.001).

The relative number of mitoK+ATP-channels in astrocytes subjected to hypoxia and hypercapnia in vitro: A, B the percentage of immunopositive cells in relation to the total number of cells under normal and hypoxic conditions. Data are presented as Me (25; 75%). *Differences compared to control (p < 0.001); #differences compared to the NbH group (p < 0.05); ##differences compared to the NbH group (p < 0.001); C, D mitoK+ATP-channels under normal and hypoxic conditions. Magnification ×175. Blue color—DAPI; Red color—primary antibodies to Kir6.2 (a subunit of the mitoK+ATP-channel) associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia; HyperH hypercapnic hypoxia
After modeling chemical hypoxia (Fig. 5B) in the control group (p < 0.001), the relative number of mitoK+ATP-channels was increased by 7 times, and in the NbH group, it was reduced by 2.4 times (p < 0.001), compared to normoxic conditions. Moreover, after the addition of sodium iodoacetate in the HyperH group, the relative number of mitoK+ATP-channels did not change when compared to the normoxic conditions and was 53% higher than that of the NbH group (p < 0.001). At the same time, both experimental groups showed significantly lower values when compared to the controls (p < 0.001).
The Number of mitoK+ ATP-Channels in the Peri-infarct Zone
The number of mitoK+ATP-channels in the peri-infarct region of the cerebral cortex (Fig. 6) in rats from the NbH group was almost 2 times higher compared to the control (p < 0.001), in the PermH group 2.6 times (p < 0.001), and in the HyperH group it is 3.4 times higher (p < 0.001). Moreover, the values of the PermH and HyperH groups were higher compared to the NbH group (p < 0.01), without showing mutual differences.

The number of mitoK+ATP-channels in the peri-infarct region of the cerebral cortex (A). Data are presented as Me (25; 75%). *Differences compared to control (p < 0.001); #differences compared to the NbH group (p < 0.001). Microphotographs of a peri-infarct region of the cerebral cortex (B). Magnification ×250. Confocal microscopy. Blue color—DAPI; red color—primary antibodies to Kir6.2 (subunit of mitoK+ATP-channel) associated with a secondary antibody conjugated to AlexaFlour555. NbH normobaric hypoxia; PermH permissive hypercapnia; HyperH hypercapnic hypoxia
General data on the effects of exposure (activation/inhibition) of permissive hypercapnia, normobaric hypoxia, and hypercapnic hypoxia on the relative number of A1-adenosine receptors in astrocytes, the number of A1-adenosine receptors in the peri-infarct region of the cerebral cortex, the relative number of mitoK+ATP-channels in astrocytes, and the number of mitoK+ATP-channels in the peri-infarct region of the cerebral cortex are presented in Table 1.
Discussion
In recent years, much attention has been paid to the neuroprotective effect of ischemic/hypoxic preconditioning (Sharp et al., 2004; Obrenovitch, 2008; Zhao et al., 2012). However, the clinical application of these methods has significant limitations (Lukyanova et al., 2009; Rybnikova & Samoilov, 2015). At the same time, the study of signaling pathways of preconditioning is important for understanding the molecular mechanisms of neuroprotection and the development of new methods of prevention/treatment of central nervous system pathology.
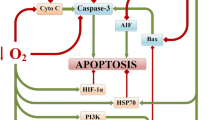
The combination of intermittent hypoxia with hypercapnia is one of the promising neuroprotective strategies for the development of resistance of the brain to ischemia. Hypercapnic hypoxia likely causes a neuroprotective effect due to increased brain perfusion (Faraco et al., 2015; Haubrich et al., 2011; Qi et al., 2012), inhibition of apoptosis in the peri-infarct region (Tao et al., 2013; Zhou et al., 2010), increased activity of the transcription factor HIF-1α (Kulikov et al., 2015), antioxidant effect (Kniffin et al., 2014; Zakynthinos et al., 2007), and increasing angiogenesis and levels of chaperone HSP-70 (Bespalov et al., 2014).
Ischemic/hypoxic preconditioning and intermittent hypoxia affect similar signaling pathways, but with different intensities (Rybnikova & Samoilov, 2015; Sharp et al., 2004), which allows us to propose a hypothesis about their neuroprotective effects. According to the classical model of preconditioning, the neuroprotection mechanism consists of a cascade of events: the release of adenosine, stimulation of A1 receptors, and opening of ATP-dependent K+-channels sensitive to sulfonylurea (Heurteaux et al., 1995; Kulinskiĭ et al., 2006; Wang et al., 2011). According to Yellon and Downey (2003), activation of A1-adenosine receptors reproduces the effect of preconditioning and increases the tolerance of myocardial cells to ischemia. Additionally, activation of ATP-dependent K+-channels in the brain causes an effect similar to ischemic preconditioning (Mayanagi et al., 2007).
