
Abstract
Objective
The aim of this study was to address the question of whether bone marrow-originated mononuclear cells (MNC) or mesenchymal stem cells (MSC) induce neural regeneration when implanted intraspinally.
Materials and Methods
The study design included 4 groups of mice: Group 1, non-traumatized control group; Groups 2, 3 and 4 spinal cord traumatized mice with 1 g force Tator clips, which received intralesionally either no cellular implants (Group 2), luciferase (Luc) (+) MNC (Group 3) or MSC (Group 4) obtained from CMV-Luc or beta-actin Luc donor transgenic mice. Following the surgery until decapitation, periodical radioluminescence imaging (RLI) and Basso Mouse Scale (BMS) evaluations was performed to monitor neural activity. Postmortem immunohistochemical techniques were used to analyze the fate of donor type implanted cells.
Results
All mice of Groups 3 and 4 showed various degrees of improvement in the BMS scores, whereas there was no change in Groups 1 and 2. The functional improvement was significantly better in Group 4 compared to Group 3 (18 vs 8, p = 0.002). The immunohistochemical staining demonstrated GFP+Luc+ neuronal/glial cells that were also positive with one or more of these markers: nestin, myelin associated glycoprotein, microtubule associated protein or myelin oligodendrocyte specific protein, which is considered as indicator of donor type neuronal regeneration. Frequency of donor type neuronal cells; Luc + signals and median BMS scores were observed 48–64 % and 68–72 %; 44–80 %; 8 and 18 within Groups III and IV respectively.
Discussion
MSCs were more effective than MNC in obtaining neuronal recovery. Substantial but incomplete functional improvement was associated with donor type in vivo imaging signals more frequently than the number of neuronal cells expressing donor markers in spinal cord sections in vitro. Our results are in favor of functional recovery arising from both donor MSC and MNCs, contributing to direct neuronal regeneration and additional indirect mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
It has long been believed that intrinsic repair is restricted after spinal cord injury (SCI) because neurogenesis rarely occurs in the nervous system. However, we wanted to challenge this dogma taking advantage of stem cell technology. Repair and replacement of the defective or damaged neuronal tissue and restoration of normal functioning is the objective of stem cell therapy for SCI [1, 2].
To demonstrate the events leading to regeneration, in vivo imaging techniques, i.e. magnetic resonance imaging or bioluminescence imaging (BLI) have been developed. These imaging techniques possess the potential to track donor type luciferase (Luc)+ cells within a recipient, which is Luc−. This is a very valuable technique for the tracking of stem cells used in regenerative medicine and enables monitoring of cellular therapy. Thus, other markers to track donor type cells in pathological specimens are required [2].
Using these advanced technologies, we initiated a research project to track donor type bone marrow mononuclear cells (MNCs) and mesenchymal stem cells (MSCs) implanted into the injured spinal cords of mice both in vivo and in vitro. Mice were also monitored for functional recovery using the Basso Mouse Scale (BMS) scoring system.
This study aimed to analyze in vivo imaging, in addition to functional and morphological assessment of neuronal regeneration, following implantation of MNCs and mesenchymal stem cells.
Materials and Methods
Animals and Injury Model
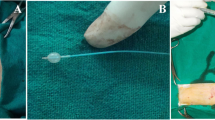
Wild type, 2-month-old mice were used for the experiments as recipients and control group. Mice in the study were traumatized with a standard 1 g closing force Tator clamp (Fig. 1) [3]. The mice in the Group 2 were only traumatized without any cell implantation. Following the trauma the mice in Group 3 and 4 received MNCs and MSCs, respectively as described below.

View of spinal cord after total laminectomy
Eighth-generation Active – Luc+ green fluorescent protein (GFP)+ transgenic mice were used for neuronal regeneration experiments as donors. All animal procedures were performed in accordance with the Guidelines for the Care and Use of Experimental Animals.
Collection of Murine Bone Marrow Mononuclear Cells
Following general anesthesia, adult transgenic mice were decapitated. Long bones (femur and tibia) were removed under sterile conditions for bone marrow cells harvesting and concentration. Mononuclear cells (MNCs) were recorded by flow cytometry. Following adjustment to the appropriate concentration, MNCs were either implanted directly or were cultured to obtain MSCs.
