
Abstract
Resolvins, belonging to the group of specialized proresolving mediators (SPMs), are metabolic products of omega-3 polyunsaturated fatty acids (ω-3 PUFAs) and are synthesized during the initial phases of acute inflammatory responses to promote the resolution of inflammation. Resolvins are produced for termination of neutrophil infiltration, stimulation of the clearance of apoptotic cells by macrophages, and promotion of tissue remodeling and homeostasis. Metabolic dysregulation due to either uncontrolled activity of pro-inflammatory responses or to inefficient resolution of inflammation results in chronic inflammation and may also lead to atherosclerosis or other chronic autoimmune diseases such as rheumatoid arthritis, psoriasis, systemic lupus erythematosus, vasculitis, inflammatory bowel diseases, and type 1 diabetes mellitus. The pathogenesis of such diseases involves a complex interplay between the immune system and, environmental factors (non-infectious or infectious), and critically depends on individual susceptibility to such factors. In the present review, resolvins and their roles in the resolution of inflammation, as well as the role of these mediators as potential therapeutic agents to counteract specific chronic autoimmune and inflammatory diseases are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute inflammation is a normal protective response when host cells are affected by injury or microbial pathogen invasion [1]. In this regard, uncontrolled inflammatory responses may lead to systemic and chronic inflammatory disorders including neurological disorders, cardiovascular diseases, periodontal diseases, asthma, rheumatoid arthritis, diabetes mellitus, as well as the inflammatory bowel disease (IBD) [1,2,3]. Recent studies have identified the resolution of inflammation as an active process, which is regulated by biochemical mediators and specialized pro-resolving mediators (SPMs), which provides a useful novel basis for our understanding of the principles of recognition and treatment of these inflammatory disorders [4].
On the whole, acute inflammatory responses appear with various signs of inflammation such as pain, swelling, redness, heat, and loss of function [5], which are then followed by the release of chemical messengers (cytokines and chemokines) and pro-inflammatory lipid mediators such as prostaglandins (PGs) and leukotrienes (LTs). The release of these molecules leads to an attack against the triggering agents and to initiation of repair of the injured tissues [6, 7]. This is accompanied by chemotaxis and recruitment of polymorphonuclear neutrophils (PMNs) and by accumulation of monocyte-derived macrophages which trigger and sustain the local or systemic inflammatory tissue reaction [1]. The initial pro-inflammatory cellular reactions also stimulate the biosynthesis of specific omega-3 polyunsaturated fatty acid-derived SPMs, which promote the resolution of inflammation [8]. In spite of the essentially protective function of acute inflammation, defective termination or prolonged acute inflammation results in chronic inflammation that can be the root cause of many disorders due to the failure of inflammation resolution [6].
The resolution of an acute inflammatory reaction is characterized by termination of PMN penetration into the infected or injured tissue and by proper apoptosis of the already infiltrated PMNs along with effective efferocytic clearance by macrophages of the apoptotic PMN, of other apoptotic cells, and of any cellular debris [7, 8]. The inflammatory process involves many mediators, of which some have pro-inflammatory (prostaglandins and leukotrienes) and others both anti-inflammatory and pro-resolving functions (resolvins, lipoxins, protectins, etc.) [4, 9]. All of these mediators could serve as potential targets for therapeutic actions. Along with lipoxins, the resolvins (resolution phase interaction products), a new family of lipid mediators, have a variety of functions including anti-inflammatory, proresolving, and neuroprotective effects against various disorders [9].
Role of Lipid Mediators in Inflammatory Responses
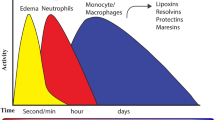
The inflammatory response to injured tissues involves the liberation of chemical signals in the form of chemokines, cytokines, and lipid-derived mediators by the cells of the innate immune system, initially the PMNs, and then macrophages, mast cells, and endothelial cells, ultimately resulting in an inflammatory defence to protect the affected cells [10]. Modulation of the initiation phase of acute inflammation is controlled by a category of lipid mediators such as PGs, LTs, and eicosanoids that are constituted from arachidonic acid (AA, omega-6) and have the efficiency to regulate endothelial permeability and PMN infiltration [11]. The initial mediators, like leukotriene B4 (LTB4) and PGs, have key roles to lead the pro-inflammatory phases with a maximum number of PMNs within seconds and minutes after the injury [10] (Fig. 1(a, b)).