This study was conducted to evaluate the role of A1-adenosine receptors and mitoK+ATP-channels in the neuroprotective effect after the combined intermittent exposures to hypercapnia and hypoxia (Tregub et al., 2015). To perform intermittent respiratory exposures, we used those parameters (gas concentrations, multiplicity, and duration of sessions) that have the maximum neuroprotective potential (Tregub et al., 2013, 2015). To assess the neuroprotective potential of the studied signaling pathways directly, we used the model of photoinduced cerebral vascular thrombosis (Barth & Mody, 2011; Pevsner et al., 2001). We also evaluated the effect of hypoxia and hypercapnia on the relative number of A1-adenosine receptors and mitoK+ATP-channels in astrocytes, which are considered as potential targets for neuroprotection and recovery in ischemic stroke (Björklund et al., 2008; Liu & Chopp, 2016).
It is established that ischemic and hypoxic preconditioning increases the level of adenosine and activates A1-adenosine receptors, which leads to an increase in the brain’s resistance to ischemia (Heurteaux et al., 1995; Obrenovitch, 2008). Moreover, Liu et al. (2011) proved that the activation of A1-adenosine receptors reduces the chemosensitivity of respiratory neurons to increased CO2, but not decreased O2. This suggests the antagonism of these receptors and permissive hypercapnia.
The data obtained in cell models in vitro (“Relative Number of A1-Adenosine Receptors in Astrocytes In Vitro” section) suggest some important conclusions. First, the stimulating effect of intermittent hypoxia, but not hypercapnia, on A1-adenosine receptors in astrocytes was detected after their isolation into a culture. This indicates the absence of a direct effect of CO2 on these receptors when combined with hypoxic exposure. In addition, this shows that intermittent hypoxia might affect the epigenetic regulation of the relative number of A1-adenosine receptors in astrocytes even after their cultivation. This can be attributed to the effects of the transcription factor HIF-1, which enhances the activity of adenosine receptors (Howell & Tennant, 2014) or to the effect of hypoxia on the G-proteins that are associated with these receptors (Jacobson & Gao, 2006).
Assessment of the relative number of A1-adenosine receptors in astrocytes under in vitro chemical hypoxia demonstrates the protective effect of the combined exposure to hypercapnia and hypoxia. This effect stabilizes the relative number of receptors in relation to the control level. It is observed both in astrocytes after exposure to hypercapnic hypoxia in vivo, and in astrocytes subjected to an equivalent in vitro exposure. Moreover, a decrease in the relative number of A1-adenosine receptors in astrocytes after exposure to normobaric hypoxia, detected under conditions of chemical hypoxia, may indicate a decrease in their adaptive potential (Obrenovitch, 2008).
It is important to note that hippocampal A1-adenosine receptors are mainly localized in neurons, in particular in synapses (Rebola et al., 2003; Tetzlaff et al., 1987). However, astrocytes were the main object of our in vitro studies because we were interested in the general patterns of the adaptive response of nervous tissue to hormetic stimuli, which were intermittent hypoxia and hypercapnia. It can be assumed that the results obtained for A1-adenosine receptors, which are less characteristic of astrocytes, will be true for neurons (Björklund et al., 2008). This is confirmed by the results from “The Number of A1-Adenosine Receptors in the Peri-infarct Zone” section, where the assessment of the density of receptors in neurons of the peri-infarct region corresponded to the trend described in “Relative Number of A1-Adenosine Receptors in Astrocytes In Vitro” section. In addition, we sought to study A1-adenosine receptors and mitoK+ATP-channels on one type of neural cells in order to correctly interpret the experimental results.