Analysis of MNCs and MSCs by Flow Cytometry
Mononuclear cells were separated by Ficoll density gradient from transgenic mice bone marrow aspirate. Cells were diluted with RPMI and analyzed by flow cytometry in order to detect the numbers of the cells within this suspension. 100 μL of cell suspension were mixed with 100 μL of calibrated beads (StemCount Fluorospheres, Beckman Coulter, Miami, USA) and 20 μL of 7-Aminoactinomycin D (7AAD). Upon 20 min incubation at room temperature, events were collected by the acquisition of 100,000 events per tube using flow cytometer (FC500). Viable nucleated cells were selected according to their FSC and SSC characteristics and their 7AAD expression. Cells were also evaluated for their green fluorescence expression in a FL1 vs SSC dot plot (Fig. 2). Cells to be implanted in the injury model were diluted in RPMI 1640 medium according to the counts achieved 10,000 cells/μL.

A Luc (+) beta-actin mouse, after intraperitoneal injection of luciferin, in vivo view
Culture of Murine Mesenchymal Stem Cells
Long bones were flushed with DMEM-LG medium, and washed twice with phosphate buffered saline (PBS) at 1500 rpm for 5 min (min). Cells were cultured in complete medium (CM) consisting of DMEM-LG (Biochrom, Germany), 20 % fetal bovine serum (FBS) (Biochrom, Germany), 2 mM glutamine, and 1 % penicillin/streptomycin at 5%CO2, 37°C and at a cell density of 3 × 105/cm2. After 48 h (h), non-adherent cells were discarded and CM was changed every 3–4 days. When cells reached subconfluency (70–80 %) in 15–20 days, they were washed with PBS, and adherent cells were removed by Cell Scraper. Cells were then cultured at 6 × 103cells/cm2 for the following passages. Analyses were done on P2 cells. Cell counts and viability were assessed routinely with an automatic cell analyzer system (Nucleocounter YC-100; Denmark) or manually. Excess cells were cryopreserved at the end of each passage in CM with 10 % DMSO (dimethyl sulfoxide). 1 × 106 cells in 100 microliters (μL) of PBS was used for injection. For adipogenic differentiation, MSCs were cultured in the presence of adipogenic medium consisting of CM with 1 μM dexamethasone, 60 μM indomethacin, 500 μM isobutyl methylxanthine (IBMX) and 5 μg/ml insulin (Sigma, St. Louis, USA) for 21 days and stained with Oil Red O (Sigma, St. Louis, USA). For osteogenic differentiation, MSCs were maintained for 21 days in osteogenic medium consisting of CM with 100 nM dexamethasone, 10 mM ß-glycerophosphate and 0.2 mM ascorbic acid (Sigma, St. Louis, USA). Cells were stained with Alizarin Red S (Sigma, St. Louis, USA) to confirm the presence of calcium phosphate deposits [4, 5].
Implantation of Murine MNCs into the Injured Spinal Cords of Mice
Bone marrow mononuclear cells were implanted into the spinal cords of Luc−GFP− recipient mice using 30-gauge (G) fine needles. Implantation was introduced in four multiple injections directly to 1 mm proximity of the lesion area in both directions. Mice were anesthetized by inhalation of 5 % halothane, which was reduced to 2 % during surgery, in combination with a mixture of nitrous oxide and oxygen (1:2, v/v). The spinal cord was exposed by laminectomy at thoracic level T8 and a 30-G needle was used at the injection site to aid penetration of the micropipette tip into the spinal cord. Donor type MNCs or MSCs were resuspended in culture medium at 10,000 cells/μL. Cell viability was assessed with 7AAD using flow cytometry, and 5 μL of the cell suspension was injected at a rate of 2.5 μL/min via a 100-mm aperture glass micropipette into the intact spinal cord at level T8. Following injection, the micropipette was left in place for 2 min to prevent backflow of cells. Mice were sacrificed 8 weeks after transplantation by lethal overdose with pentobarbital and transcardially perfused with 4 % paraformaldehyde in PBS. Tissue was cryoprotected in 30 % sucrose, and a rostrocaudal segment of the spinal cord 1.0 cm in length encompassing the transplant site was removed, embedded in optimal cutting temperature (OCT) compound, and cryosectioned longitudinally into 20 μ serial sections.
In Vivo Imaging of Engrafted Donor Type Cells
For in vivo imaging of mice, IVIS 3-dimensional bioluminescence/fluorescence optical imaging system IVRS50 (Xenogen, Alameda) was used. Five minutes after 150 mg/kg Luc injection into the peritoneum, mice were sedated with sebofluorein and placed in the camera revealing the operation site visible to the IVS 100 camera. Camera imaging was used for detection of BLIs and captured pictures (Fig. 3). Images were taken immediately in the perioperative period and after sutures were completed.