Pro-resolving mediators in inflammatory responses. Tissue injury and trauma trigger an acute inflammatory cascade as a host defensive mechanism. PMN infiltration is an early step that provokes monocytes and macrophages to remove apoptotic PMNs. At the stage of resolution phase, adaptive immune responses (T and B cells) are initiated simultaneously with tissue regeneration (a). Trauma, tissue injury, and infections initiate the inflammatory process, which comprises of cells like neutrophils and the release of many mediators, such as PGE2, PGI2, and LTB4 along with many cytokines and chemokines. The release of PGs leads to the migration of neutrophils from capillary venules. Concurrent with the accumulation of neutrophils, switching of the lipid mediator class takes place, so that the appearance of lipoxin results in the recruitment of monocytes. Other SPMs, like resolvins and protectins, limit PMN tissue infiltration and stimulate macrophage phagocytosis and efferocytosis of cellular debris. SPMs and resolvins promote the resolution of inflammation and resolve exudates. Also, SPMs and resolvins suppress chronic inflammation and improve tissue regeneration and wound healing. Dysregulation of any of these mechanisms may cause chronic inflammatory diseases, such as diabetes, atherosclerosis, and rheumatoid arthritis (b). Prostaglandin (PG), leukotriene (LT), specialized pro-resolving mediator (SPM), polymorphonuclear neutrophil (PMN)
In the early phase of infectious inflammation, activated neutrophils kill bacteria extracellularly by either generating neutrophil extracellular traps (NETs) or phagocytosing them. The selection of neutrophils to produce NETs, instead of turning on their phagocytotic machinery, seems to depend on the combination of various signals involved in the metabolic, adhesive, and activating conditions of the neutrophils involved, on the impetus from the extracellular micro-environment and, more vitally, on the signals derived from the stimulating particles themselves [12]. Also, the size of the stimulating particles contributes to the choice of the neutrophil whether to form NETs (induced primarily by large particles like parasites) or eliminate the microbe by phagocytosis (induced primarily by small particles such as bacteria and viruses) [12,13,14]. Under certain circumstances, also bacteria and viruses stimulate the formation of NETs [15, 16].
It is well established that an association exists between the availability of the two neutrophil key enzymes, the neutrophil elastase (NE) and myeloperoxidase (MPO), when they function in the cytosol, in the nucleus, and in phagolysosomes during phagocytosis and NETosis. In phagocytosis, both enzymes are recruited to the phagolysosomes, so keeping them away from the nucleus. In contrast, during NETosis, the release of these enzymes into the cytosol leads to the degradation of actin cytoskeleton, which is required to allow their translocation to the nucleus with ensuing chromatin decondensation, which, again, is required for the phagocyte to generate NETs. Finally, the sequestration of NE and MPO into the nucleus and the cytoskeletal degradation obstructs the phagocytotic function of the activated neutrophil [12]. Thus, the dichotomous choice of neutrophils, i.e., to phagocytose or make NETs, has potential consequences for the development of autoimmune and inflammatory diseases, such as systemic lupus erythematosus (SLE), characterized by defective phagocytic clearance, or lupus nephritis and systemic sclerosis, in which pathogenesis that is the microparticle-triggered NET generation has been considered to play a role [12].
The resolution of inflammation is a secondary defence mechanism that can be controlled by the initial pro-inflammatory agents at the onset of the inflammation phase. The resolution step is controlled by the local release of different SPMs such as lipoxins, resolvins, protectins, and maresins [15, 16]. Thus, the biosynthesis of these mediators depends on the initial pro-inflammatory stimuli that trigger compensatory mechanisms for the resolution of inflammation [10].
Resolvins and protectins can be generated along different pathways by various immune cells, such as macrophages and PMNs, from the following polyunsaturated fatty acids (PUFAs): eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids [10, 17]. Changing the phase to resolution of inflammation, some eicosanoids such as PGE2 and PGD2, provokes the biosynthesis of pro-resolving lipid mediators such as lipoxin A4 (LXA4), deriving from arachidonic acid, that acts as an endogenous “stop” signal to control the resolution phase within hours up to days [18, 19]. At this stage, the produced SPMs can halt PMN infiltration, reduce the generation of pro-inflammatory mediators, enhance phagocytosis of apoptotic PMNs (“efferocytosis”) by macrophages, and promote bacterial clearance [19, 20]. Actually, phagocytosis of dead cells by macrophages results in the biosynthesis of SPMs, which are involved in the processing of apoptotic cell clearance, similar to “find me” and “eat me” signals including ATP, adenosine, and CX3CL during apoptotic cell engulfment [21, 22].
SPMs, by binding to G protein-coupled receptors (GPCRs), induce specialized biological actions (Fig. 2). It has been clarified that GPCRs, such as chemR23/ERV, are activated by RVE1 and RVE2, while ALX/FPR2 and GPR32/DRV are stimulated by LXA4 and RVD1 [23]. Furthermore, GPR32 is activated by RVD5 and RVD3 [24]. On the other hand, RVE1 and RVE2 are considered as kinds of endogenous receptor antagonists for the LTB4 receptor BLT1, which has the capacity to organize PMN trafficking and NF-κB activation at the site of inflammation. RVE1 is considered as an agonist of chemR23 on monocytes and dendritic cells and thereby stereo-selectively transduces signals to these cells [23], along with the capability to reduce the expression of IL-6 and IFN-γ and to inhibit the migration of dendritic cells and cytokine secretion [19]. Besides the capacity to inhibit PMN recruitment and promotion of macrophage phagocytosis, SPMs are also able to enhance the host defence through the efflux of phagocytes from inflamed tissues to the lymphatics [25]. Either a hyperactive pro-inflammatory responses or an inefficiency to stimulate resolution, which results from the disordered function of mediators, are prototypical of chronic inflammatory disorders such as atherosclerosis, diabetes, and arthritis [26,27,28].