The functioning of K+ATP-channels depends on the redox state of the active groups of protein channels, and redox agents modulate their activity (Garlid & Beavis, 1986). Under conditions of hypoxia and ischemia, an increase in the concentration of reactive oxygen species (Mironova et al., 2010), a change in the ratios of GSH/GSSG and NAD+/NADH, which lead to the modification of thiol groups of cysteines in membrane structures (Obrenovitch, 2008) have been demonstrated. Moreover, an increase in the intracellular level of reactive oxygen radicals leads to a pronounced activation of mitoK+ATP-channels (Zhang et al., 2001). This is also facilitated by an increase in the synthesis of NO with the formation of peroxynitrite (Dahlem et al., 2004; Lacza et al., 2003; Sasaki et al., 2000) and subsequent activation of protein kinase C (Krenz et al., 2002). In addition, there is evidence that the selective activation of mitochondrial ATP-dependent K+ channels in astrocytes induces delayed preconditioning (Rajapakse et al., 2003) and increases the uptake of glutamate in culture, which may provide an additional protective advantage (Sun et al., 2008).
Evaluation of the relative number of mitoK+ATP-channels in astrocytes (“The Relative Number of mitoK+ATP-Channels in Astrocytes In Vitro” section) after exposure to in vivo normobaric hypoxia and permissive hypercapnia indicates the possibility of regulating their number through epigenetic mechanisms. This may be due to O2-deficient oxidative stress (Mironova et al., 2010) and activation of protein kinase C, caused by an increase in the concentration of CO2/bicarbonate and an increase in intracellular Ca2+ (Obrenovitch, 2008). In addition, the effect on sodium iodoacetate astrocytes demonstrates the stabilizing effect of hypercapnia on the regulation of the relative number of mitoK+ATP-channels. This effect is to maintain the number of channels after the exposure to hypercapnic hypoxia at the control level, which can be interpreted as an indicator of ischemic/hypoxic tolerance (Obrenovitch, 2008). This may be due to the antioxidant effects of carbon dioxide (Barth et al., 1998; Goss et al., 1999; Zakynthinos et al., 2007; Zhao et al., 1998) and the subsequent limitation of oxidative stress in astrocytes.
Modeling the focal ischemic stroke (“The Number of A1-Adenosine Receptors in the Peri-infarct Zone” and “The Number of mitoK+ATP-Channels in the Peri-infarct Zone” sections) revealed trends similar to in vitro results. We found that permissive hypercapnia inhibits A1 receptors in cells surrounding stroke region and reduces their number when combined with normobaric hypoxia. At the same time, permissive hypercapnia increases the number of mitoK+ATP-channels both in isolated exposure and when combined with normobaric hypoxia (Fig. 7). These data, in the context of higher neuroprotective potential of hypercapnic hypoxia, can be considered as a protective mechanism against excessive induction of the adenosine signaling link with an emphasis on the final preconditioning effectors—mitoK+ATP-channels (Heurteaux et al., 1995; Obrenovitch, 2008). This assumption is also supported by data that prolonged activation of adenosine A1 receptors may have a negative role in the pathogenesis of neurological disorders (Stockwell et al., 2017).

The role of A1-adenosine receptors and mitoK+ATP-channels in the molecular mechanisms of the neuroprotective efficacy of hypercapnia and hypercapnic hypoxia. Red lines—inhibition; Green lines—activation/induction. References to primary sources are indicated in square brackets
The results of this study are consistent with the previously obtained data (Tregub et al., 2014), which showed that application of A1-adenosine receptor blockers and mitoK+ATP-channels before exposure to hypercapnic hypoxia precludes the formation of brain’s resistance to hypoxia. At the same time, blocking adenosine receptors did not affect the neuroprotective efficacy of permissive hypercapnia, in contrast to normobaric hypoxia. However, blocking mitoK+ATP-channels had an inhibitory effect on the formation of resistance to hypoxia for both permissive hypercapnia and normobaric hypoxia. Also, Lindauer et al. (2003) showed that exposure to hypercapnia stimulates Ca2+-activated and ATP-dependent potassium channels in the brain vessels.
The observed effect of the combined exposure to moderate doses of hypoxia and hypercapnia can be discussed from the perspective of the hormesis paradigm (Leak et al., 2018). The main reason for this is the fact that both a deficiency of oxygen and carbon dioxide and their excess in the body causes serious damage. Therefore, the positive effects found in the intermittent effects of moderate doses of hypoxia and hypercapnia will be relevant only for a certain therapeutic range of gas concentrations, duration of sessions, and frequency of courses. It is also important to emphasize that a hypothesis has already been formulated regarding hypercapnia and hypoxia reflecting the possibility of their use as a hormetic stimulus (Pruimboom & Muskiet, 2018).