Weekly imaging of rats in Group 4 showing continuous donor type of cell presence
To examine the viability of transplanted cells in the injured spinal cord, contusion injury was induced at the 8th thoracic vertebra level and transplantation of neural stem and progenitor cells (NSPCs) was performed after injury. Images were obtained daily for 1 week, then weekly over 8-week period. We observed drastic reductions (nearly 80 %) in signal intensity within the first 4 days after transplantation, then a relatively stable bioluminescent signal for the next 8 weeks. Cell distribution and signal intensity were assessed in living animals using BLI. An intense focal spot of bioluminescence was observed at the transplantation site without leaking to the surrounding tissue.
Monitoring Functional Recovery following Implantation of Cellular Products Using Basso Mouse Scale Open Field Locomotor Scoring System
The recovery of locomotor function was evaluated for 8 weeks using the generally utilized BMS scoring system (Tables 1, 2, 3 and 4).
Immunocytochemical Analysis of Green Fluorescent Protein Expression in Recipients of Cell Transplantation for Tracking of Donor Type Regeneration
The green fluorescent protein (GFP) transgenic mice were sacrificed 8 weeks following the injury. Animals were deeply anesthetized with an intraperitoneal injection of sodium pentobarbital and were transcardially perfused with 4 % paraformaldehyde in 0.1 M PBS, pH 7.4. The spinal cords were dissected and fixed in 4 % paraformaldehyde for 8 h and embedded in Cryomatrix (Shandon Cryomatrix compound VWR International; Mississauga, Ontario, Canada) and stored in −80°C freezer. Immunoflourescent staining was performed on eight micrometer cryostat sections placed on positive charged glass slides (Superfrost Fisher Scientific; Ottawa, Ontario, Canada). The cryostat sections were dried and stored at −20°C freezer until antibody incubation.
Immunostaining of GFP/Luc Expressing Donor Cells in Postmortem Spinal Cord Sections with Neuronal Markers
For immunohistochemistry, the cryostat sections were thawed and rehydrated in PBS for 5 min. The sections were then incubated with protein blocking solution for 30 min. The primary antibodies were performed for demonstration of different cell types. For demonstration of neural differentiation, mouse monoclonal microtubule associated protein (MAP) 2a and 2b (Abcam-ab3096), for oligodendroglial and Schwann cell differentiation, mouse monoclonal myelin oligodendrocyte specific protein (MOSP) (Abcam-ab3094), and for neural stem cell differentiation mouse monoclonal Nestin (Abcam-ab6142) were used. As luciferase was one of the proteins transfected to the donor mice, primary antibody against luciferase was used on tissue sections for demonstration and amplification the signals of the donor originated differentiation. The cocktails of polyclonal anti-goat luciferase antibody (Novus -NB100-1677) and primary antibodies against one of the differentiation antigens were applied. The antibody cocktail combinations (and dilutions) were performed as: Luciferase (1/1000) with Nestin (1/300), Luciferase (1/1000) with MAP2 (1/150), and Luciferase (1/1000) with MOSP (1/150) mentioned dilutions. The primary antibody cocktail incubations were performed overnight at +4°C. The slides were rinsed in PBS for 10 min at the end of the incubation. The secondary antibody coctails Texas red conjugated rabbit anti-mouse IgG (Pierce biotech 31610) (1/200) and FITC conjugated anti-goat IgG (Sigma F 9012) (1/25) were incubated for 2 h at room temperature. The slides were rinsed in PBS for 10 min and 4′-6-diamidino-2-phenylindole (DAPI) was performed for nuclear counterstaining. Microscopic examination was performed by using SpectrumGreen, SpectrumOrange, DAPI and triple filters. Green signals were interpreted as transfected cell origin, whereas red signals were interpreted as the final differentiation signal. The differentiated implanted cell signals were demonstrated as yellow.
Statistical Analysis
Kruskal-Wallis test was used to test the difference between groups. SPSS for Windows 11.5 was used for statistical analysis. A p value less than 0.05 were considered as significant.