Proresolving receptors and biological functions of SPMs. LXA4 and RVD1 are agonists for ALX/FPR2. Lipoxin A4, as well as the D-series resolvins, RVD1, RVD3, and RVD5 are agonists for DRV/GPR32. The E-series resolvins, RVE1 and RVE2, are agonists for ERV/chemR23 and antagonists for the leukotriene B4 receptor, BLT1. ALX/FPR2: LXA4 receptor, DRV/GPR32: RVD1 receptor, ERV/ChemR23: chemerin/RVE1 receptor, BLT1: LTB4 receptor 1. ALX A lipoxin, FPR2 formyl peptide receptor 2, GPR32 G protein-coupled receptor 32, ChemR23 Chemerin Receptor 23, RVD resolvin D, RVE resolvin E
Resolvins and Their Targets
Serhan and his co-workers pioneered the concept that the local response to acute inflammation activates specific biochemical and cellular programs of resolution [7, 29,30,31]. These mediators include the D-series resolvins (RVD1, RVD2, RVD3, RVD4, and RVD5) derived from DHA [32, 33] and the E-series resolvins (RVE1, RVE2, and RVE3) that are derived from EPA [34, 35] (Fig. 3). The detection of resolvins along with other lipid mediators such as maresins and protectins has been particularly important since these lipid mediators have been demonstrated to support human health [36] by mitigating inflammation [37] in various pathological conditions, notably in diseases with predominantly autoimmune etiopathogenesis [13, 36, 38] and in cardiovascular diseases [38], type 2 diabetes mellitus [39], and cancer [40].

Biosynthetic pathways of resolvin generation. Different kinds of resolvins are derived from n-3 PUFA as EPA and DHA. The biosynthesis of E-series resolvins from EPA is initiated by insertion of molecular oxygen at the 18th carbon site, converting it to bioactive E-series members resolvinE1, resolvinE2, and resolvinE3. D-series resolvin biosynthesis is along with the insertion of molecular oxygen at the 17th carbon site of DHA. The 17-sHPDHA precursor is changed to different intermediates of D-series resolvins including resolvinD1, resolvinD2, resolvinD3, and resolvinD4, each one of which has a specific function. See [4]. PUFA polyunsaturated fatty acid, DHA docosahexaenoic acid, EPA eicosapentaenoic acid
Two receptors from the G protein family (chemR23 and BLT1) are capable of binding to RVE1. The chemR23, which has a high binding affinity for RVE1, is expressed on the surface of macrophages and dendritic cells and is able to stimulate phagocytosis of apoptotic PMNs by macrophages. Herrera et al. found that RVE1 could reduce the penetration and migration of PMNs and inhibit their rolling [39]. Furthermore, RVE1 is able to increase the CCR5 expression and reduce the NF-κB signalling [39]. Herova et al. indicated that the expression of chemR23 is limited to the surface of naïve macrophages and classical M1 macrophages, while these receptors are not expressed on the surface of M2 macrophages [40]. In addition to that, expression of chemR23 on the surface of macrophages increases their ability to phagocytose as well as to secrete the anti-inflammatory cytokine IL-10, thereby boosting their capacity to resolve inflammation [40]. In a study on a mouse model of pneumonia, the usage of RVE1 reduced pulmonary PMN accumulation, lessened secretion of inflammatory cytokines, improved the clearance of bacterial cells, and enhanced the survival of mice [41]. Elkebir et al. (2012) argued that infection was eliminated by RVE1 in mice with acute lung inflammation [42]. Via the LTB4 receptor, BLT1, RVE1 not only is able to produce NADPH oxidases and reactive oxygen species but it also increases neutrophil apoptosis leading to their efferocytosis by the macrophages, resulting in the production of phagocytosis-derived metabolic products, such as 19-hydroxy-RVE1, 20-hydroxy-RVE1, and 20-carboxy-RVE with pro-resolution properties in vivo [43]. The ability of RVE2s to control PMN penetration and their anti-inflammatory properties was first discovered by Oh et al. in zymosan-induced rat model of peritonitis, in which they reported that RVE2 was able to inhibit PMN chemotaxis, increase phagocytosis, and produce Il-10 [44].
The D-series of resolvin family (the RvDs) is another group of lipid-proresolving molecules with the ability to inhibit PMN penetration and to regulate their activities to enhance phagocytosis by macrophages and to remove and clear apoptotic cells and bacteria in both in vivo and in vitro environments [45,46,47]. A study on the murine model of arterial neointima formation (carotid ligation) showed that the use of RVD2 in these mice recruited macrophages and regulated PMNs. In vitro studies have confirmed a direct impact of SPMs on vascular smooth muscle cells (VSMCs) that includes lower migration in response to platelet-derived growth factor (PDGF) and altered reaction to inflammatory stimuli (e.g. TNF-α) [48]. According to these results, changes in the interactions between circulating leukocytes and the vessel wall and direct effects on the phenotype of VSMCs are the probable mechanisms through which SPMs affect the development of neointima in this murine model [48]. The treatment by RVD2 and MaR1 on the reduction of infiltrating monocytes/macrophages and PMNs, as well as changes in the macrophage phenotype towards M2, leads to the resolution of inflammation and to the reduction of neointima after vascular injury [44, 48].
RVDs can also affect target cells through binding to expressed receptors (GPR32-G protein in coupled receptor, ALX/FPR2) on the surface of immune cells [49, 50]. The overexpression of both receptors improves the RVD1macrophage phagocytosis of PMNs, and conversely, the inactivity of these two receptors weakens this action [49]. Park et al. approved the inhibitory impact of RVD2 for transient receptor potential (TRP) subtype ankyrin 1 (TRPA1) and vanilloid 1 (TRPV1) that play significant roles in the inflammatory pain responses [51]. Another benefit of using RVD1 has been observed in acute lung injury, in which the lesions are caused by LPS [52, 53]. In this model, RVD1 inhibits the activity of the inflammatory factor NF-κB and thereby inhibits its effect on peroxisome proliferator-activated receptor (PPAR) γ and the ensuing inflammatory downstream signalling pathway.