Changes in the relative number of A1-adenosine receptors and mitoK+ATP-channels affect many cellular functions and should be considered as a target for novel preventive and therapeutic methods in hypoxic/ischemic damage of the central nervous system. The combination of hypercapnic-hypoxic respiratory exposures with pharmacological modulators (2-chloroadenosine, diazoxide) is a promising approach (Li & Roth, 1999; Sun et al., 2012). At the same time, combination of pharmacological modulators with the exposure to hypercapnic hypoxia and drugs that affect other adaptogenic signaling pathways can be most effective (Baillieul et al., 2017). This is confirmed by the data on the increase in the neuroprotective effects of hypoxia in combination with a NOS inhibitor (Malyshev et al., 1999), as well as an increase in the cardioprotective effect when combining hypoxic preconditioning with opioid receptor activators (Maslov et al., 2009).
Conclusions
According to the results of the study, we can conclude that hypercapnia, unlike hypoxia, does not have a stimulating effect on the relative number of A1-adenosine receptors. Moreover, hypercapnia, regardless of hypoxia, causes an increase in the relative number of mitoK+ATP-channels when they are combined, which is an important component of the mechanism of neuroprotective effectiveness of hypercapnic hypoxia. These results open prospects for the development of new neuroprotective techniques based on a combination of hypercapnic-hypoxic exposures and pharmacological modulators of the signaling pathways in preconditioning.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Code Availability
Not applicable.
References
Ahmet, I., Krawczyk, M., Heller, P., Moon, C., Lakatta, E. G., & Talan, M. I. (2004). Beneficial effects of chronic pharmacological manipulation of beta-adrenoreceptor subtype signaling in rodent dilated ischemic cardiomyopathy. Circulation, 110(9), 1083–1090.
Baillieul, S., Chacaroun, S., Doutreleau, S., Detante, O., Pépin, J. L., & Verges, S. (2017). Hypoxic conditioning and the central nervous system: A new therapeutic opportunity for brain and spinal cord injuries? Experimental Biology and Medicine (Maywood, N.J.), 242(11), 1198–1206.
Barth, A. M., & Mody, I. (2011). Changes in hippocampal neuronal activity during and after unilateral selective hippocampal ischemia in vivo. Journal of Neuroscience, 31, 851–860.
Barth, A., Bauer, R., Gedrange, T., Walter, B., Klinger, W., & Zwiener, U. (1998). Influence of hypoxia and hypoxia/hypercapnia upon brain and blood peroxidative and glutathione status in normal weight and growth-restricted newborn piglets. Experimental and Toxicologic Pathology, 50(4–5), 402–410.
Bespalov, A. G., Tregub, P. P., Kulikov, V. P., Pijanzin, A. I., & Belousov, A. A. (2014). The role of VEGF, HSP-70 and protein S-100B in the potentiation effect of the neuroprotective effect of hypercapnic hypoxia. Patologicheskaia Fiziologiia i Eksperimentalnaia Terapiia, 2, 24–27.
Björklund, O., Shang, M., Tonazzini, I., Daré, E., & Fredholm, B. B. (2008). Adenosine A1 and A3 receptors protect astrocytes from hypoxic damage. European Journal of Pharmacology, 596(1–3), 6–13.
Cao, T., Ma, T., Xu, Y., Tian, Y., Cai, Q., Li, B., & Li, H. (2019). Caffeine treatment promotes differentiation and maturation of hypoxic oligodendrocytes via counterbalancing adenosine 1 adenosine receptor-induced calcium overload. Medical Science Monitor, 25, 1729–1739.
Coppi, E., Dettori, I., Cherchi, F., Bulli, I., Venturini, M., Lana, D., Giovannini, M. G., Pedata, F., & Pugliese, A. M. (2020). A2B adenosine receptors: When outsiders may become an attractive target to treat brain ischemia or demyelination. International Journal of Molecular Sciences, 21(24), 9697.
Chen, W. J., Chen, H. W., Yu, S. L., Huang, C. H., Wang, T. D., Chen, J. J., Chien, C. T., Chen, H. Y., Yang, P. C., & Lee, Y. T. (2005). Gene relative number profiles in hypoxic preconditioning using cDNA microarray analysis: Altered relative number of an angiogenic factor, carcinoembryonic antigen-related cell adhesion molecule 1. Shock, 24, 124–131.