Results
According to the data that obtained, BMS score in Group 3 increased to a median of 8, and in Group 4, to 18. The recovery started on the 7th and the 5th days in Group 3 and Group 4, respectively. The highest values were reached on the 21st and 17th days. Images of the Luc+GFP+ cells applied intralesionally to mice in Groups 3 and 4 were taken during the operation and following the simultaneous luciferase enzyme given intraperitoneally with real-time in vivo imaging system demonstrated brightness proportional to stem cell density in the application region (Fig. 4).

First minute view of transgenic mouse after stem cell implantation
The stem cells transplanted in Groups 3 and 4 due to GFP content of the spines extracted were green. Goat antiluciferase antibody was used as the transfected cell indicator. The signal from the cells containing luciferase was amplified. Whether these cells transformed to neural stem cells or to neurons were investigated with Nestin and MAP, respectively. In addition, Schwann cells around myelinated fibers were examined to determine whether or not they contained luciferase.
In Group 3 (MNCs), histopathological evaluation revealed that there was no transformation of transfected cells to any nerve tissue cells in any of the 3 test subjects. In Group 4 (MSCs), there was evidence that the transfected cells expressed Nestin and neural cell markers changed to neurons in 5 subjects, and changed to Schwann cells in 3 (Figs. 5, 6 and 7). No signal of change was observed in the Group 2. The comparative results obtained by in vivo imaging, immunocytochemistry and BMS evaluation is summarized in Table 1. The functional recovery was observed both in the presence and absence of neuronal cells expressing Luc/GFP partially. Also in vivo Luc signals was not always correlated with the presence of such cells. However BMS scores correlated with in vivo signals more often.

Nestin positive (spectrum orange) (a) and luciferaz (b) positive (spectrum green) transfected origined cell may reveal neural stem cell diferantiation. The double labeling with yellow signal (c) reveals the trasfered stem cell origin of the nestin positive cell. Neural stem cell phenotype in view of the yellow cytoplasm is observed (Blue signal nuclear dye DAPI) (100× objective, Zeiss Axioskop 40)

Double labelled cell with orange-yellow color cytoplasm, which is the combination of Microtubule-associated protein (MAP2) (red) and luciferaz (green) signals accepted as an indicator of neural differentiation. Blue color reflects DAPI stained nucleus (100× objective, Zeiss Axioskop 40, with triple filter for FITC, TR, DAPI)

a and b Schwan cell signals in red color on a double stained case with anti MOSP and Luciferase. This Picture represents no diferentiation of the transfected cells to schwann cells in that case. c The schwann cells in yellow color of another case represents the double labelled signals revealing the differentiation of the transfected cells to scwann cell. (A 40× objective, B&C 100× objective, Zeiss Axioskop 40, with triple filter for FITC, TR, DAPI)
Discussion
Improved recovery was found in both MNC and MSC transplantation groups compared with the no cell-injected control group, and statistically significant differences between the control and cellular therapy groups were determined using the BMS scoring (p < 0.001) (Table 2). It has been suggested that cellular implants such as bone marrow-derived MNCs and MSCs can help to overcome some of the barriers to regeneration [6]. BMS values were analyzed using Kruskal Wallis variance test and p value was determined as <0.001. This result indicated statistically significant clinical improvement in group 3 and 4. It was remarkable that mean BMS score in group 3 was 8 while 18 in group 4. BMS scores were analyzed with Mann–Whitney U test and p value was determined as 0.002. This result indicated a better improvement with MSC than MNC.
In some previous studies, in vivo fluorescence tracking with GFP has suggested that mouse MSCs transplanted into the mouse spinal cord migrate towards the injury site within 4 weeks after transplant, and some of these cells express neuronal or astrocytic markers. MSCs have two phenotypes: nestin-positive MSCs which are able to integrate some extrinsic signals when co-cultured with neurons leading to a differentiation into astrocyte-like cells and nestin-negative MSCs which are unable to adopt a neural phenotype but remain able to differentiate into adipocytes, chondrocytes or osteocytes [7]. Nestin-positive MSCs are able to stimulate astroglial fate in striatal progenitor cultures and to repress neuronal and oligodendroglial fate through the release of diffusible factor. Functional improvement was also observed in a mouse model, either with or without expression of markers of neurons and astrocytes [8–11]. Chopp et al. found that intramedullary transplantation of MSCs 1 week after SCI improved functional outcome over a five-week period, with a few cells expressing neural markers [12]. Wu et al. also showed functional recovery after MSC transplantation at the time of injury and attributed this to enhanced differentiation of endogenous neural stem cells as indicated by in vitro coculture experiments [13]. In contrast, some authors observed functional recovery only when MSCs were transplanted in a delayed fashion after 1 week, and suggested that the strands formed by MSCs may provide axon guidance to explain the improvement rather than cellular replacement [14, 15]. Others failed to find functional recovery in rats, but agreed that MSCs were associated with preservation of host tissue including the white matter, and provided directional guidance to regenerating axons [15–17].