Resolvins and Autoimmune Diseases
Autoimmune diseases are the conditions arising from an abnormal immune response against healthy cells, tissues, and organs. Complex mechanisms, involving T cells, the thymus, the bone marrow, lead to the breaking of the immune tolerance [54]. In the presence of genetic predisposition and/or environmental triggers, autoreactive B and T cells and autoantibodies could become involved in a pathological inflammatory response, leading to tissue damage [54]. Activation of inflammatory mediators contributes to the development of autoimmune diseases such as SLE, rheumatoid arthritis, autoimmune hepatitis, and type 1 diabetes mellitus [55]. Through the stimulation of a number of signalling pathways, resolvins might favourably impact this process. Resolvins could also inhibit leukocyte recruitment to inflammation site by stimulating non-inflammatory monocyte recruitment and by activating macrophages to increase the efferocytic capacity towards apoptotic neutrophils [56]. Inhibition of leukocyte recruitment leads to resolution of inflammation, repair and regeneration of damaged tissue, and pain relief [57].
Defects in the resolution of inflammation and inflammatory signals increase the risk of autoimmune diseases [58]. The onset of the inflammation process causes the production of some pro-inflammatory cytokines such as interleukin-1 (IL-1β), IL-6, tumour necrosis factor-α (TNF-α), interferons (IFNs), macrophage migration inhibitory factor (MIF), and high mobility group B1 (HMGB1) protein, which predominate the production of anti-inflammatory cytokines such as IL-10, IL-4, and TGF-β [59, 60]. Indeed, an imbalance between pro- and anti-inflammatory cytokines results in tissue damage and inflammatory disorders, and the inability of the immune system to resolve the inflammation results in the continuation of inflammatory processes in autoimmune diseases such as SLE [61, 62]. In these diseases, the process of repairing damaged tissues is delayed even after taking anti-inflammatory drugs and immunosuppressive agents. Therefore, the use of resolution-inducing agents such as PUFAs and the metabolites derived from them, such as resolvins, lipoxins, and protectins, has the potential to restore the normal physiological function of damaged tissues and organs [62].
Macrophages and monocytes have GPCRs on their surfaces; the subtypes P2ry2, P2ry6, and Edg5 have a particularly important role in the regulation of inflammation and cell-mediated immunity [63]. Many studies on PUFAs and the derived-metabolites, including resolvins and lipoxins, indicate that these lipid mediators act as agonists of G protein that plays an important role in the inflammation, leukocyte recruitment, lipid synthesis, and glucose homeostasis [57]. It can be thus concluded that PUFAs and derivatives thereof, such as the anti-inflammatory and cytoprotective resolvins, lipoxins, and protectins, are capable of inhibiting the pro-inflammatory MIF, IL-2, IL-6, TNF-a, and HMGB1, thus inhibiting inflammation in autoimmune diseases such as rheumatoid arthritis and SLE [62].
Furthermore, since leukotriene B4 (LTB4) is involved in the pathogenesis of inflammatory bowel disease, lipid mediators such as resolvins could be hypothesized as a potential treatment since studies on rats with colitis have shown that RVE1 increases their survival, sustains body weight, and reduces the severity of histological lesions [34]. Studies also indicated that resolvins reduced both the expression of pro-inflammatory genes such as those coding for IL-12, TNF-α, NF-κB, and decreased inducible NO synthesis in models of inflammatory bowel disease [64].
In addition, many studies indicated roles for dendritic cells in the pathogenesis of inflammatory bowel disease because these cells could lead the orientation of T cells towards the inflammation during maturation and increase the inflammation and resistance of T cells through the production of inflammatory cytokines such as IL-12, IL-23, and TNF-α [65, 66]. Interestingly, it has been found that lipid mediators, such as the resolvins and lipoxins, could play roles in the function of dendritic cells, especially through different receptors, and prevent their orientation towards inflammation. For instance, EPA-derived RVE1 induced the expression of its specific receptor chemR23 on the surface of dendritic cells and inhibited the secretion of IL-12, thereby impeding inflammation [19].
The secretion of pro-inflammatory molecules, such as IL-1beta and LTB4, is a leading contributor to inflammation and autoimmune diseases such as rheumatoid arthritis and psoriasis [67, 68]. Trials indicate that the nutritional use of DHA and EPA, abundantly found in fish oil, can reduce the secretion of inflammatory mediators and thus might be useful for treating inflammatory and autoimmune diseases [37, 69]. Very recent human data support this notion, as high doses of EPA significantly reduced major cardiovascular events in patients at high cardiovascular risk [70].