Dahlem, Y., Horn, T., Butinas, L., Gonoi, T., Wolf, T., & Siemen, D. (2004). The human mitochondrial KATP channel is modulated by calcium and nitric oxide: A patch-clamp approach. Biochimica et Biophysica Acta, 1656, 46–56.
Dell, R. B., Holleran, S., & Ramakrishnan, R. (2002). Sample size determination. ILAR Journal, 43, 207–213.
Deryagin, O. G., Gavrilova, S. A., Gainutdinov, K. L., Golubeva, A. V., Andrianov, V. V., Yafarova, G. G., Buravkov, S. V., & Koshelev, V. B. (2017). Molecular bases of brain preconditioning. Frontiers in Neuroscience, 11, 427.
Faraco, C. C., Strother, M. K., Siero, J. C., Arteaga, D. F., Scott, A. O., Jordan, L. C., & Donahue, M. J. (2015). The cumulative influence of hyperoxia and hypercapnia on blood oxygenation and R2. Journal of Cerebral Blood Flow and Metabolism, 35(12), 2032–2042.
Garlid, K. D., & Beavis, A. D. (1986). Evidence for the existence of an inner membrane anion channel in mitochondria. Biochimica et Biophysica Acta, 273, 13578–13582.
Goss, S. P., Singh, R. J., & Kalyanaraman, B. (1999). Bicarbonate enhances the peroxidase activity of Cu, Zn-superoxide dismutase. Role of carbonate anion radical. Journal of Biological Chemistry, 274(40), 28233–28239.
Haubrich, C., Steiner, L., Kasprowicz, M., Diedler, J., Carrera, E., Diehl, R. R., Smielewski, P., & Czosnyka, M. (2011). Short-term moderate hypocapnia augments detection of optimal cerebral perfusion pressure. Journal of Neurotrauma, 28(7), 1133–1137.
Heurteaux, C., Lauritzen, I., Widmann, C., & Lazdunski, M. (1995). Essential role of adenosine, adenosine A1 receptors, ATP-sensitive K+ channels in cerebral ischemic preconditioning. Proceedings of the National Academy of Sciences USA, 92(10), 4666–4670.
Howell, N. J., & Tennant, D. (2014). The role of HIFs in ischemia-reperfusion injury. Hypoxia, 2, 107–115.
Ilie, A., Ciocan, D., Zagrean, A. M., Nita, D. A., Zagrean, L., & Moldovan, M. (2006). Endogenous activation of adenosine A1 receptors accelerates ischemic suppression of spontaneous electrocortical activity. Journal of Neurophysiology, 96(5), 2809–2814.
Jacobson, K. A., & Gao, Z. G. (2006). Adenosine receptors as therapeutic targets. Nature Reviews Drug Discovery, 5, 247–264.
Kniffin, C. D., Burnett, L. E., & Burnett, K. G. (2014). Recovery from hypoxia and hypercapnic hypoxia: Impacts on the transcription of key antioxidants in the shrimp Litopenaeus vannamei. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 170, 43–49.
Krenz, M., Oldenburg, O., Wimpee, H., Cohen, M. V., Garlid, K. D., Critz, S. D., Downey, J. M., & Benoit, J. N. (2002). Opening of ATP-sensitive potassium channels causes generation of free radicals in vascular smooth muscle cells. Basic Research in Cardiology, 97, 365–373.
Kulikov, V. P., Tregub, P. P., Kovzelev, P. D., Dorokhov, E. A., & Belousov, A. A. (2015). Hypercapnia-alternative hypoxia signal incentives to increase HIF-1α and erythropoietin in the brain. Patologicheskaia Fiziologiia i Eksperimentalnaia Terapiia, 3, 34–37.
Kulinskiĭ, V. I., Gavrilina, T. V., Minakina, L. N., & Kovtun, VIu. (2006). Biochemical and pharmacological mechanisms of different types of hypoxic preconditioning in cerebral ischemia in mice. Biomeditsinskaya Khimiya, 52(3), 309–316.
Kuvacheva, N. V., Morgun, A. V., Komleva, Y. K., Khilazheva, E. D., Gorina, Y. V., Lopatina, O. L., Arutyunyan, S. A., & Salmina, A. B. (2015). In vitro modeling of brain progenitor cell development under the effect of environmental factors. Bulletin of Experimental Biology and Medicine, 159(4), 546–549.
Lacza, Z., Snipes, J., Kis, B., Szabo, C., Grover, G., & Busija, D. (2003). Investigation of the subunit composition and the pharmacology of the mitochondrial ATP-dependent K+ channel in the brain. Brain Research, 994, 27–36.