MSCs have an immunosuppressive role in the injured spinal cord [18, 19]. Syková [20] reported that immunosuppressive properties may combine to reduce the acute inflammatory response, and therefore, reduce cavity formation, as well as decrease astrocyte and microglia/macrophage reactivity. In addition, Vaquero et al. [17] intravenously administered rat MSCs directly into the injured rat spinal cord and reported formed bundles and reduced cavity formation in all rats. These studies have shown the immunosuppressive properties of MSCs. In addition the immunsuppressive effect of MSCs may promote axonal regeneration or may encourage functional plasticity by establishing an environment that supports axonal growth. Wright et al. [21] reported that MSCs may promote axonal regeneration or encourage functional plasticity by establishing an environment that supports axonal growth, for example, by abrogating the inhibitory influence of the glial scar. MSCs synthesize brain-derived neurotrophic factor, nerve growth factor (NGF), and vascular endothelial growth factor (VEGF), and these neurotrophic cytokines stimulate the nerve growth [22]. Dasari et al. [23] reported that MSCs reduce apoptosis within a few weeks in order to restore neurological functions.
The fluorescence image revealed that few transfected cells were differentiated to neurons according to the yellow signals obtained on MAP-2 stained sections. When viewed with direct fluorescence, native GFP expression was not detectable in the grafted cells on tissue sections. Antiluciferase antibody was thus used for signal amplification.
Nestin is a neural stem cell marker, but its expression by reactive astrocytes has been shown [24]. Thus, the neural differentiation in this study was interpreted reliably by demonstration of MAP-2 positivity on GFP+, Luc + cells.
Oligodendrocytic but not neuronal differentiation has also been observed following intravenous transplantation into a rat model of SCI. Transplanted cells survived without immunosuppression and resulted in functional recovery at 3 and 4 weeks [25]. Human MSCs transplanted into the injured spinal cord may survive long–term, and after transplantation into the injured cord, very few cells may remain 11 weeks. Persistence of the transplanted cells may not be required for functional recovery [26].
There are several limitations to the current dataset that require further investigation. To avoid a discussion about the quantity and the quality of cells implanted, pre-defined numbers of MNCs (1 × 106) and of MSCs (1 × 105) were injected in the injury sites. In addition, cells were analyzed by flow cytometry and quantitated. The best method of administering MSCs is being explored, and while direct administration results in the highest number of cells in the injury site.
Using both BLI and histological examination, we investigated the relation between the timing of transplantation and cell viability, migration, morphology, and function, by observing differences in NSPCs when transplanted at different times relative to injury. The methods in the present study can be widely applied to various research fields of regenerative medicine, including cell transplantation therapy and the study of primary cultured cells [27–31].
The interpretation of differentiation was done with the help of transfected proteins. The in vivo tracking of the transfected stem cells was done by luciferase. For this purpose, we performed double immunofluorescent technique. Antiluciferase antibody and the primary antibody against the antigens present in the final differentiation were used. The green signal demonstrated the stem cell origin of the cell whereas the red signal showed the type of final differentiated cell. The double-labelled cells were detected in yellow with the combination of green and red signals. However we cannot exclude the possibility of phagocytosis of cytoplasmic material from damaged donor marrow cells by neuronal cells at the injury site. This may explain the lack of positive correlation between functional recovery and presence of Luc/GFP positive neuronal cells. The better outcome following implantation of MSCs compared to BMNCs may result from the stromal cell behavior of MSCs, specificity not observed among adult BM cells. Thus MSCs may secret growth factors promoting endogenous neuronal regeneration. Only dynamic in vitro imaging may solve the underlying mechanism and prove if trans differentiation is occurring in this model. However, as seen in our study in vivo imaging is not the optimal but the best tool among the ones used in our experiments for detection of neuronal regeneration.