Human acute inflammatory demyelinating polyradiculoneuropathy (AIDP) disease, the major reason of acute autoimmune neuromuscular paralysis, is provoked by autoimmune invasion to the peripheral nervous system [71, 72]. The experimental autoimmune neuritis (EAN), an animal model of AIDP, has been applied to determine the therapeutic and functional principles of AIDP [71, 72]. This disease occurs due to the impaired phagocytosis of apoptotic T cells. Normally, the apoptosis of autoreactive T cells is a process leading to the end of inflammatory responses in EAN and is necessary following the clearance of apoptotic cells by macrophages to prevent the accumulation of damaged cells and the spread of infection and inflammation (secondary necrosis) [73]. In addition, the clearance of apoptotic cells caused by active macrophages leads to a change in the anti-inflammatory phenotype inducing Th cells towards the Treg phenotype due to the production of anti-inflammatory cytokines [74,75,76]. Studies have indicated that Treg cells and anti-inflammatory macrophages play important roles in the resolution of inflammation. Therefore, there is a need to find ways to enhance the phagocytosis of apoptotic cells in the inflamed tissue to resolve the neural inflammation in AIDP [77, 78].
Through the presence of two receptors, the ALX/FPR2 and GPR32, on the surface of neutrophils and macrophages, RVD1 is able to regulate the migration of neutrophils, increase phagocytosis of apoptotic T cells by macrophages (in an ALX/FPR2-dependent manner), and thus reduce the accumulation of apoptotic T cells [32]. Thus, suchRVD1-induced changes increase the expression of TGF-β via anti-inflammatory phenotype of macrophages, reduce the pro-inflammatory factors such as TNF-α, and enhance the number of Treg cells, consequently leading to the resolution of inflammation in autoimmune diseases such as the EAN and AIDP [32, 79].
Concluding Remarks
The activation of inflammation by a dysregulated immune system is a central mechanism in the development of autoimmune diseases. In the absence of an efficient inhibition of inflammation, the adaptive immune system of the host elicits a strong proinflammatory response. Therefore, inflammation should be controlled at its early stages. Such an early control can prevent the occurrence of chronic inflammation, can lead to repair and regeneration of the damaged tissue parts, and might thereby delay or counteract the clinical onset of autoimmune diseases. Indeed, as an inherent component of inflammation, the proinflammatory processes are counteracted by resolution-promoting lipid mediators, such as the resolvins derived from PUFAs. These agents can prevent the spread of inflammation and its chronicity in various ways, notably by acting on different receptors expressed on the surface of macrophages, PMNs, and dendritic cells.
The cannabinoid receptor type 2 (CB2) are class A serpentine receptors that couple primarily to Gi/o proteins to adjust an array of signalling pathways [80]. CB2 receptors, which are expressed on fibroblasts, endothelial cells, muscle cells, and activated immune cells [81], have been the subject of significant attention owing to their immunomodulatory effects which have potential utility in the treatment of different pathologies [82]. Lenabasum (a composition of ajulemic acid; formerly called Anabasum) is a synthetic, orally active cannabinoid-derived small molecular weight drug that acts primarily as an agonist of the CB2 receptor [83]. In experimental studies, lenabasum has been shown to stimulate the production of SPMs, leading to bacterial clearance and resolution of inflammation without immunosuppression [84,85,86]. Animal studies have shown that lenabasum has beneficial effects on lung and skin inflammation, as well as on fibrosis in systemic sclerosis [87, 88], inflammation in cystic fibrosis, and joint inflammation in rheumatoid arthritis [85].
The therapeutic use of the PUFA-derived lipid mediators during the inflammation process to resolve inflammation and prevent the development of chronic inflammation could be a useful therapeutic option. On the other hand, since incomplete efferocytosis is one cause of several abnormalities including autoimmune diseases [89,90,91,92], by facilitating efferocytosis and activating macrophage cells to efficiently clear of apoptotic cells and cell debris, the lipid mediators can prevent the progression of inflammation and subsequent inflammatory diseases, in particular autoimmune diseases [89].
The connection between lipid mediators and the clearance of leukocytes via apoptosis has been detected by identifying that the release of lysophosphatidylcholine from apoptotic cells could specifically assemble phagocytes to the resolution site [93]. The presence of RVE1 receptors on dendritic cells and neutrophils, as well as the essential role of these cells in inflammatory responses, could support the link between the biosynthesis of lipid mediators and their engagement in cellular trafficking and regulation of inflammatory disorders such as inflammatory bowel disease [19, 34].
Owing to their ability to stereoselectively regulate and reduce inflammation, the SPMs and their intricate and highly specific molecular pathways are attracting an ever-increasing attention among many researchers. The PUFA-derived lipid mediators as pro-resolving agents are effective and sustained factors that stimulate leukocyte recruitment, PMN apoptosis at the damaged site, and clearance of damaged tissue parts during the inflammation. Therefore, these agents are excellent candidates for the treatment of diseases associated with the defective inflammation resolution and also for the facilitation of recovery from primary and secondary infections.
Taken together, the SPMs play an important role in the prevention of chronic inflammation, which is the root cause of various chronic diseases. Therefore, an accurate understanding of the molecular mechanisms by which these pro-resolving compounds may prevent or even cure such pathological conditions, should help designing novel strategies to prevent and treat inflammatory and autoimmune diseases without compromising the ability of the host cells to be in charge of the natural host defence.