Laffey, J. G., Tanaka, M., Engelberts, D., Luo, X., Yuan, S., Tanswell, A. K., Post, M., Lindsay, T., & Kavanagh, B. P. (2000). Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. American Journal of Respiratory and Critical Care Medicine, 162, 2287–2294.
Leak, R. K., Calabrese, E. J., Kozumbo, W. J., Gidday, J. M., Johnson, T. E., Mitchell, J. R., Ozaki, C. K., Wetzker, R., Bast, A., Belz, R. G., Bøtker, H. E., Koch, S., Mattson, M. P., Simon, R. P., Jirtle, R. L., & Andersen, M. E. (2018). Enhancing and extending biological performance and resilience. Dose Response, 16, 1559325818784501. https://doi.org/10.1177/1559325818784501
Li, B., & Roth, S. (1999). Retinal ischemic preconditioning in the rat: Requirement for adenosine and repetitive induction. Investigative Ophthalmology & Visual Science, 40(6), 1200–1216.
Li, H. L., Zaghloul, N., Ahmed, I., Omelchenko, A., Firestein, B. L., Huang, H., & Collins, L. (2019). Caffeine inhibits hypoxia-induced nuclear accumulation in HIF-1α and promotes neonatal neuronal survival. Experimental Neurology, 317, 66–77.
Lindauer, U., Vogt, J., Schuh-Hofer, S., Dreier, J. P., & Dirnagl, U. (2003). Cerebrovascular vasodilation to extraluminal acidosis occurs via combined activation of ATP-sensitive and Ca2+-activated potassium channels. Journal of Cerebral Blood Flow and Metabolism, 23(10), 1227–1238.
Liu, Z., & Chopp, M. (2016). Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Progress in Neurobiology, 144, 103–120.
Liu, C., Cao, Y., Malhotra, A., & Ling, L. (2011). Sleep fragmentation attenuates the hypercapnic (but not hypoxic) ventilatory responses via adenosine A1 receptors in awake rats. Respiratory Physiology & Neurobiology, 175(1), 29–36.
Lukyanova, L. D., Germanova, E. L., & Kopaladze, R. A. (2009). Development of resistance of an organism under various conditions of hypoxic preconditioning: Role of the hypoxic period and reoxygenation. Bulletin of Experimental Biology and Medicine, 147(4), 400–404.
Martí Navia, A., Dal Ben, D., Lambertucci, C., Spinaci, A., Volpini, R., Marques-Morgado, I., Coelho, J. E., Lopes, L. V., Marucci, G., & Buccioni, M. (2020). Adenosine receptors as neuroinflammation modulators: Role of A1 agonists and A2A antagonists. Cells, 9(7), 1739.
Malyshev, I. Y., Zenina, T. A., Golubeva, L. Y., Saltykova, V. A., Manukhina, E. B., Mikoyan, V. D., Kubrina, L. N., & Vanin, A. F. (1999). NO-dependent mechanisms of adaptation to hypoxia. Nitric Oxide, 3, 105–113.
Maslov, L. N., Lishmanov, Y. B., Oeltgen, P. R., Barzakh, E. I., Krylatov, A. V., Govindaswami, M., & Brown, S. A. (2009). Activation of peripheral δ2 opioid receptors increases cardiac tolerance to ischemia/reperfusion injury: Involvement of protein kinase C, NO-synthase, KATP channels and the autonomic nervous system. Life Sciences, 84, 657–663.
Mahmoud, S., Gharagozloo, M., Simard, C., & Gris, D. (2019). Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells, 8(2), 184.
Mayanagi, K., Gáspár, T., Katakam, P. V., Kis, B., & Busija, D. W. (2007). The mitochondrial K(ATP) channel opener BMS-191095 reduces neuronal damage after transient focal cerebral ischemia in rats. Journal of Cerebral Blood Flow and Metabolism, 27(2), 348–355.
Mironova, G. D., Shigaeva, M. I., Gritsenko, E. N., Murzaeva, S. V., Gorbacheva, O. S., Germanova, E. L., & Lukyanova, L. D. (2010). Functioning of the mitochondrial ATP-dependent potassium channel in rats varying in their resistance to hypoxia. Involvement of the channel in the process of animal’s adaptation to hypoxia. Journal of Bioenergetics and Biomembranes, 42(6), 473–481.