Fluorescent protein reporters such as GFP are advantageous because GFP fluorescence can be visualized directly, without further processing [2]. The cloning of GFP was the first example of a gene-based fluorescent protein reporter that is intrinsically fluorescent. A number of methods can be used to introduce the reporter gene, including pronuclear injection, retroviral- mediated gene transfer, or gene transfer in embryonic stem cells. The classic and most widely used route for the production of transgenics is through the introduction of a DNA construct linking a promoter/enhancer element to the reporter gene into zygotes by pronuclear injection [2]. The phenotype of the transplanted GFP-expressing cells was evaluated with cell type–specific antibodies within each cell lineage to show that the transgene marker is useful for tracking all cell types within a transplant. These data show that cultured cells generated from GFP-expressing mouse can be easily visualized and tracked in vivo after transplantation into the spinal cord with direct fluorescence or anti-GFP immunohistochemistry [27, 32–38].
The advantage of generating cells from GFP-expressing mice for transplantation into mouse hosts is that it is technically simpler to perform transplants in rats than in mice for studies such as spinal cord regeneration, and this allows for syngenic transplant studies without the potential concerns of donor cell rejection. We have shown that utilizing the transgenic mouse strains that constitutively express GFP as a marker for identifying transplanted cells in situ has a number of distinct advantages, including the specificity of fluorescence signal and the stable and retained expression of GFP in vivo for long-term studies. We have shown that GFP-expressing stem/progenitor cells derived from the spinal ependymal region can be detected for at least 42 days after transplantation into the spinal cord and that these differentiate into astrocytes and oligodendrocytes. However, we cannot exclude that there is partial silencing of the transgene after transplantation. We have also determined that generating cells for transplantation from the GFP-expressing W mouse strain is advantageous compared with the SD strain, because GFP expression of cells derived from the W mouse strain is readily detected by direct fluorescence both in vitro and in vivo, without the need for immunohistochemical amplification of the GFP signal. Also, GFP-expressing transplanted cells derived from the W strain can be readily identified, their migration can be tracked in situ, and their phenotype can be determined by immunostaining with cell type–specific antibodies [21, 39–41].
Conclusion
In summary, this study was able to show that intralesional implantation of MSCs may result in durable engraftment more frequently than MNCs. Both in vivo BLI and in vitro immunocytochemistry supplied proof suggesting the contribution of donor cells to functional improvement either directly by inducing donor type GFP− and Luc + neuronal differentiation or indirectly leading to improvement in BMS scores and in vivo BLI signals in the absence of such cells.
The clinical applicability of our results is still immature unless studies analyzing the optimal timing of implantation that allows preparation of autologous MSCs are completed.
References
Duda, J., Karimi, M., Negrin, R. S. (2007). Methods for ımaging cells fates in hematopoiesis. In M. Beksac (Ed.), Bone marrow and stem cell transplantation (pp. 17–34).
Leeb, C., Jurga, M., McGuckin, C., Moriggl, R., & Kenner, L. (2010). Promising new sources for pluripotent stem cells. Stem Cell Reviews and Reports, 6(1), 15–26.
Rivlin, A. S., & Tator, C. H. (1978). Effect of duration of acute spinal cord compression in a new acute cord injury model in the rat. Surgical Neurology, 10(1), 39–43.
Zurita, M., Otero, L., Aguayo, C., Bonilla, C., Ferreira, E., Parajón, A., et al. (2010). Cell therapy for spinal cord repair: optimization of biologic scaffolds for survival and neural differentiation of human bone marrow stromal cells. Cytotherapy, 12(4), 522–537.
Pittenger, M. F., Mackay, A. M., Beck, S. C., Jaiswal, R. K., Douglas, R., Mosca, J. D., et al. (1999). Multilineage potential of adult human mesenchymal stem cells. Science, 284(5411), 143–147.
Parr, A. M., Tator, C. H., & Keating, A. (2007). Bone marrow-derived mesenchymal stromal cells for the repair of central nervous system injury. Bone Marrow Transplantation, 40(7), 609–619.
Wislet-Gendebien, S., Bruyère, F., Hans, G., Leprince, P., Moonen, G., & Rogister, B. (2004). Nestinpositive mesenchymal stem cells favour the astroglial lineage in neuralprogenitors and stem cells by releasing active BMP4. BMC Neuroscience, 15, 5–33.
Lee, J., Kuroda, S., Shichinohe, H., Ikeda, J., Seki, T., Hida, K., et al. (2003). Migration and differentiation of nuclear fluorescence-labeled bone marrow stromal cells after transplantation into cerebral infarct and spinal cord injury in mice. Neuropathology, 23(3), 169–180.