References
Kumar V, Abbas AK, Fausto N, Aster JC (2014) Robbins and Cotran pathologic basis of disease, professional edition e-book. elsevier health sciences
Libby P, Ridker PM, Maseri A (2002) Inflammation and atherosclerosis. Circulation 105(9):1135–1143
Hansson GK, Libby P (2006) The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol 6(7):508–519
Serhan CN, Petasis NA (2011) Resolvins and protectins in inflammation resolution. Chem Rev 111(10):5922–5943. https://doi.org/10.1021/cr100396c
Majno G, Joris I (2004) Cells, tissues, and disease: principles of general pathology. Oxford University Press
Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL (2007) Resolution of inflammation: state of the art, definitions and terms. FASEB J 21(2):325–332
Serhan CN (2004) A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol 122(4):305–321
Serhan CN, Ward PA, Gilroy DW (2010) Fundamentals of inflammation. Cambridge University Press
Schwab JM, Serhan CN (2006) Lipoxins and new lipid mediators in the resolution of inflammation. Curr Opin Pharmacol 6(4):414–420
Crean D, Godson C (2015) Specialised lipid mediators and their targets. In: Seminars in immunology, vol 3. Elsevier, pp 169–176
Samuelsson B, Dahlen S-E, Lindgren JA, Rouzer CA, Serhan CN (1987) Leukotrienes and lipoxins: structures, biosynthesis, and biological effects. Science 237(4819):1171–1176
Manfredi AA, Ramirez GA, Rovere-Querini P, Maugeri N (2018) The neutrophil’s choice: phagocytose vs make neutrophil extracellular traps. Front Immunol 9:288
Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V (2014) Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15(11):1017–1025
Warnatsch A, Tsourouktsoglou T-D, Branzk N, Wang Q, Reincke S, Herbst S, Gutierrez M, Papayannopoulos V (2017) Reactive oxygen species localization programs inflammation to clear microbes of different size. Immunity 46(3):421–432
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P (2012) Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12(3):324–333
Kim S-J, Jenne CN (2016) Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. In: Seminars in immunology, vol 6. Elsevier, pp 546–554
Chiurchiù V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, Serhan CN (2016) Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med 8(353):353ra111
Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN (2001) Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2(7):612–619
Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, Yang R, Petasis NA, Serhan CN (2005) Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med 201(5):713–722. https://doi.org/10.1084/jem.20042031
Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN (2012) Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484(7395):524–528
Han CZ, Ravichandran KS (2011) Metabolic connections during apoptotic cell engulfment. Cell 147(7):1442–1445
Köröskényi K, Duró E, Pallai A, Sarang Z, Kloor D, Ucker DS, Beceiro S, Castrillo A, Chawla A, Ledent CA (2011) Involvement of adenosine A2A receptors in engulfment-dependent apoptotic cell suppression of inflammation. J Immunol 1002284
Serhan CN, Chiang N (2013) Resolution phase lipid mediators of inflammation: agonists of resolution. Curr Opin Pharmacol 13(4):632–640
Dalli J, Winkler JW, Colas RA, Arnardottir H, Cheng C-YC, Chiang N, Petasis NA, Serhan CN (2013) Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem Biol 20(2):188–201
Schwab JM, Chiang N, Arita M, Serhan CN (2007) Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447(7146):869–874
Spite M, Serhan CN (2010) Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 107(10):1170–1184
Nathan C (2002) Points of control in inflammation. Nature 420(6917):846–852
Back M, Yurdagul A Jr, Tabas I, Oorni K, Kovanen PT (2019) Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol 16:389–406. https://doi.org/10.1038/s41569-019-0169-2
Serhan CN (2007) Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol 25:101–137. https://doi.org/10.1146/annurev.immunol.25.022106.141647
Bannenberg G, Serhan CN (2010) Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta 1801(12):1260–1273. https://doi.org/10.1016/j.bbalip.2010.08.002
Serhan CN (2010) Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol 177(4):1576–1591. https://doi.org/10.2353/ajpath.2010.100322
Sun YP, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, Serhan CN (2007) Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem 282(13):9323–9334. https://doi.org/10.1074/jbc.M609212200
Spite M, Norling LV, Summers L, Yang R, Cooper D, Petasis NA, Flower RJ, Perretti M, Serhan CN (2009) Resolvin D2 is a potent regulator of leukocytes and controls microbial sepsis. Nature 461(7268):1287–1291. https://doi.org/10.1038/nature08541
Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, Blumberg RS, Serhan CN (2005) Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci U S A 102(21):7671–7676. https://doi.org/10.1073/pnas.0409271102
Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K (2000) Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 192(8):1197–1204
Fetterman JW Jr, Zdanowicz MM (2009) Therapeutic potential of n-3 polyunsaturated fatty acids in disease. Am J Health Syst Pharm 66(13):1169–1179. https://doi.org/10.2146/ajhp080411