Neckár, J., Papousek, F., Nováková, O., Ost’ádal, B., & Kolár, F. (2002). Cardioprotective effects of chronic hypoxia and ischaemic preconditioning are not additive. Basic Research in Cardiology, 97(2), 161–167.
Obrenovitch, T. P. (2008). Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiological Reviews, 88, 211–247.
Pevsner, P. H., Eichenbaum, J. W., Miller, D. C., Pivawer, G., Eichenbaum, K. D., Stern, A., Zakian, K. L., & Koutcher, J. A. (2001). A photothrombotic model of small early ischemic infarcts in the rat brain with histologic and MRI correlation. Journal of Pharmacological and Toxicological Methods, 45, 227–233.
Pruimboom, L., & Muskiet, F. A. J. (2018). Intermittent living: The use of ancient challenges as a vaccine against the deleterious effects of modern life—A hypothesis. Medical Hypotheses, 120, 28–42.
Qi, L., Meng, L., Li, Y., & Qu, Y. (2012). Arterial carbon dioxide partial pressure influences CYP4A distribution in the rat brain. Histology and Histopathology, 27(7), 897–903.
Rajapakse, N., Kis, B., Horiguchi, T., Snipes, J., & Busija, D. (2003). Diazoxide pretreatment induces delayed preconditioning in astrocytes against oxygen glucose deprivation and hydrogen peroxide-induced toxicity. Journal of Neuroscience Research, 73, 206–214.
Rebola, N., Pinheiro, P. C., Oliveira, C. R., Malva, J. O., & Cunha, R. A. (2003). Subcellular localization of adenosine A(1) receptors in nerve terminals and synapses of the rat hippocampus. Brain Research, 987(1), 49–58.
Rybnikova, E., & Samoilov, M. (2015). Current insights into the molecular mechanisms of hypoxic pre- and postconditioning using hypobaric hypoxia. Frontiers in Neuroscience, 9, 388.
Sasaki, N., Sato, T., Ohler, A., O’Rourke, B., & Marban, E. (2000). Activation of mitochondrial ATP-dependent potassium channels by nitric oxide. Circulation, 101, 439–445.
Stockwell, J., Jakova, E., & Cayabyab, F. S. (2017). Adenosine A1 and A2A receptors in the brain: Current research and their role in neurodegeneration. Molecules, 22(4), 676.
Smith, C. O., Nehrke, K., & Brookes, P. S. (2017). The Slo(w) path to identifying the mitochondrial channels responsible for ischemic protection. The Biochemical Journal, 474(12), 2067–2094.
Sharp, F. R., Ran, R., Lu, A., Tang, Y., Strauss, K. I., Glass, T., Ardizzone, T., & Bernaudin, M. (2004). Hypoxic preconditioning protects against ischemic brain injury. NeuroRx, 1(1), 26–35.
Shatilo, V. B., Korkushko, O. V., Ischuk, V. A., Downey, H. F., & Serebrovskaya, T. V. (2008). Effects of intermittent hypoxia training on exercise performance, hemodynamics, and ventilation in healthy senior men. High Altitude Medicine & Biology, 9(1), 43–52.
Siafakas, N. M., Jordan, M., Wagner, H., Breen, E. C., Benoit, H., & Wagner, P. D. (2001). Diaphragmatic angiogenic growth factor mRNA responses to increased ventilation caused by hypoxia and hypercapnia. European Respiratory Journal, 17, 681–687.
Sun, X. L., Zeng, X. N., Zhou, F., Dai, C. P., Ding, J. H., & Hu, G. (2008). KATP channel openers facilitate glutamate uptake by gluts in rat primary cultured astrocytes. Neuropsychopharmacology, 33, 1336–1342.
Sun, J., Tong, L., Luan, Q., Deng, J., Li, Y., Li, Z., Dong, H., & Xiong, L. (2012). Protective effect of delayed remote limb ischemic postconditioning: Role of mitochondrial K(ATP) channels in a rat model of focal cerebral ischemic reperfusion injury. Journal of Cerebral Blood Flow and Metabolism, 32(5), 851–859.
Szeto, V., Chen, N. H., Sun, H. S., & Feng, Z. P. (2018). The role of KATP channels in cerebral ischemic stroke and diabetes. Acta Pharmacologica Sinica, 39(5), 683–694.