Yano, S., Kuroda, S., Lee, J. B., Shichinohe, H., Seki, T., Ikeda, J., et al. (2005). In vivo fluorescence tracking of bone marrow stromal cells transplanted into a pneumatic injury model of rat spinal cord. Journal of Neurotrauma, 22(8), 907–918.
Koda, M., Okada, S., Nakayama, T., Koshizuka, S., Kamada, T., Nishio, Y., et al. (2005). Hematopoietic stem cell and marrow stromal cell for spinal cord injury in mice. Neuroreport, 16(16), 1763–1767.
Wang, Y., Mah, N., Prigione, A., Wolfrum, K., Andrade-Navarro, M. A., & Adjaye, J. (2010). A transcriptional roadmap to the ınduction of pluripotency in somatic cells. Stem Cell Reviews and Reports, 6(2), 282–296.
Chopp, M., & Li, Y. (2002). Treatment of neural injury with marrow stromal cells. Lancet Neurology, 1(2), 92–100.
Wu, S., Suzuki, Y., Ejiri, Y., Noda, T., Bai, H., Kitada, M., et al. (2003). Bone marrow stromal cells enhance differentiation of cocultured neurosphere cells and promote regeneration of injured spinal cord. Journal of Neuroscience Research, 72(3), 343–351.
Scheper, W., & Copray, S. (2009). The molecular mechanism of ınduced pluripotency: a two-stage switch. Stem Cell Reviews and Reports, 5(3), 204–223.
Moviglia, G. A., Fernandez, V. R., Brizuela, J. A., Saslavsky, J., Vrsalovic, F., Varela, G., et al. (2006). Combined protocol of cell therapy for chronic spinal cord injury. Report on the electrical and functional recovery of two patients. Cytotherapy, 8(3), 202–209.
De Haro, J., Zurita, M., Ayllon, L., & Vaquero, J. (2005). Detection of 111 ınoxine-labeled bone marrow stromal cells after intravenous or intralesional administration in chronic paraplegic rats. Neuroscience Letters, 377(1), 7–11.
Vaquero, J., Zurita, M., Oya, S., & Santos, M. (2006). Cell therapy using bone marrow stromal cells in chronic paraplegic rats: systemic or local administration? Neuroscience Letters, 398(1–2), 129–134.
Deng, W., Obrocka, M., Fischer, I., & Prockop, D. J. (2001). In vitro differentiation of human marrow stromal cells into early progenitors of neural cells by conditions that increase intracellular cyclic AMP. Biochemical and Biophysical Research Communications, 282(1), 148–152.
Di Nicola, M., Carlo-Stella, C., Magni, M., Milanesi, M., Longoni, P. D., Matteucci, P., et al. (2002). Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood, 99(10), 3838–3843.
Syková, E., & Jendlová, P. (2005). Magnetic resonance tracking of implanted adult and embryonic stem cells in injured brain and spinal cord. Annals of the New York Academy of Sciences, 1049, 146–160.
Wright, K. T., El Masri, W., Osman, A., Chowdhury, J., & Johnson, W. E. (2011). Concise review: bone marrow for the treatment of spinal cord injury: mechanisms and clinical applications. Stem Cells, 29(2), 169–178.
Hernándeza, J., Torres-Espína, A., & Navarro, X. (2011). Adult stem cell transplants for spinal cord injury repair: current state in preclinical research. Current Stem Cell Research & Therapy, 6(3), 273–287.
Dasari, V. R., Spomar, D. G., Cady, C., Gujrati, M., Rao, J. S., & Dinh, D. H. (2007). Mesenchymal stem cells from rat bone marrow downregulate caspase-3-mediated apoptotic pathway after spinal cord injury in rats. Neurochemical Research, 32(12), 2080–2093.
Frisen, J., Johansson, C. B., Torok, C., Risling, M., & Lendahl, U. (1995). Rapid, widespread, and longlasting induction of nestin contributes to the generation of glial scar tissue after CNS injury. The Journal of Cell Biology, 131(2), 453–464.
Sykova, E., Jendelova, P., Urdzikova, L., Lesný, P., & Hejcl, A. (2006). Bone marrow stem cells and polymer hydrogels-two strategies for spinal cord injury repair. Cellular and Molecular Neurobiology, 26(7–8), 1113–1129.