Simopoulos AP (2002) Omega-3 fatty acids in inflammation and autoimmune diseases. J Am Coll Nutr 21(6):495–505
Harper CR, Jacobson TA (2001) The fats of life: the role of omega-3 fatty acids in the prevention of coronary heart disease. Arch Intern Med 161(18):2185–2192
Herrera BS, Hasturk H, Kantarci A, Freire MO, Nguyen O, Kansal S, Van Dyke TE (2015) Impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infect Immun 83(2):792–801
Herová M, Schmid M, Gemperle C, Hersberger M (2015) ChemR23, the receptor for chemerin and resolvin E1, is expressed and functional on M1 but not on M2 macrophages. J Immunol 1402166
Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, Miyasho T, Takamiya R, Asano K, Ishizaka A (2010) The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol 184(2):836–843
El Kebir D, Gjorstrup P, Filep JG (2012) Resolvin E1 promotes phagocytosis-induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci 201206641
Hong S, Porter TF, Lu Y, Oh SF, Pillai PS, Serhan CN (2008) Resolvin E1 metabolome in local inactivation during inflammation-resolution. J Immunol 180(5):3512–3519
Titos E, Rius B, González-Périz A, López-Vicario C, Morán-Salvador E, Martínez-Clemente M, Arroyo V, Clària J (2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 1100225
Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510(7503):92–101
Kohli P, Levy BD (2009) Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol 158(4):960–971
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273
Akagi D, Chen M, Toy R, Chatterjee A, Conte MS (2015) Systemic delivery of proresolving lipid mediators resolvin D2 and maresin 1 attenuates intimal hyperplasia in mice. FASEB J 29(6):2504–2513
Krishnamoorthy S, Recchiuti A, Chiang N, Yacoubian S, Lee C-H, Yang R, Petasis NA, Serhan CN (2010) Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci 107(4):1660–1665
Krishnamoorthy S, Recchiuti A, Chiang N, Fredman G, Serhan CN (2012) Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol 180(5):2018–2027
Park C-K, Xu Z-Z, Liu T, Lü N, Serhan CN, Ji R-R (2011) Resolvin D2 is a potent endogenous inhibitor for transient receptor potential subtype V1/A1, inflammatory pain, and spinal cord synaptic plasticity in mice: distinct roles of resolvin D1, D2, and E1. J Neurosci 31(50):18433–18438
Wang B, Gong X, J-y W, Zhang L, Zhang Z, Li H-z, Min S (2011) Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm Pharmacol Ther 24(4):434–441
Liao Z, Dong J, Wu W, Yang T, Wang T, Guo L, Chen L, Xu D, Wen F (2012) Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respir Res 13(1):110
Wang L, Wang FS, Gershwin ME (2015) Human autoimmune diseases: a comprehensive update. J Intern Med 278(4):369–395
Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN (2007) TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13(5):543–551
Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, Petasis NA, Erwig L, Rees AJ, Savill J (2002) Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol 13(10):2497–2507
Milligan G, Stoddart LA, Brown AJ (2006) G protein-coupled receptors for free fatty acids. Cell Signal 18(9):1360–1365. https://doi.org/10.1016/j.cellsig.2006.03.011
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nat Rev Endocrinol 5(4):219–226
Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac R-L (2002) Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med 196(8):1025–1037
Chiang N, Arita M, Serhan CN (2005) Anti-inflammatory circuitry: lipoxin, aspirin-triggered lipoxins and their receptor ALX. Prostaglandins Leukot Essent Fat Acids 73(3–4):163–177
Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142(5):687–698
Das UN (2011) Lipoxins as biomarkers of lupus and other inflammatory conditions. Lipids Health Dis 10:76. https://doi.org/10.1186/1476-511x-10-76
Lattin JE, Schroder K, Su AI, Walker JR, Zhang J, Wiltshire T, Saijo K, Glass CK, Hume DA, Kellie S, Sweet MJ (2008) Expression analysis of G protein-coupled receptors in mouse macrophages. Immunome Res 4:5. https://doi.org/10.1186/1745-7580-4-5
Weylandt KH, Kang JX, Wiedenmann B, Baumgart DC (2007) Lipoxins and resolvins in inflammatory bowel disease. Inflamm Bowel Dis 13(6):797–799. https://doi.org/10.1002/ibd.20109
Kelsall BL, Leon F (2005) Involvement of intestinal dendritic cells in oral tolerance, immunity to pathogens, and inflammatory bowel disease. Immunol Rev 206:132–148. https://doi.org/10.1111/j.0105-2896.2005.00292.x
Baumgart DC, Metzke D, Schmitz J, Scheffold A, Sturm A, Wiedenmann B, Dignass AU (2005) Patients with active inflammatory bowel disease lack immature peripheral blood plasmacytoid and myeloid dendritic cells. Gut 54(2):228–236. https://doi.org/10.1136/gut.2004.040360
Allen B (1991) Fish oil in combination with other therapies in the treatment of psoriasis. In: Health effects of omega 3 polyunsaturated fatty acids in seafoods, vol 66. Karger Publishers, pp 436–445
Cleland L, James M (1997) Rheumatoid arthritis and the balance of dietary N-6 and N-3 essential fatty acids. Br J Rheumatol 36(5):513–514
Kremer JM (2000) N-3 fatty acid supplements in rheumatoid arthritis. Am J Clin Nutr 71(1 Suppl):349s–351s. https://doi.org/10.1093/ajcn/71.1.349s
Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT Jr, Juliano RA, Jiao L, Granowitz C (2019) Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med 380(1):11–22
Hughes RA, Cornblath DR (2005) Guillain-Barre syndrome. Lancet (London, England) 366(9497):1653–1666. https://doi.org/10.1016/s0140-6736(05)67665-9
Soliven B (2012) Autoimmune neuropathies: insights from animal models. J Peripher Nerv Syst 17:28–33
Yun JH, Henson PM, Tuder RM (2008) Phagocytic clearance of apoptotic cells: role in lung disease. Exp Rev Respir Med 2(6):753–765. https://doi.org/10.1586/17476348.2.6.753
Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S (2008) The phenotype of human Th17 cells and their precursors, the cytokines that mediate their differentiation and the role of Th17 cells in inflammation. Int Immunol 20(11):1361–1368. https://doi.org/10.1093/intimm/dxn106
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM (1998) Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101(4):890–898
Kimura A, Naka T, Kishimoto T (2007) IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc Natl Acad Sci 104(29):12099–12104. https://doi.org/10.1073/pnas.0705268104
Luchting B, Rachinger-Adam B, Heyn J, Hinske LC, Kreth S, Azad SC (2015) Anti-inflammatory T-cell shift in neuropathic pain. J Neuroinflammation 12:12. https://doi.org/10.1186/s12974-014-0225-0
Zhang Z, Zhang ZY, Schluesener HJ (2009) Compound A, a plant origin ligand of glucocorticoid receptors, increases regulatory T cells and M2 macrophages to attenuate experimental autoimmune neuritis with reduced side effects. J Immunol 183(5):3081–3091. https://doi.org/10.4049/jimmunol.0901088
Luo B, Han F, Xu K, Wang J, Liu Z, Shen Z, Li J, Liu Y, Jiang M, Zhang ZY, Zhang Z (2016) Resolvin D1 programs inflammation resolution by increasing TGF-beta expression induced by dying cell clearance in experimental autoimmune neuritis. J Neurosci 36(37):9590–9603. https://doi.org/10.1523/jneurosci.0020-16.2016
Dhopeshwarkar A, Mackie K (2014) CB2 cannabinoid receptors as a therapeutic target—what does the future hold? Mol Pharmacol 86(4):430–437
Rom S, Persidsky Y (2013) Cannabinoid receptor 2: potential role in immunomodulation and neuroinflammation. J NeuroImmune Pharmacol 8(3):608–620
Turcotte C, Blanchet M-R, Laviolette M, Flamand N (2016) The CB 2 receptor and its role as a regulator of inflammation. Cell Mol Life Sci 73(23):4449–4470
Tepper MA, Zurier RB, Burstein SH (2014) Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg Med Chem 22(13):3245–3251
Motwani MP, Bennett F, Norris PC, Maini AA, George MJ, Newson J, Henderson A, Hobbs AJ, Tepper M, White B (2018) Potent anti-inflammatory and pro-resolving effects of anabasum in a human model of self-resolving acute inflammation. Clin Pharmacol Ther 104(4):675–686
Zurier RB, Rossetti RG, Lane JH, Goldberg JM, Hunter SA, Burstein SH (1998) Dimethylheptyl-THC-11 OIC acid: a nonpsychoactive antiinflammatory agent with a cannabinoid template structure. Arthritis Rheum 41(1):163–170
Zurier RB, Sun Y-P, George KL, Stebulis JA, Rossetti RG, Skulas A, Judge E, Serhan CN (2009) Ajulemic acid, a synthetic cannabinoid, increases formation of the endogenous proresolving and anti-inflammatory eicosanoid, lipoxin A4. FASEB J 23(5):1503–1509
Gonzalez EG, Selvi E, Balistreri E, Akhmetshina A, Palumbo K, Lorenzini S, Lazzerini PE, Montilli C, Capecchi PL, Lucattelli M (2012) Synthetic cannabinoid ajulemic acid exerts potent antifibrotic effects in experimental models of systemic sclerosis. Ann Rheum Dis 71(9):1545–1551
Lucattelli M, Fineschi S, Selvi E, Gonzalez EG, Bartalesi B, De Cunto G, Lorenzini S, Galeazzi M, Lungarella G (2016) Ajulemic acid exerts potent anti-fibrotic effect during the fibrogenic phase of bleomycin lung. Respir Res 17(1):49
Abdolmaleki F, Farahani N, Gheibi Hayat SM, Pirro M, Bianconi V, Barreto GE, Sahebkar A (2018) The role of efferocytosis in autoimmune diseases. Front Immunol 9:1645. https://doi.org/10.3389/fimmu.2018.01645
Gheibi Hayat SM, Bianconi V, Pirro M, Sahebkar A (2019) Efferocytosis: molecular mechanisms and pathophysiological perspectives. Immunol Cell Biol 97(2):124–133. https://doi.org/10.1111/imcb.12206
Tajbakhsh A, Gheibi Hayat SM, Butler AE, Sahebkar A (2019) Effect of soluble cleavage products of important receptors/ligands on efferocytosis: their role in inflammatory, autoimmune and cardiovascular disease. Ageing Res Rev 50:43–57. https://doi.org/10.1016/j.arr.2019.01.007
Tajbakhsh A, Rezaee M, Kovanen PT, Sahebkar A (2018) Efferocytosis in atherosclerotic lesions: malfunctioning regulatory pathways and control mechanisms. Pharmacol Ther 188:12–25. https://doi.org/10.1016/j.pharmthera.2018.02.003
Lauber K, Blumenthal SG, Waibel M, Wesselborg S (2004) Clearance of apoptotic cells: getting rid of the corpses. Mol Cell 14(3):277–287
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
No informed consent was required to prepare the manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abdolmaleki, F., Kovanen, P.T., Mardani, R. et al. Resolvins: Emerging Players in Autoimmune and Inflammatory Diseases. Clinic Rev Allerg Immunol 58, 82–91 (2020). https://doi.org/10.1007/s12016-019-08754-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-019-08754-9