Tao, T., Liu, Y., Zhang, J., Xu, Y., Li, W., & Zhao, M. (2013). Therapeutic hypercapnia improves functional recovery and attenuates injury via antiapoptotic mechanisms in a rat focal cerebral ischemia/reperfusion model. Brain Research, 1533, 52–62.
Tetzlaff, W., Schubert, P., & Kreutzberg, G. W. (1987). Synaptic and extrasynaptic localization of adenosine binding sites in the rat hippocampus. Neuroscience, 21(3), 869–875.
Tregub, P., Kulikov, V., & Bespalov, A. (2013). Tolerance to acute hypoxia maximally increases in case of joint effect of normobaric hypoxia and permissive hypercapnia in rats. Pathophysiology, 3, 165–170.
Tregub, P. P., Kulikov, V. P., Stepanova, L. A., Zabrodina, A. S., & Nagibaeva, M. E. (2014). The role of adenosine A1 receptors and mitochondrial K+ATP channels in the mechanism of increasing the resistance to acute hypoxia in the combined effects of hypoxia and hypercapnia. Patologicheskaia Fiziologiia i Eksperimentalnaia Terapiia, 4, 48–52.
Tregub, P., Kulikov, V., Motin, Y., Bespalov, A., & Osipov, I. (2015). Combined exposure to hypercapnia and hypoxia provides its maximum neuroprotection effect during focal ischemic injury in the brain. Journal of Stroke and Cerebrovascular Diseases, 24(2), 381–387.
Wang, L., Zhu, Q. L., Wang, G. Z., Deng, T. Z., Chen, R., Liu, M. H., & Wang, S. W. (2011). The protective roles of mitochondrial ATP-sensitive potassium channels during hypoxia-ischemia-reperfusion in brain. Neuroscience Letters, 491(1), 63–67.
Yang, C. C., Lin, L. C., Wu, M. S., Chien, C. T., & Lai, M. K. (2009). Repetitive hypoxic preconditioning attenuates renal ischemia/reperfusion induced oxidative injury via upregulating HIF-1 alpha-dependent bcl-2 signaling. Transplantation, 88, 1251–1260.
Yellon, D. M., & Downey, J. M. (2003). Preconditioning the myocardium: From cellular physiology to clinical cardiology. Physiological Reviews, 83(4), 1113–1151.
Zakynthinos, S., Katsaounou, P., Karatza, M. H., Roussos, C., & Vassilakopoulos, T. (2007). Antioxidants increase the ventilatory response to hyperoxic hypercapnia. American Journal of Respiratory and Critical Care Medicine, 175, 62–68.
Zhang, D., Chen, Y., Campbell, W., Zou, A., Gross, G., & Li, P. (2001). Characteristics and suproxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channel. Circulation Research, 89, 1177–1183.
Zhao, Z. S., Khan, S., & O’Brien, P. J. (1998). Catecholic iron complexes as cytoprotective superoxide scavengers against hypoxia/reoxygenation injury in isolated hepatocytes. Biochemical Pharmacology, 56(7), 825–830.
Zhao, Y. D., Cheng, S. Y., Ou, S., Xiao, Z., He, W. J., Jian-Cui, & Ruan, H. Z. (2012). Effect of hypobaric hypoxia on the P2X receptors of pyramidal cells in the immature rat hippocampus CA1 sub-field. Brain Injury, 26(3), 282–290.
Zhou, Q., Cao, B., Niu, L., Cui, X., Yu, H., Liu, J., Li, H., & Li, W. (2010). Effects of permissive hypercapnia on transient global cerebral ischemia–reperfusion injury in rats. Anesthesiology, 112, 288–297.
Acknowledgements
The study was supported by a grant from the Russian Science Foundation (Project No. 18-75-00016). We wish to thank Prof. Alla Salmina and Dr. Elizaveta Boytsova for great help in the work on this study.
Funding
The study was supported by a grant from the Russian Science Foundation (Project No. 18-75-00016).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical Approval
Animal experiments were approved by the Bioethical Commission of the local Ethics Committee of Krasnoyarsk State Medical University. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed (EU Directive 2010/63/EU for animal experiments).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tregub, P.P., Malinovskaya, N.A., Osipova, E.D. et al. Hypercapnia Modulates the Activity of Adenosine A1 Receptors and mitoK+ATP-Channels in Rat Brain When Exposed to Intermittent Hypoxia. Neuromol Med 24, 155–168 (2022). https://doi.org/10.1007/s12017-021-08672-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-021-08672-0