Himes, B. T., Neuhuber, B., Coleman, C., Kushner, R., Swanger, S. A., Kopen, G. C., et al. (2006). Recovery of function following grafting of human bone marrow-derived stromal cells into the injured spinal cord. Neurorehabilitation and Neural Repair, 20(2), 278–296.
Lois, C., Hong, E. J., Pease, S., et al. (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science, 295, 868–872.
Hakamata, Y., Tahara, K., Uchida, H., Sakuma, Y., Nakamura, M., Kume, A., et al. (2001). Green fluorescent proteintransgenic rat: a tool for organ transplantation research. Biochemical and Biophysical Research Communications, 286(4), 779–785.
Hadjantonakis, A. K., Gertsenstein, M., Ikawa, M., Okabe, M., & Nagy, A. (1998). Generating green fluorescent mice by germline transmission of green fluorescent ES cells. Mechanisms of Development, 76(1–2), 79–90.
Onifer, S. M., White, L. A., Whittemore, S. R., & Holets, V. R. (1993). In vitro labeling strategies for identifying primary neural tissue and a neuronal cell line after transplantation in the CNS. Cell Transplantation, 2(2), 131–149.
Parr, A. M., Kulbatski, I., Zahir, T., Wang, X., Yue, C., Keating, A., et al. (2008). Transplanted adult spinal cord-derived neural stem/progenitor cells promote early functional recovery after rat spinal cord injury. Neuroscience, 155(3), 760–770.
Vroemen, M., Aigner, L., Winkler, J., & Weidner, N. (2003). Adult neural progenitor cell grafts survive after acute spinal cord injury and integrate along axonal pathways. European Journal of Neuroscience, 18(4), 743–751.
Parr, A. M., Kulbatski, I., & Tator, C. H. (2007). Transplantation of adult rat spinal cord stem/progenitor cells for spinal cord injury. Journal of Neurotrauma, 24(5), 835–845.
Attar, A., Ayten, M., Ozdemir, M., Ozgencil, E., Bozkurt, M., Kaptanoglu, E., et al. (2011). An attempt to treat patients who have injured spinal cords with intralesional implantation of concentrated autologous bone marrow cells. Cytotherapy, 13(1), 54–60.
Inoue, H., Ohsawa, I., Murakami, T., Kimura, A., Hakamata, Y., Sato, Y., et al. (2005). Development of new inbred transgenic strains of rats with LacZ or GFP. Biochemical and Biophysical Research Communications, 329(1), 288–295.
Mujtaba, T., Han, S. S., Fischer, I., Sandgren, E. P., & Rao, M. S. (2002). Stable expression of the alkaline phosphatase marker gene by neural cells in culture and after transplantation into the CNS using cells derived from a transgenic rat. Experimental Neurology, 174(1), 48–57.
Prasher, D. C., Eckenrode, V. K., Ward, W. W., Prendergast, F. G., & Cormier, M. J. (1992). Primary structure of the Aequorea victoria green-fluorescent protein. Gene, 111(2), 229–233.
Ikawa, M., Tanaka, N., Kao, W. W., & Verma, I. M. (2003). Generation of transgenic mice using lentiviral vectors: a novel preclinical assessment of lentiviral vectors for gene therapy. Molecular Therapy, 8(4), 666–673.
Nakanishi, T., Kuroiwa, A., Yamada, S., Isotani, A., Yamashita, A., Tairaka, A., et al. (2002). FISH analysis of 142 EGFP transgene integration sites into the mouse genome. Genomics, 80(6), 564–574.
Tsien, R. Y. (1998). The green fluorescent protein. Annual Review of Biochemistry, 67, 509–544.
Mignone, J. L., Kukekov, V., Chiang, A. S., Steindler, D., & Enikolopov, G. (2004). Neural stem and progenitor cells in nestin-GFP transgenic mice. The Journal of Comparative Neurology, 469(3), 311–324.
Acknowledgment
This study was supported by grants from the Turkish Scientific Research Council TUBİTAK, Ankara University Research Resources (SBAG) and the Turkish Academy of Sciences.
Conflicts of Interest
The authors declare no potential conflicts of interest.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Ozdemir, M., Attar, A., Kuzu, I. et al. Stem Cell Therapy in Spinal Cord Injury: In Vivo and Postmortem Tracking of Bone Marrow Mononuclear or Mesenchymal Stem Cells. Stem Cell Rev and Rep 8, 953–962 (2012). https://doi.org/10.1007/s12015-012-9376-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-012-9376-5